

Summary
Granulomas play a key role in host protection against mycobacterial pathogens, with their breakdown contributing to exacerbated disease. To better understand the initiation and maintenance of these structures, we employed both high-resolution multiplex static imaging and intravital multiphoton microscopy of Mycobacterium bovis BCG-induced liver granulomas. We found that Kupffer cells directly capture blood-borne bacteria and subsequently nucleate formation of a nascent granuloma by recruiting both uninfected liver-resident macrophages and blood-derived monocytes. Within the mature granuloma, these myeloid cell populations form a relatively immobile cellular matrix that interacts with a highly dynamic effector T cell population. The efficient recruitment of these T cells is highly dependent on TNFα-derived signals, which also maintain the granuloma structure through preferential effects on uninfected macrophage populations. By characterizing the migration of both innate and adaptive immune cells throughout the process of granuloma development, these studies provide a new perspective on the cellular events involved in mycobacterial containment and escape.
Introduction
A protective immune response against pathogenic mycobacterial infections depends on the de novo formation of organized inflammatory lesions termed granulomas. The microenvironment that develops within the granuloma fosters interactions between innate and adaptive immune system components critical for mycobacterial containment and maintenance of an asymptomatic state. The static spatial localization of cells within mycobacteria-induced granulomas has been extensively studied by standard histologic and immunohistochemical methods, leading to speculation about the role(s) played by intra-granuloma cell distribution and interactions in effective host defense (Saunders and Britton, 2007; Ulrichs and Kaufmann, 2006). However, existing studies have not provided information on cellular dynamics within granulomas on the scale of minutes to hours, preventing an appreciation of how active cell immigration, emigration, and intra-lesional migration might contribute to the development and maintenance of these protective structures. Likewise, information is lacking on the connection between the earliest events in bacterial uptake, the processes that underlie nascent granuloma formation and maturation, and the loss of host protection that is seen in some individuals.
Infection of mice with Mycobacterium bovis Bacillus Calmette-Guérin (BCG) represents a well established model of cellular immunity and granuloma formation (Kaufmann et al., 1995). Intravenous (i.v.) infections with BCG result in a granulomatous response in the liver, with the first of these structures being clearly delineated at the histological level approximately 2 weeks post-infection (p.i.) (Khalil et al., 1975). Critical to the generation and maintenance of a protective mycobacterial granuloma is the crosstalk between antigen-specific CD4+ αβ T cells and infected macrophages, producing an effector response in the later cell type that can prevent replication and/or mediate killing of intracellular bacteria (Ladel et al., 1995a; Ladel et al., 1995b). TNFα is a primary mediator of granuloma development, with the granulomatous response being severely impaired in mice deficient in this gene product or its receptor (Algood et al., 2003; Lin et al., 2007). Importantly, blockade of TNFα signaling in animals with established BCG granulomas results in a loss in granuloma cellularity and protective function (Kindler et al., 1989), demonstrating the critical role played by TNFα-derived signals in the maintenance of these structures. The central role of TNFα seen in experimental granuloma models can explain recent observations of M. tuberculosis reactivation in some patients treated with antagonists of the TNFα pathway (Wallis et al., 2004), providing a strong clinical rationale for further studies of the interplay between this cytokine and the cells involved in granuloma-dependent anti-microbial activity.
The ability to observe directly the behavior of individual cell types during immune responses using in situ and intravital imaging techniques has revealed the highly dynamic migratory behavior of many hematopoietic cells, uncovered fundamental mechanisms regulating cellular recruitment to inflamed tissues, and permitted description of the cell-cell interactions involved in the generation of effector responses and immunological memory (Bajenoff et al., 2007; Halin et al., 2005). While early studies examined the process of cellular recruitment to inflammatory sites through blood vessels, recent experiments have focused on the behavior of immune cells extravasating out of the vasculature and entering the tissue parenchyma (Mempel et al., 2004). However, the majority of these latter studies have focused on lymphoid tissues, with only a handful examining inflammatory responses in non-lymphoid organs (Auffray et al., 2007; Boissonnas et al., 2007; Geissmann et al., 2005; Kawakami et al., 2005; Mrass et al., 2006) and still fewer looking at immune responses to pathogens (Davis et al., 2002; Frevert et al., 2005). Thus, the behavior of tissue-resident immune cells or those recruited from the blood during an inflammatory response to an infectious agent remains largely unexplored. Because the liver is amenable to intravital microscopy (IVM) (Frevert et al., 2005; Geissmann et al., 2005; McCuskey et al., 1983), the BCG infection model represents an ideal system to examine host-pathogen interactions and cellular dynamics throughout the process of mycobacterial granuloma formation.
Here we describe experiments utilizing a combination of high-resolution, multiplexed static immunofluorescence microscopy and dynamic multiphoton intravital imaging techniques to examine the initiation, development, and dissolution of mycobacterial granulomas. We find that BCG is rapidly cleared from the blood through direct capture and phagocytosis by liver resident macrophages [Kupffer cells (KCs)]. These infected phagocytes persist within the liver for at least several weeks following infection, nucleating the formation of granulomas through the attraction of uninfected KCs and blood monocytes, both of which are dependent on TNFα-derived signals for their maintenance within the structure. In contrast to the slow movement of these myeloid populations, which occurs over days to weeks, activated CD4+ T cells readily enter but do not leave the granuloma, within which they migrate rapidly in close apposition to the relatively static myeloid cell network. Together, our observations indicate that maintenance of a functional protective granuloma environment involves the TNFα-dependent maintenance of a quasi-static myeloid cell scaffold in intimate contact with a highly dynamic effector T cell population.
Results
Direct uptake and clearance of blood-borne mycobacteria by Kupffer cells
We first sought to identify the cellular components involved in the initial interactions with BCG immediately following infection. Consistent with the role of the liver as a primary site for clearance of systemic infections (Benacerraf et al., 1959), mycobacteria were rapidly depleted from the blood and became associated with perfused liver tissue within 5 minutes of i.v. infection (Figure S1). Liver-mediated clearance of blood-borne pathogens is typically dependent on KCs attached to the luminal walls of liver sinusoids, but may also involve other cell types such as granulocytes (Gregory and Wing, 2002; Knolle and Gerken, 2000). To investigate this issue, we utilized hepatic two-photon IVM to observe directly the initial interactions between host cellular components and blood-borne BCG. Imaging was performed on the surgically exposed livers of animals acutely infected through an i.v. catheter with genetically modified BCG expressing a red fluorescent protein variant (BCG-RFP). Visualization of innate immune cell components was accomplished using either of two enhanced green fluorescent protein (EGFP)-expressing mouse lines. Gene-targeted mice expressing EGFP under the control of the endogenous lysozyme M promoter (LysM-EGFP) have fluorescent populations primarily consisting of neutrophils, macrophages, and monocytes (Faust et al., 2000) while animals bearing EGFP recombined into the MHC class II I-Ab locus (MHCII-EGFP) contain fluorescent macrophages, dendritic cells (DCs), and B cells (Boes et al., 2002). As expected, in both lines KCs expressed EGFP and represented the primary fluorescent cell population within the liver sinusoids as analyzed by IVM (Figure 1) or conventional tissue immunohistology (Figure S2). Within one minute of i.v. infection, BCG became associated with the fluorescent KC population (Figure 1a and b; Movies S1 and S2). While neutrophils (identified by their high levels of EGFP expression) were occasionally seen passively flowing through the sinusoidal spaces in the LysM-EGFP mice, they did not appear to make direct contact with the mycobacteria nor did they frequently take up this organism (Movie S2). Indeed, BCG appeared in intracellular compartments at 15 minutes p.i., suggesting direct phagocytosis from the blood-stream (Figure 1c; Movie S3). Even at 3 hours p.i., when numerous neutrophils were migrating along the sinusoidal walls in response to mycobacteria-induced inflammatory signals, we were unable to observe any direct interactions between BCG and neutrophils (Movie S4), suggesting that these cells do not play a critical role in bacterial clearance by the liver during the initial stages of infection.
Figure 1. Rapid uptake of systemic BCG by Kupffer cells.
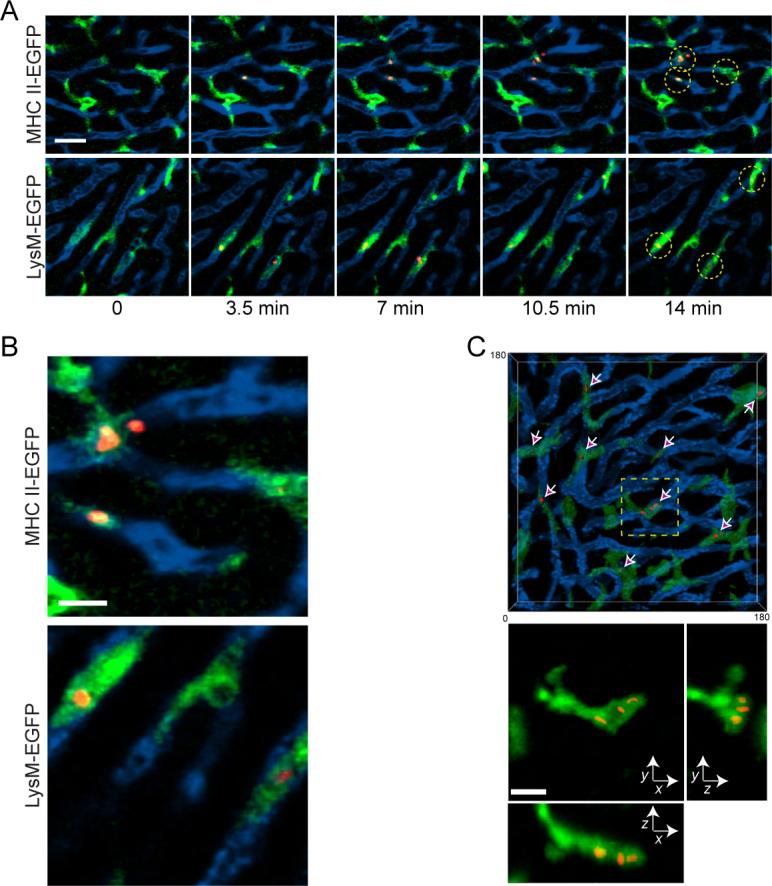
A) MHC II-EGFP (top panels) or LysM-EGFP (bottom panels) animals were subject to intravital hepatic imaging, capturing a single 2 dimensional image every 1 second. One minute after beginning image capture, 1×107 BCG-RFP bacteria were injected through an intravenous catheter. Data show intravital snapshots of the association between mycobacteria (red) and EGFP-expressing Kupffer cells (green) lining sinusoidal vessels that were demarcated by an intravenous injection of QD705 (blue). Scale bar, 25μm. See also Movies S1 and S2. B) Enlarged regions from the last time points of the image series in part A. Scale bar, 10μm. C) Volumetric surface rendering from an intravital 3D image stack of a field of Kupffer cells (green) in a LysM-EGFP mouse 15 minutes after infection with BCG-RFP bacteria (red). The animal had been injected with QD705 (blue) prior to imaging. The transparency of the Kupffer cell rendering was set to 50% to allow visualization of the intracellular BCG (arrows). Bottom panel represents maximum intensity projection images across X, Y and Z dimensions magnified from the boxed region in the top panel. Scale bar, 10μm. See also Movie S3.
Kupffer cells persist following BCG infection and redistribute into granulomas
Having demonstrated that KCs are responsible for direct uptake of mycobacteria from the blood, we then examined the fate of the KC population during granuloma formation. As there are no unique markers for KCs that enable their distinction from newly recruited monocyte/macrophage populations, we developed a system to track a small percentage of these cells during the course of BCG infection based on their relatively slow rate of turnover in bone marrow radiation chimeras (Kennedy and Abkowitz, 1997). LysM-EGFP mice were lethally irradiated and reconstituted with wild-type non-fluorescent bone marrow. Four weeks after reconstitution we were unable to detect any EGFP-expressing cells (neturophils and monocytes) in the blood of these chimeric animals, demonstrating complete engraftment by non-fluorescent donor-derived bone marrow (Figure 2a). In contrast, EGFP-expressing KCs were still readily detected in these chimeric mice (Figure 2a) and represented approximately 20% of the total F4/80+ cell population in the liver (data not shown). Thus, this system permitted us to follow the fate of a defined population of EGFP-expressing KCs during granuloma formation, because any newly recruited blood-derived monocyte/macrophage population would be non-fluorescent.
Figure 2. Kupffer cells persist and spatially redistribute during mycobacterial granuloma formation.

A-C) EGFP-expressing cell populations in the blood and liver of infected (3 weeks p.i.) and uninfected LysM-EGFP bone marrow chimeras were analyzed by flow cytometry (A) and the absolute number of F4/80+ (B) and F4/80+ EGFP+ (C) cells in the liver was quantified (3 mice/group, mean +/− SEM). Data are representative of at least 3 independent experiments. D) Representative images of liver sections from LysM-EGFP bone marrow chimeras (green) harvested at the indicated time points after infection with BCG-RFP bacteria (red) and stained for the macrophage marker F4/80 (blue). Right panels present a magnified view from the boxed region in left panels. Left panel scale bar, 50μm, right panel scale bar, 20μm. Arrows indicate EGFP+ cells that contain BCG. E) Quantification of EGFP+ cell redistribution into granulomas over time obtained from images of liver sections. The graph represents the total number of EGFP+ cells divided by the number of EGFP+ cells inside the granulomas from 842 cells at 2 weeks and 537 cells at 3 weeks (mean +/− SEM of 5 mice per time point compiled from 3 independent experiments).
Using the LysM-EGFP chimeric animals, we first examined whether BCG infection increased the rate of KC turnover, as might be expected based on evidence for elevated levels of macrophage apoptosis following BCG infection (Klingler et al., 1997; Matsunaga and Ito, 2000). LysM-EGFP chimeras were infected with BCG and 3 weeks later, a time when large numbers of granulomas are present in the liver, the number of EGFP-expressing KCs was determined using flow cytometry. As shown in Figure 2, EGFP-expressing cells were still readily detectable in the liver of infected animals, although they represented a smaller percentage of the cells than in uninfected mice. However, because the total number of macrophages in the liver increased by ∼15 fold as a result of infection (Figure 2b), the absolute number of KCs in the liver remained constant between uninfected and infected animals (Figure 2c). These data suggest that infection with BCG does not result in the death of the infected KC population followed by replacement with donor-derived, non-fluorescent macrophages. However, an alternative explanation is that BCG infection induces proliferation of the KC population that precisely compensates for a substantial degree of BCG-induced KC death. To distinguish between these possibilities, we used BrdU incorporation to determine whether infection with BCG induced proliferation of the EGFP-expressing KC population. These studies revealed no significant differences in the proliferation of EGFP-expressing KCs between control animals and infected animals during the first 2 weeks of granuloma development (Figure S3). Thus, BCG infection does not change the intrinsic turnover rate of the EGFP-expressing KC population. Given these findings, we conclude that the fluorescent cells in the liver at 3 weeks p.i. represent the same population that was present prior to infection.
We next examined the behavior of these persisting KCs during the process of granuloma formation. Livers from infected and uninfected chimeric animals were harvested at weekly intervals during the first 3 weeks following infection and analyzed using multicolor high-resolution confocal imaging to look for changes in the spatial distribution of KCs. Consistent with previous reports, distinct hepatic granulomas were not observed until 2 weeks p.i. (Pelletier et al., 1982). Between 2 and 3 weeks we observed a gradual redistribution of EGFP-expressing KCs into granulomas and a corresponding reduction in the number of these cells in the surrounding tissue (Figures 2d and e). These cells also underwent dramatic morphological changes over the course of infection, appearing in the mature lesion as large cells with numerous membrane extensions (Movie S5). The granulomas appeared to be composed of both uninfected and infected EGFP-expressing KCs, suggesting that resident macrophages present in the liver are recruited into the granuloma structures irrespective of infection status. Interestingly, the overall density of F4/80+ cells in the surrounding sinusoids did not decrease, suggesting that KCs that have migrated into granulomas can be replaced through recruitment of monocytes from the blood. These blood-derived monocytes also contribute to the cellularity of the granuloma itself, because the total number of F4/80+ cells in the liver markedly exceeds the number present in an uninfected animal and there is no large excess of such cells in the non-granulomatous hepatic parenchyma at late time points (> 2 weeks p.i.).
Preferential retention of mycobacteria-infected Kupffer cells following anti-TNFα-mediated granuloma dissolution
As noted above, TNFα plays a central role in promoting granuloma formation as well as in maintaining the cellularity and protective function of established granulomas (Kindler et al., 1989). Having shown that liver granuloma formation involves recruitment of tissue-resident and blood-derived myeloid cells to foci of initially infected KCs, we next examined the effects of anti-TNFα on the cellular composition of these granulomas. Time course experiments revealed that 2 to 4 days following anti-TNFα treatment of animals bearing established granulomas there was a profound decrease in both the number of macrophages and the size of granulomas (Figure S4). At these early time points anti-TNFα had no effect on mycobacterial burden in the liver but did increase bacterial levels two weeks following the start of treatment (data not shown). This delayed effect in altering mycobacterial burden may stem from the relatively slow growth rate characteristic of BCG.
Based on these findings, livers of LysM-EGFP chimeras bearing established granulomas were examined 4 days following the start of anti-TNFα treatment, using immunofluorescence histology. Consistent with our earlier findings, the size of liver granulomas was dramatically reduced in anti-TNFα treated mice as compared to control treated animals (Figure 3a). However, despite this reduction in cellularity, many granulomas in the anti-TNFα-treated animals continued to contain EGFP-expressing KCs (Figures 3a and b). While there was a significant reduction in the total number of EGFP-expressing cells in the liver following anti-TNFα treatment in comparison to control-treated animals, the number of BCG-infected EGFP-expressing cells was comparable between the two groups (Figure 3b). Thus, in anti-TNFα-treated animals, a higher percentage of the remaining EGFP-expressing cell population was infected with BCG (Figure 3c). These data indicate that the initial stages of anti-TNFα-mediated granuloma dissolution involve the preferential loss of uninfected macrophages (whether derived from blood monocytes or recruited uninfected KCs) and the retention of KCs that captured mycobacteria at the start of infection and nucleated granuloma formation prior to anti-TNFα treatment.
Figure 3. Preferential loss of uninfected macrophage populations following TNFα blockade.

A) Representative images of liver sections from LysM-EGFP bone marrow chimeras (green) that had been infected with BCG-RFP bacteria (white) for 3 weeks before being treated with anti-TNFα or control IgG every other day for 4 days. Sections were stained with anti-F4/80 (red). B) Quantification of liver section images showing the absolute number of EGFP+ cells and infected EGFP+ cells remaining after anti-TNFα or control IgG treatment. Graph shows the mean +/− SEM of 365 cells for the anti-TNFα group and 546 cells for the control IgG group compiled from a total of 5 mice obtained from 3 independent experiments (A). The percentage of infected cells was obtained by dividing the number of infected EGFP+ cells by the total number of EGFP+ cells (C).
Limited macrophage and mycobacterial dynamics during granuloma development
The preceding experiments examining the recruitment and redistribution of monocytes/macrophages during granuloma development and the loss of these populations during granuloma dissolution suggest a certain degree of macrophage mobility. However, whether these myeloid populations possessed the dynamic behavior and rapid motility characteristic of many hematopoietic cell populations in secondary lymphoid organs remained an open question. To examine macrophage and mycobacterial dynamics during granuloma development, LysM-EGFP mice were examined by hepatic 2P-IVM at various time points following infection with BCG-RFP. At no time following infection were we able to observe a significant degree of either uninfected or infected tissue macrophage migration or a consistent pattern of influx by blood-derived monocytes (Figure 4; Movies S6, S7, S8, and S9). In general, the immature granulomas present two weeks p.i. were composed of loosely associated LysM-EGFP-expressing cells that dynamically extended and retracted membrane processes (Figure 4b; Movie S7), while the more mature structures at three weeks p.i. contained a dense network of macrophages that showed little mobility compared with the actively migrating neutrophils and monocytes in the surrounding tissue (Figure 4c; Movies S8 and S9). To examine cell motility within the mature granuloma in greater detail, attempts were made to monitor migration of individual cells within these structures. However due to the high density of amorphously shaped, EGFP-expressing cells within a given granuloma, individual cell tracking was not possible. Instead, an image subtraction-based analysis was used to detect movement of labeled structures within the granuloma which resulted in the preferential removal of signal derived from static, but not from moving objects (Figure S7). Using this method, dynamic populations of EGFP-expressing neutrophils and monocytes were easily visualized in the sinusoidal spaces surrounding each granuloma, but the majority of fluorescence signal derived from macrophages present in the core of the granuloma was lost, indicating a lack of motility over these time scales (Figures 4d; Movie S10). Consistent with the lack of macrophage movement within lesions, individual mycobacteria in the liver also appeared relatively immobile throughout granuloma development (Movies S6, S7, and S8). We thus conclude that the macrophage aggregation process initiated by mycobacterial infection occurs over long time periods that are not readily visualized using the techniques presented in these studies and that once incorporated into the granuloma structure, myeloid cells show little net cellular displacement.
Figure 4. Limited migrational dynamics of macrophages during granuloma development.
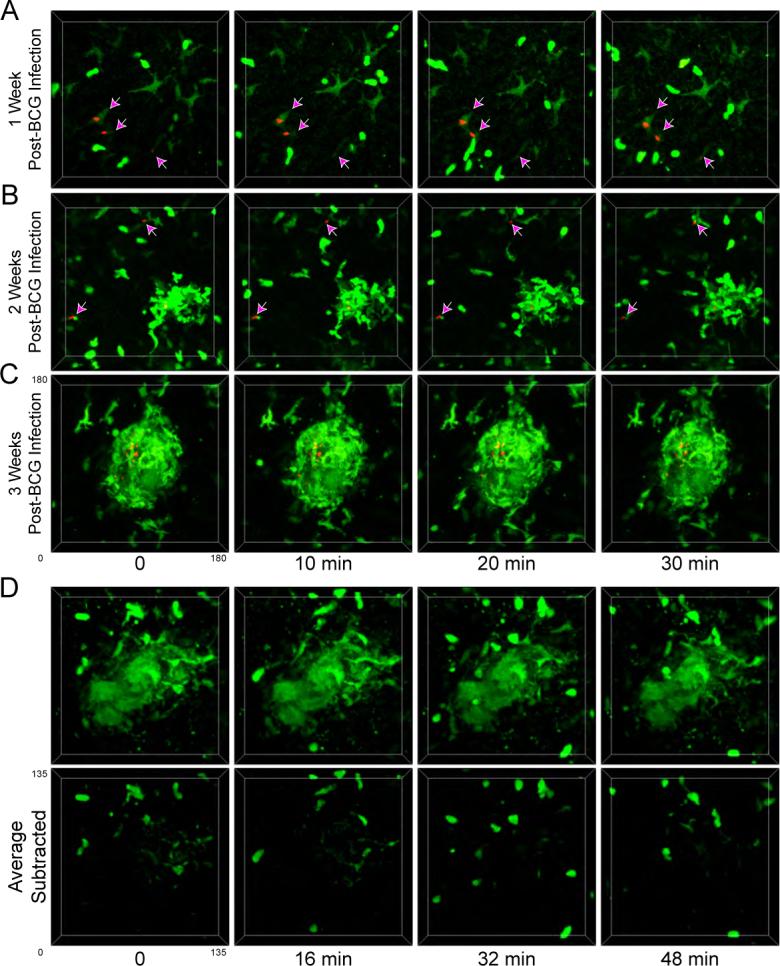
A-C) LysM-EGFP mice (green) that had been infected with BCG-RFP bacteria (red) for 1 (A), 2 (B), or 3 (C) weeks were subject to hepatic IVM. Individual time points from 4D image series are shown. Arrows in A and B point to extra-granuloma macrophages associated with mycobacteria. See also Movies S6, S7, S8, and S9. D) 4D image series of a granuloma from a LysM-EGFP mouse (green) at three weeks p.i. with BCG-RFP. The top panel shows the raw images while the bottom panel shows the same data after subtracting fluorescence intensity at each time point by a mean intensity time projection image obtained from the same data set, thus preferentially removing objects with minimal spatial displacement. See also Movie S10.
T cells display rapid motility but restricted migration inside granulomas
The granuloma microenvironment is thought to foster interactions between innate and adaptive immune system components that are critical for its protective function. Thus, we sought to understand how T cells behave within the context of the relatively static myeloid structure that was observed in the preceding experiments. To facilitate imaging and tracking of individual T cells, we reconstituted RAG-1-deficient mice with a mixture of polyclonal EGFP-expressing CD4+ T cells and non-fluorescent CD4+ T cells, CD8+ T cells, and B cells, thus allowing visualization of a fraction (∼30−60%) of the CD4+ T cell population. Animals were allowed to reconstitute fully over a period of at least 4 weeks and were subsequently infected with BCG-RFP. RAG-deficient animals are normally unable to control BCG infection, but reconstitution of the αβ T cell compartment of these animals in this manner enables them to mount a protective immune response characterized by bacterial clearance (Feng and Britton, 2000; Kaufmann et al., 1995; Ladel et al., 1995b).
Three weeks following BCG-RFP infection animals were analyzed using 2P-IVM. The location and extent of individual granulomas could be easily identified by the disruption created in the regular sinusoidal pattern, visualized using an injected fluorescent blood tracer (Figure 5a, left panel). This observation is consistent with immunofluorescence histology performed on liver sections from infected LysM-EGFP mice, which revealed that sinusoids abruptly terminate at the border of granulomas (Figure S5b). Large numbers of EGFP-expressing T cells were seen within both the confines of individual granulomas, as well as within the surrounding sinusoidal vessels (Figure 5a). Comparing the migratory dynamics of the intra- and extra-lesional T cells revealed that those within granulomas display only a slightly lower velocity but a dramatically reduced spatial displacement over time when compared with cells within the sinusoids (Figures 5b and d; Movie S11). The finding that the displacement of T cells inside of granulomas reached a plateau much sooner than sinusoidal T cells is consistent with their significantly lower confinement ratio (Figure 5c), indicating constrained migration.
Figure 5. Rapid but restricted migration of granuloma-associated T cells.
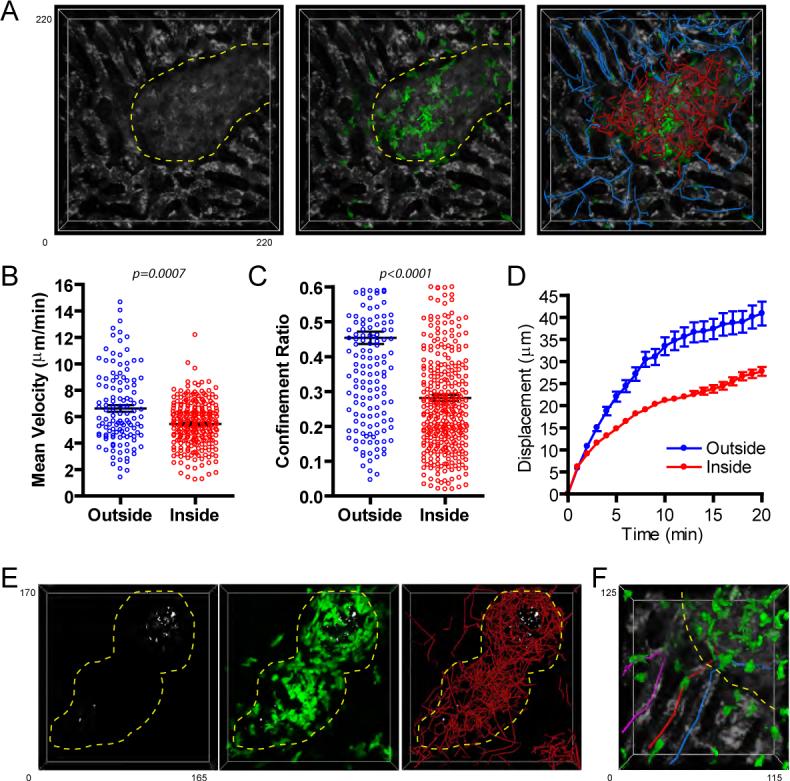
A) RAG1-deficient animals reconstituted with EGFP-expressing and non-expressing CD4+ T cells (∼30% of CD4+ T cells expressing EGFP in reconstituted animal) were infected with BCG-RFP bacteria for 3 weeks prior to performing hepatic IVM. Animals were injected with BSA-TR (white) immediately before imaging to visualize the sinusoidal network. A representative image from a 4D data set showing the disruption in the sinusoidal network caused by the granuloma lesion (left), the location of T cells within these structures (middle), and the migration paths of T cell inside (red) and outside (blue) the granuloma (right). See also Movie S11. B-D) Quantification of velocity (B), confinement ratio (total displacement/cumulative path length) (C) and displacement over time (D) for T cells located inside (red) and outside (blue) of granulomas. For B and C, data points represent individual cells compiled from 4 individual experiments. Graphs show mean +/− SEM. The p values from a Mann-Whitney test are shown. E) Granuloma-associated EGFP-expressing T cells (green) in a reconstituted RAG animal (∼60% of CD4+ T cells expressing EGFP in reconstituted animal) can be seen swarming around a core of mycobacteria (white). Right panel shows migration paths of individual T cells within the granuloma. See also Movie S12. F) Enlarged region of the granuloma from part A showing T cells entering (red track), exiting (blue track), and turning at the border (purple track) of a granuloma.
TNFα blockade had only marginal effects on T cell behavior in the granuloma. While four days following the start of anti-TNFα treatment, granuloma-associated T cells in RAG-reconstituted animals did display a decrease in their overall displacement over time when compared to control treated animals (Figure S6a and b), this likely resulted from the dramatic reduction in granuloma size. We also observed a small but significant decrease in T cell velocities after anti-TNFα treatment (Figure S6c), however whether this difference results from changes in the chemokine, cytokine, or adhesive environment that the T cells encounter or simply an increase in the density of the granuloma through which the T cell must travel is unresolved at present.
Visualizing the migration of EGFP-expressing T cells relative to the mycobacterial populations within mature granulomas revealed the swarming behavior of T cells around a central core of myeloid-cell resident BCG (Figure 5e; Movie S12), with an occasional T cell division event (Movie S13). Overall, these data reveal that T cells inside of granulomas are highly motile, but display a restricted migration pattern, tending to remain localized within the borders of the granuloma rather than rapidly exiting and re-entering the structure.
Exogenous effector T cells are rapidly recruited into and retained within granulomas
Imaging of T cell dynamics in granulomas revealed only an occasional cell entering or exiting the lesion (Figure 5f). Quantifying granuloma-associated T cell turnover by 2P-IVM of BCG-infected RAG-1-deficient animals that had been reconstituted with a mixture of polyclonal EGFP-expressing CD4+ T cells and non-fluorescent CD8+ T cells and B cells (100% of CD4+ T cells expressing EGFP) revealed that on average T cells enter into and exit from a granuloma at a rate of 5.7±2.1 and 5.6±1.6 (mean ±SD) cells per hour, respectively. Because of their extremely high density and rapid rate of motility, it was difficult to obtain accurate counts of granuloma-associated T cells in these animals; however rough estimates suggest that these entry and exit rates correspond to less than 7% of the total T cell population being replaced per hour.
These relatively low rates of T cell entry and egress from the granuloma could indicate that a mature lesion represents a relatively closed structure, with physical and/or chemical barriers that restrict free movement of lymphoid cells into and out of the lesion. Alternatively, among the T cells traversing the liver, only a small fraction may posses the molecular profile (chemokine receptors, integrins, selectins, etc.) needed for granuloma entry and once inside the granuloma these cells could be retained in the structure based on adhesive or chemotactic mechanisms. To differentiate between these possibilities, we developed a system to examine the permeability of granulomas based on their ability to recruit exogenous effector T cells acutely.
2P-IVM was performed on the livers of granuloma-bearing mice that had been infected 3 weeks earlier with BCG-RFP. During the imaging session, pre-activated, CMTPX-labeled OT-II TCR transgenic T cells on a RAG1-deficient background (OTII-RAG) were adoptively transferred into the recipient animal through an i.v. catheter. As T cells can localize to granulomas in the absence of antigen recognition (Co et al., 2006), we hypothesized that these OTII-RAG cells would serve as a source of T cells with the potential to enter the granuloma. Using a fluorescent blood tracer to delineate the borders of individual granulomas, we observed a rapid recruitment of activated T cells into these structures beginning 20 minutes post-T cell transfer, with increasing numbers of cells accumulating within the granuloma over the next 2 hours (Figures 6a and b; Movie S14). The relative ease with which T cells entered the granuloma is consistent with immunofluorescence staining of fixed liver sections for collagen IV, which failed to reveal a defined extracellular matrix barrier on the periphery of the granuloma that might block entry into these structures. Rather, much of the collagen structure within the granuloma appeared to come from the remnants of sinusoidal vessels that had been invaded by macrophages during granuloma development (Figure S5a).
Figure 6. T cells are rapidly recruited to and retained within granuloma structures.
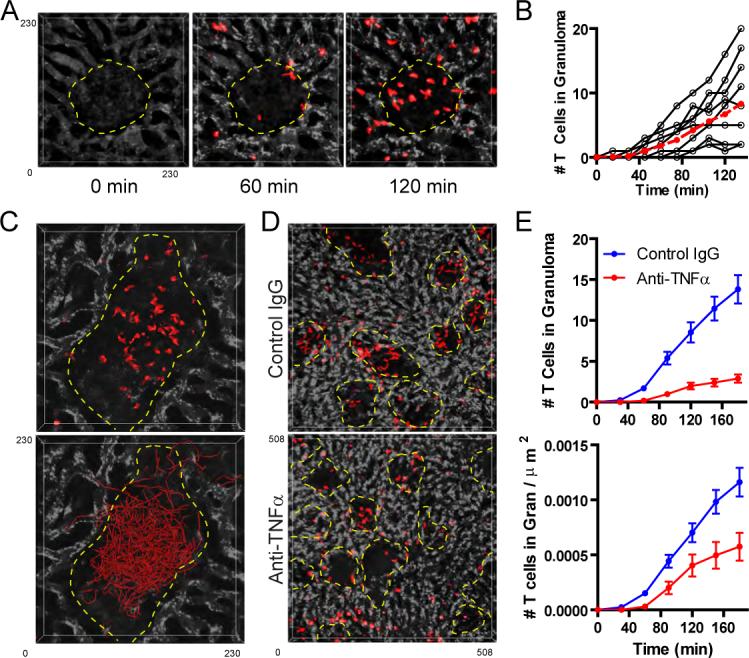
A) Mice that had been infected with BCG-RFP bacteria 3 weeks earlier were injected with BSA-647 (white) to visualize the sinusoidal network and subjected to hepatic IVM. Fifteen minutes after the start of the imaging session, CMTPX-labeled in vitro stimulated OT-II T cells (red) were injected i.v. through a catheter to look at T cell entry into the granulomas. See also Movie S14. B) Quantification of T cell recruitment over time into 10 granulomas (black lines=individual granulomas, red line= mean values). Data were compiled from 3 individual experiments. C) CMTPX-labeled in vitro stimulated OT-II T cells (red) were transferred into mice that had been infected with BCG-RFP bacteria 3 weeks earlier. 12 hours later the animals were injected with BSA-647 (white) to visualize the sinusoidal network and the migration of T cells within individual granulomas was visualized using hepatic IVM. Tracks of individual granuloma-associated T cells are shown (red). See also Movie S15. D) Mice that had been infected with BCG-RFP bacteria for 3 weeks were treated with control IgG or anti-TNFα for 4 days and subsequently subjected to hepatic IVM. Five minutes after the start of imaging, CMTPX-labeled in vitro stimulated OT-II T cells (red) were injected i.v. through a catheter to examine acute T cell entry into the granulomas. Images represent a single time point from a 4D image series captured after 3 hours of continuous imaging. E) Quantification of both the average rate of T cell recruitment into granulomas over time (top panel) and the average rate of recruitment adjusted for granuloma size (bottom panel) (mean +/− SEM from 45 control IgG-treated and 39 anti-TNFα-treated granulomas compiled from 2 independent experiments).
The extent of T cell recruitment varied widely between individual granulomas (Figure 6b), suggesting heterogeneity in either the vascular delivery of T cells to a granuloma or in the ability of an individual granuloma to attract T cells. In either case, examining animals that had been given dye-labeled T cells 12−16 hours prior to imaging revealed that many granulomas contained large numbers of T cells exhibiting active but spatially restricted migration (Figure 6c; Movie S15). These data demonstrate that over relatively short periods of time effector T cells can freely enter, but not exit, the mature granuloma.
We also examined the effect of blocking TNFα-derived signals on the efficiency of effector T cell recruitment into granulomas. Animals bearing established granulomas were treated with anti-TNFα or control IgG for 4 days and subsequently transferred with in vitro stimulated, dye-labeled effector T cells. Monitoring of the acute recruitment of T cells into granulomas was performed using 2P IVM (Figure 6d). Quantification of this recruitment revealed an impaired ability of granulomas to efficiently recruit T cells following TNFα blockade, which was only partially the result of the smaller granuloma size in the TNFα-treated animals. These data suggest that the activation status of granulomas is a critical factor in their ability to attract effector T cells.
Restricted migration of granuloma-associated T cells is defined by a macrophage-delineated border
The preceding experiments revealed the disparate dynamic behavior between granuloma-associated macrophages and T cells, with the former being largely sessile and the latter actively migratory. In lymph nodes, we have reported that rapidly migrating lymphocytes preferentially move along fixed stromal cell networks (Bajenoff et al., 2006), which likely involves the chemokinetic stimulation of the T cells through stromal cell-associated chemokines such as CCL21 (Asperti-Boursin et al., 2007; Okada and Cyster, 2007; Worbs et al., 2007). Given these prior observations and the possible parallel of these lymph node findings to the results reported here for lymphocyte vs. macrophage elements in granulomas, we focused on the physical relationship between these two populations. In vitro stimulated, dye-labeled OTII-RAG T cells were transferred into LysM-EGFP mice bearing established granulomas and fixed liver sections from these animals were examined by immunofluorescence static imaging. We observed both endogenous and exogenous CD4+ T cell populations tightly packed between macrophage cell junctions in the granuloma interior (Figure 7a). To characterize the dynamics of these transferred T cells relative to the dense macrophage network, dye-labeled effector T cells were again transferred into granuloma-bearing LysM-EGFP mice and 2P-IVM was performed 12−16 hours later. These data revealed that as a T cell migrates through a granuloma it actively displaces macrophage membrane processes so it can pass through these very closely juxtaposed cells (Movie S16). Strikingly, granuloma-associated T cell migration also appeared to be strictly limited by the myeloid cell-defined border (Figures 7b and 7c; Movie S17). In some granulomas, macrophages could be seen extending out from the periphery of the structure. In these cases, T cells were often observed migrating along these cellular projections (Figure 7d and Movies S18 and S19). Interestingly, in vitro generated effector T cells that were transferred into LysM-EGFP animals bearing immature granulomas (12 days post-infection) were also recruited to these nascent structures and actively migrated in close apposition to the macrophage aggregates (Movie S20). Taken together, these data suggest that direct interactions between T cells and macrophages may play a critical role in defining the limits of T cell migration once these lymphocytes have entered the granuloma, and raise the intriguing possibility that T cell movement is actively promoted by signals physically associated with this underlying myeloid network.
Figure 7. T cell migration within granulomas is defined by a macrophage-delineated border.

A) CMTPX-labeled in vitro stimulated OT-II T cells (red) were transferred into LysM-EGFP mice (green) that had been infected with BCG-RFP bacteria 3 weeks earlier. Twelve hours later, livers were fixed, sectioned and stained with an antibody to CD4 (white) and a nuclear dye (blue). Scale Bar, 15μm B) CMTPX-labeled in vitro stimulated OT-II T cells (red) were transferred into LysM-EGFP mice (green) that had been infected with BCG-RFP bacteria 3 weeks earlier. Twelve hours later, BSA-647 (white) was injected into the animals to visualize the sinusoidal network and hepatic IVM was performed. A single time point from a 4D data set showing T cells in relation to sinusoidal network (left panel), T cells in relation to macrophages (middle panel), and T cell migration paths (blue) in relation to macrophages (right panel). See also Movie S17. C) XY (top panel) and XZ (bottom panel) projection images from the upper right quadrant of the granuloma from part A showing T cell paths (blue) in relation to macrophages (green). D) Intravital snapshots of a T cell migrating along macrophage processes that extend from the granuloma. See also Movies S18 and S19.
Discussion
The development of a mycobacterial granuloma involves the spatial (re)organization of diverse cell types within the infected tissue, culminating in the generation of an inflammatory lesion that is the primary effector site for the host anti-pathogen response. The structural architecture of the granuloma is presumably critical for its protective function, both by creating physical barriers to prevent bacterial dissemination as well as by facilitating interactions between key cell types such as antigen-specific T cells and macrophages. While static histological observations and studies of gene-targeted mice have provided substantial insight into the mechanisms involved in granuloma formation and host defense, fundamental questions remain regarding the behavior of individual cell types at various stages of the mycobacterial response and the corresponding impact of these behaviors on granuloma function.
The data presented here reveal the mycobacterial granuloma to be a highly dynamic structure containing cell types with unique migrational behaviors. Within the liver, the primary target organ after systemic mycobacterial infection, clearance of the pathogen is mediated through direct capture and phagocytosis of blood-borne bacteria by KCs. Both infected and uninfected KCs appear to persist throughout the subsequent process of granuloma development, with the uninfected KCs gradually redistributing from the surrounding tissue into individual granulomas that contain the initially infected, surviving KCs. These two KC populations and additional myeloid cells derived from the blood all undergo dramatic morphological changes over time, from round or stellate cells to large, tightly packed epithelioid forms that presumably correspond to the large giant cells seen in standard histological preparations. Given the long-term persistence of the initially infected macrophage population, their central location in the mature structure, and the selective persistence of these infected KCs during granuloma dissolution in the presence of anti-TNFα, it appears that these infected cells nucleate granuloma formation, perhaps by serving as an early source of chemoattractant that facilitates initial macrophage agglomeration (Flynn and Chan, 2001; Peters and Ernst, 2003). These data also suggest that uninfected macrophages have a unique requirement for TNFα-derived signals for survival and/or retention within the granuloma structure that is not shared by infected macrophages. Overall, these data demonstrate that the mycobacterial granuloma is comprised of a diverse set of recruited monocytes/macrophages and infected and uninfected tissue resident macrophages, each of which may play a unique role in mycobacterial containment.
Examination of the macrophage population in mature granulomas by intravital microscopy revealed dynamic motion of membrane processes, but little net cellular displacement. These data are consistent with a previous study that examined the dynamics of macrophages after infection of zebrafish embryos with M. marinum (Davis et al., 2002). However, in contrast to our observations, this earlier report also demonstrated significant macrophage migration outside of granulomas at various stages of the granulomatous response. Indeed, using an M. marimum super-infection model in adult frogs and zebrafish, a more recent report by this same group demonstrated that newly infected macrophages can home to established granulomas as early as 3 days following re-infection (Cosma et al., 2004). While our histological studies using the LysM-EGFP chimeric animals clearly suggest migration of motile macrophage populations into granulomas, the time frame of this process (>1 week) may be too slow to detect by intravital imaging techniques limited to data collection of <3 hrs. Indeed, macrophage aggregation begins in zebrafish embryos within 3 days p.i., suggesting that the entire process of macrophage accretion may be greatly accelerated in this species. Alternatively, intrinsic differences in the pathogenicity of the infecting organisms, the tissues in which granuloma formation was analyzed, and/or the specific monocyte/macrophage populations recruited during infection may account for these differences. Interestingly, a recent report has demonstrated rapid migration of murine microglial populations in response to brain injury (Davalos et al., 2005), suggesting that the degree of macrophage motility in mammals may be at least partially tissue and/or stimulus dependent.
The limited dynamics exhibited by macrophages during the mycobacterial response was in direct contrast to the rapid motility characteristic of granuloma-associated T cells. A particularly striking observation was that despite this dynamic behavior, these T cells were still efficiently retained in the granuloma. What mechanisms might function to restrict the ability of T cells to leave the lesion? One possibility is a physical barrier at the surface of the structure. Findings from several studies have revealed that recently activated antigen non-specific T cells can enter established granulomas (Co et al., 2006; Hogan et al., 2007; Sewell et al., 2003), but given that in these previous studies T cells had the opportunity to enter the lesion over a relatively long time period (1 or more weeks), the permeability of the granuloma border was unclear. We found that T cells can enter granulomas within minutes of localizing to their borders and subsequently accumulate within these structures, displaying rapid motility, highly restricted migration, and limited egress into the surrounding sinusoidal space. While our data do not rule out the existence of a passive physical barrier that would act as a one-way valve, allowing T cells to freely enter the granuloma but retarding their subsequent egress, we feel this is unlikely and favor the hypothesis that retention of migrating T cells within the granuloma results from interactions between T cells and the granuloma macrophage network. This same macrophage network may also be important for sustaining T cell motility within the granuloma. These ideas are consistent with our data showing that the extent of T cell migration within the granuloma appears to be restricted to a macrophage-delineated border and that granuloma-associated migrating T cells must constantly squeeze through narrow cell junctions created by a dense macrophage network. Upon entering the granuloma, T cells could become attached to the macrophage network through specific adhesive interactions between proteins expressed on the two cell types (van den Berg et al., 1992) and/or immobilized chemokines that are recognized by T cells and presented on the surface of macrophages, perhaps through interactions with plasma membrane-bound gylcosaminoglycans (Slimani et al., 2003). Interestingly, production of T cell-specific chemokines by macrophages is thought to be dependent on TNFα-derived signals (Algood et al., 2003), which may explain our data showing that granulomas in animals treated with anti-TNFα for short periods of time are less efficient at recruiting T cell from the surrounding tissue. Our observations that granuloma-associated T cells migrate in close apposition to and within the confines of a macrophage scaffold are reminiscent of our recent findings that naïve T cells specifically migrate along a stromal cell network that defines the paracortical region of the lymph node (Bajenoff et al., 2006). Thus, an intriguing possibility is that T cells in different activation states and in different tissue microenvironments utilize related mechanisms for both locomotion and to define spatial territories.
The data presented here provide insight into how dynamic cellular processes in both innate and adaptive immune system components contribute to processes of granuloma formation, maintenance, and dissolution. In particular, our findings emphasize that a protective granuloma environment involves the TNFα-dependent maintenance of a static myeloid cell scaffold that supports the migration of a highly motile population of effector T cells. The use of intravital imaging techniques such as those employed here, coupled with genetic or chemical disruption of key candidate signaling molecules involved in cell migration and effector activity, should provide a greater understanding of the specific pathways utilized to regulate cell behavior within the granuloma and thereby help to reveal fundamental mechanisms of mycobacterial control.
Experimental Procedures
Mice and Animal Procedures
C57BL/6 LysM-EGFP knock-in mice (Faust et al., 2000) were a gift from Stephan Graf (Albert Einstein University, NY). C57BL/6 IAb-EGFP knock-in mice (Boes et al., 2002) were a gift from Hidde Ploegh (Harvard University, MA). C57BL/6 mice and transgenic mice expressing EGFP under the control of the human ubiquitin promoter (UbEGFP) (Schaefer et al., 2001) were obtained from Jackson Laboratories. C57BL/6 RAG1-deficient (Mombaerts et al., 1992) and C57BL/6 CD45.2 congenic mice were obtained from Taconic Laboratories through a special contract with the NIAID. OTII TCR transgenic (Barnden et al., 1998) RAG1-deficient mice were obtained from Taconic Laboratories through the NIAID exchange program.
For the generation of bone marrow chimeras, C57BL/6 LysM-EGFP mice were γ-irradiated with two doses of 500 rads from a cesium source and subsequently reconstituted with 2×106 C57BL/6 CD45.2 congenic bone marrow cells. For anti-TNFα-induced granuloma dissolution, BCG-infected animals were injected i.p. with 1mg of anti-TNFα (XT22.11) or control anti-β-galactosidase antibody (GL113) every other day until the time of analysis. For RAG reconstitutions, CD4+ T cells were purified from the LNs of UbEGFP mice using a MACs negative selection kit (Miltenyi Biotech). Biotinylated anti-NK1.1 (PK136) (BD Biosciences) was added to the negative depletion cocktail to remove NK and NKT cells. EGFP-expressing CD4+ T cells were mixed with unpurified B6 lymphocytes and injected i.v. into RAG1-deficient recipient animals to obtain animals with a fraction of fluorescent CD4+ T cells. Animals were allowed to reconstitute for a minimum of 4 weeks prior to infecting with BCG. All animal procedures used in this study were approved by the Animal Care and Use Committee, NIAID, NIH.
Bacterial Cultures, Infections and Quantification
M. bovis BCG strain Pasteur expressing the red fluorescent protein dsRed (BCG-RFP) has been previously described (Abadie et al., 2005). BCG-RFP or non-recombinant M. bovis BCG strain Pasteur were expanded to log phase in Middlebrook 7H9 liquid medium supplemented with ADC (Difco), washed, aliquoted in PBS and stored at −80°C until further use. BCG-RFP bacteria were grown in the presence of Hygromycin (Sigma). Quantification of bacterial stocks for infection and bacterial loads in organs was performed by culture on 7H11 agar supplemented with OADC (Difco). For mycobacterial infections, animals were inoculated i.v. with 2×106 BCG, unless otherwise specified.
In Vitro T Cell Stimulation and Adoptive Transfers
OTII-RAG-deficient TCR transgenic LN cells were stimulated with 2μM ovalbumin 323−339 peptide plus mitomycin C-treated, T cell depleted C57BL/6 splenocytes. Two days following stimulation, cells were expanded in the presence of 30 U/ml recombinant human IL-2 (R&D systems) and used for experiments 7 days post-activation. In vitro generated effector T cells were labeled with 2μM CMTPX or 2μM TAMRA-SE (Invitrogen) as described (Qi et al., 2006). 1×107 cells were injected i.v. into animals bearing established granulomas either before or during imaging, as indicated.
Immunofluorescence Microscopy
Livers were fixed by portal vein perfusion with 4% paraformaldehyde/PBS, excised from the animals, and fixed for an additional 12 hours followed by dehydration in 30% sucrose/PBS prior to embedding in OCT freezing media. 16μm sections were cut on a CM3050s cryostat (Leica) and adhered to Superfrost Plus slides (VWR). Sections were permeablized and blocked in PBS containing 0.3% Triton X-100 (Sigma) and 10% goat serum (Jackson Immunoresearch) followed by staining with AlexaFluor 647-conjugated anti-CD3 (clone 17A2), unconjugated anti-MHC class II (clone 2G9) (all from BD Biosciences), AlexaFluor 647-conjugated anti-F4/80 (clone BM8) (Caltag), rabbit polyclonal anti-LYVE-1 (Novus Biologicals), rabbit polyclonal anti-Collagen IV (Abcam). Unconjugated primary antibodies were stained with AlexaFluor conjugated secondary antibodies (Invitrogen). Stained slides were mounted with Prolong Gold or Prolong Gold plus DAPI (Invitrogen) and 3D image stacks were acquired on a LSM 510 confocal microscope (Carl Zeiss Microimaging). Images are displayed as 2D maximum intensity projections.
Two Photon Intravital Hepatic Imaging and Image Analysis
Mice were anesthetized using isoflurane (Baxter) as previously described (Qi et al., 2006), the abdomen of the animal was shaved, and a midline and subcostal incision was made through the dermis. A high temperature cautery was used to open the peritoneum. The falciform ligament was resected and the liver gently exteriorized onto a glass coverslip attached to a custom made steel imaging platform. The liver was covered with plastic wrap, the incision was packed with moist gauze and the imaging platform was sealed to prevent dehydration. In some experiments, a catheter was placed into the tail vein using a 30GA needle attached to PE-10 tubing (Becton Dickinson) and secured using Durapore Tape (Fischer Scientific). Liver sinusoids were visualized by injecting Texas Red-conjugated bovine serum albumin (BSA-TR), AlexaFluor-647-conjugated BSA (BSA-647) or non-targeted Quantum Dots 705 (QD705) (Invitrogen) i.v. immediately prior to imaging. Approximate molecular weights for BSA conjugates and QD705 are 66kDa and 1500kDa, respectively.
Images were acquired using an inverted LSM 510 NLO multiphoton microscope (Carl Zeiss Microimaging) enclosed in a custom-built environmental chamber that was maintained at 35°C using heated air. This system had been custom fitted with 3 external non-descanned PMT detectors in the reflected light path. Images were acquired using either a 20×/0.8 NA air objective or a 40×/1.2 NA water immersion objective. Fluorescence excitation was provided by a Chamelon XR Ti:Sapphire laser (Coherent). EGFP and dsRed were excited at 920nm while CFSE and CMTPX were excited at 820nm. Typical voxel dimensions were 0.9 × 0.9 × 2−3μm using the 20× objective and 0.45 × 0.45 × 1−2μm using the 40× objective. For 4-dimensional (4D) data sets, 3-dimensional stacks were captures every 1 minute, unless otherwise specified. For images displayed as volumetric perspective renderings, the dimensions of the imaging frame in μm are shown in the figure.
Raw imaging data were processed with Imaris (Biplane) with a Gaussian filter used for noise reduction. Automatic 3D object tracking using Imaris Spots was aided with manual corrections to retrieve cell spatial coordinates over time. These data were imported into MatLab (MathWorks) for calculating cell velocities, displacements, and confinement ratios (total displacement/cumulative path length). For calculating T cell recruitment into granulomas over time, the number of T cell inside individual granulomas was counted at multiple time points throughout 4D image series. Area normalized data was obtained by dividing the number of T cells inside a granuloma at a given time point by the 2D area of the granuloma in the XY dimension. For dynamic display of imaging data sets, image sequences exported from Imaris were composed in AfterEffects (Adobe) to produce video clips.
Intrahepatic Mononuclear Cell (IHMC) and Peripheral Blood Leukocyte (PBL) Isolation and Analysis
Kupffer cells were quantified by isolating IHMC as previously described (Berenzon et al., 2003), with slight modification. Briefly, livers were perfused through the portal vein with 0.5mg/ml of Liberase CI (Roche) and 0.05% DNase I (Bohreinger Manheim) in PBS containing calcium and magnesium and subsequently excised, digested for an additional 45 minutes at 37°C, and passed through a 100μm cell strainer (Falcon). Cell suspensions were washed, resuspended in a 33% Percoll solution (Amersham Biosciences) and centrifuged at 800g for 20 minutes. Cells were analyzed by flow cytometry. Peripheral blood was collected from mice through a cardiac puncture into EDTA containing tubes. Erythrocytes were lysed using ACK lysis buffer (Biowhittaker), cells were washed and analyzed by flow cytometry. Blood and liver derived cell populations were stained with anti-GR1-PE (BD Bioscience) and anti-F4/80-Alexa 647 (Caltag). Dead cells were gated out of the analysis using a 7AAD (BD Bioscience) counter-stain. Flow cytometric data were collected on a FACSCalibur or an LSR II (BD Biosciences) and analyzed with FlowJo software (TreeStar).
Supplementary Material
Acknowledgements
We are grateful to Hai Qi, Marc Bajénoff, Jennifer Ziskin, Katrin Mayer, and Matyas Sandor for helpful discussion and/or comments on the manuscript and Sandy White, Ina Ifrim, and Austin Rinker for research assistance. This research was supported by the Intramural Research Program of NIAID, NIH, DHHS. The authors declare that they have no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abadie V, Badell E, Douillard P, Ensergueix D, Leenen PJ, Tanguy M, Fiette L, Saeland S, Gicquel B, Winter N. Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood. 2005;106:1843–1850. doi: 10.1182/blood-2005-03-1281. [DOI] [PubMed] [Google Scholar]
- Algood HM, Chan J, Flynn JL. Chemokines and tuberculosis. Cytokine Growth Factor Rev. 2003;14:467–477. doi: 10.1016/s1359-6101(03)00054-6. [DOI] [PubMed] [Google Scholar]
- Asperti-Boursin F, Real E, Bismuth G, Trautmann A, Donnadieu E. CCR7 ligands control basal T cell motility within lymph node slices in a phosphoinositide 3-kinase-independent manner. J Exp Med. 2007;204:1167–1179. doi: 10.1084/jem.20062079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- Bajenoff M, Egen JG, Koo LY, Laugier JP, Brau F, Glaichenhaus N, Germain RN. Stromal cell networks regulate lymphocyte entry, migration, and territoriality in lymph nodes. Immunity. 2006;25:989–1001. doi: 10.1016/j.immuni.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenoff M, Egen JG, Qi H, Huang AY, Castellino F, Germain RN. Highways, byways and breadcrumbs: directing lymphocyte traffic in the lymph node. Trends in immunology. 2007;0:0. doi: 10.1016/j.it.2007.06.005. [DOI] [PubMed] [Google Scholar]
- Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- Benacerraf B, Sebestyen MM, Schlossman S. A quantitative study of the kinetics of blood clearance of P32-labelled Escherichia coli and Staphylococci by the reticuloendothelial system. J Exp Med. 1959;110:27–48. doi: 10.1084/jem.110.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenzon D, Schwenk RJ, Letellier L, Guebre-Xabier M, Williams J, Krzych U. Protracted protection to Plasmodium berghei malaria is linked to functionally and phenotypically heterogeneous liver memory CD8+ T cells. J Immunol. 2003;171:2024–2034. doi: 10.4049/jimmunol.171.4.2024. [DOI] [PubMed] [Google Scholar]
- Boes M, Cerny J, Massol R, Op den Brouw M, Kirchhausen T, Chen J, Ploegh HL. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature. 2002;418:983–988. doi: 10.1038/nature01004. [DOI] [PubMed] [Google Scholar]
- Boissonnas A, Fetler L, Zeelenberg IS, Hugues S, Amigorena S. In vivo imaging of cytotoxic T cell infiltration and elimination of a solid tumor. J Exp Med. 2007;204:345–356. doi: 10.1084/jem.20061890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Co DO, Hogan LH, Karman J, Heninger E, Vang S, Wells K, Kawaoka Y, Sandor M. Interactions between T cells responding to concurrent mycobacterial and influenza infections. J Immunol. 2006;177:8456–8465. doi: 10.4049/jimmunol.177.12.8456. [DOI] [PubMed] [Google Scholar]
- Cosma CL, Humbert O, Ramakrishnan L. Superinfecting mycobacteria home to established tuberculous granulomas. Nat Immunol. 2004;5:828–835. doi: 10.1038/ni1091. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- Davis JM, Clay H, Lewis JL, Ghori N, Herbomel P, Ramakrishnan L. Real-Time Visualization of Mycobacterium-Macrophage Interactions Leading to Initiation of Granuloma Formation in Zebrafish Embryos. 2002;17:693–702. doi: 10.1016/s1074-7613(02)00475-2. [DOI] [PubMed] [Google Scholar]
- Faust N, Varas F, Kelly LM, Heck S, Graf T. Insertion of enhanced green fluorescent protein into the lysozyme gene creates mice with green fluorescent granulocytes and macrophages. Blood. 2000;96:719–726. [PubMed] [Google Scholar]
- Feng CG, Britton WJ. CD4+ and CD8+ T cells mediate adoptive immunity to aerosol infection of Mycobacterium bovis bacillus Calmette-Guerin. J Infect Dis. 2000;181:1846–1849. doi: 10.1086/315466. [DOI] [PubMed] [Google Scholar]
- Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- Frevert U, Engelmann S, Zougbede S, Stange J, Ng B, Matuschewski K, Liebes L, Yee H. Intravital observation of Plasmodium berghei sporozoite infection of the liver. PLoS Biol. 2005;3:e192. doi: 10.1371/journal.pbio.0030192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Cameron TO, Sidobre S, Manlongat N, Kronenberg M, Briskin MJ, Dustin ML, Littman DR. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3:e113. doi: 10.1371/journal.pbio.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory SH, Wing EJ. Neutrophil-Kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J Leukoc Biol. 2002;72:239–248. [PubMed] [Google Scholar]
- Halin C, Rodrigo Mora J, Sumen C, von Andrian UH. In vivo imaging of lymphocyte trafficking. Annu Rev Cell Dev Biol. 2005;21:581–603. doi: 10.1146/annurev.cellbio.21.122303.133159. [DOI] [PubMed] [Google Scholar]
- Hogan LH, Co DO, Karman J, Heninger E, Suresh M, Sandor M. Virally activated CD8 T cells home to Mycobacterium bovis BCG-induced granulomas but enhance antimycobacterial protection only in immunodeficient mice. Infect Immun. 2007;75:1154–1166. doi: 10.1128/IAI.00943-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann SH, Ladel CH, Flesch IE. T cells and cytokines in intracellular bacterial infections: experiences with Mycobacterium bovis BCG. Ciba Found Symp. 1995;195:123–132. doi: 10.1002/9780470514849.ch9. discussion 132−126. [DOI] [PubMed] [Google Scholar]
- Kawakami N, Nagerl UV, Odoardi F, Bonhoeffer T, Wekerle H, Flugel A. Live imaging of effector cell trafficking and autoantigen recognition within the unfolding autoimmune encephalomyelitis lesion. J Exp Med. 2005;201:1805–1814. doi: 10.1084/jem.20050011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy DW, Abkowitz JL. Kinetics of central nervous system microglial and macrophage engraftment: analysis using a transgenic bone marrow transplantation model. Blood. 1997;90:986–993. [PubMed] [Google Scholar]
- Khalil A, Bourut C, Halle-Pannenko O, Mathe G, Rappaport H. Histologic reactions of the thymus, spleen, liver and lymph nodes to intravenous and subcutaneous BCG injections. Biomedicine. 1975;22:112–121. [PubMed] [Google Scholar]
- Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell. 1989;56:731–740. doi: 10.1016/0092-8674(89)90676-4. [DOI] [PubMed] [Google Scholar]
- Klingler K, Tchou-Wong KM, Brandli O, Aston C, Kim R, Chi C, Rom WN. Effects of mycobacteria on regulation of apoptosis in mononuclear phagocytes. Infect Immun. 1997;65:5272–5278. doi: 10.1128/iai.65.12.5272-5278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol Rev. 2000;174:21–34. doi: 10.1034/j.1600-0528.2002.017408.x. [DOI] [PubMed] [Google Scholar]
- Ladel CH, Daugelat S, Kaufmann SH. Immune response to Mycobacterium bovis bacille Calmette Guerin infection in major histocompatibility complex class I- and II-deficient knock-out mice: contribution of CD4 and CD8 T cells to acquired resistance. Eur J Immunol. 1995a;25:377–384. doi: 10.1002/eji.1830250211. [DOI] [PubMed] [Google Scholar]
- Ladel CH, Hess J, Daugelat S, Mombaerts P, Tonegawa S, Kaufmann SH. Contribution of alpha/beta and gamma/delta T lymphocytes to immunity against Mycobacterium bovis bacillus Calmette Guerin: studies with T cell receptor-deficient mutant mice. Eur J Immunol. 1995b;25:838–846. doi: 10.1002/eji.1830250331. [DOI] [PubMed] [Google Scholar]
- Lin PL, Plessner HL, Voitenok NN, Flynn JL. Tumor necrosis factor and tuberculosis.. The journal of investigative dermatology; Symposium proceedings; the Society for Investigative Dermatology, Inc. 2007. pp. 22–25. [DOI] [PubMed] [Google Scholar]
- Matsunaga K, Ito M. Quantitative analysis of apoptotic cell death in granulomatous inflammation induced by intravenous challenge with Cryptococcus neoformans and bacillus Calmette-Guerin vaccine. Pathol Int. 2000;50:206–218. doi: 10.1046/j.1440-1827.2000.01025.x. [DOI] [PubMed] [Google Scholar]
- McCuskey RS, Urbaschek R, McCuskey PA, Urbaschek B. In vivo microscopic observations of the responses of Kupffer cells and the hepatic microcirculation to Mycobacterium bovis BCG alone and in combination with endotoxin. Infect Immun. 1983;42:362–367. doi: 10.1128/iai.42.1.362-367.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mempel TR, Scimone ML, Mora JR, von Andrian UH. In vivo imaging of leukocyte trafficking in blood vessels and tissues. Curr Opin Immunol. 2004;16:406–417. doi: 10.1016/j.coi.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Mrass P, Takano H, Ng LG, Daxini S, Lasaro MO, Iparraguirre A, Cavanagh LL, von Andrian UH, Ertl HC, Haydon PG, Weninger W. Random migration precedes stable target cell interactions of tumor-infiltrating T cells. J Exp Med. 2006;203:2749–2761. doi: 10.1084/jem.20060710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Cyster JG. CC chemokine receptor 7 contributes to Gi-dependent T cell motility in the lymph node. J Immunol. 2007;178:2973–2978. doi: 10.4049/jimmunol.178.5.2973. [DOI] [PubMed] [Google Scholar]
- Pelletier M, Forget A, Bourassa D, Gros P, Skamene E. Immunopathology of BCG infection in genetically resistant and susceptible mouse strains. J Immunol. 1982;129:2179–2185. [PubMed] [Google Scholar]
- Peters W, Ernst JD. Mechanisms of cell recruitment in the immune response to Mycobacterium tuberculosis. Microbes Infect. 2003;5:151–158. doi: 10.1016/s1286-4579(02)00082-5. [DOI] [PubMed] [Google Scholar]
- Qi H, Egen JG, Huang AY, Germain RN. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science. 2006;312:1672–1676. doi: 10.1126/science.1125703. [DOI] [PubMed] [Google Scholar]
- Saunders BM, Britton WJ. Life and death in the granuloma: immunopathology of tuberculosis. Immunol Cell Biol. 2007;85:103–111. doi: 10.1038/sj.icb.7100027. [DOI] [PubMed] [Google Scholar]
- Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. Observation of antigen-dependent CD8+ T-cell/ dendritic cell interactions in vivo. Cell Immunol. 2001;214:110–122. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
- Sewell DL, Reinke EK, Co DO, Hogan LH, Fritz RB, Sandor M, Fabry Z. Infection with Mycobacterium bovis BCG diverts traffic of myelin oligodendroglial glycoprotein autoantigen-specific T cells away from the central nervous system and ameliorates experimental autoimmune encephalomyelitis. Clin Diagn Lab Immunol. 2003;10:564–572. doi: 10.1128/CDLI.10.4.564-572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slimani H, Charnaux N, Mbemba E, Saffar L, Vassy R, Vita C, Gattegno L. Interaction of RANTES with syndecan-1 and syndecan-4 expressed by human primary macrophages. Biochim Biophys Acta. 2003;1617:80–88. doi: 10.1016/j.bbamem.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Ulrichs T, Kaufmann SH. New insights into the function of granulomas in human tuberculosis. J Pathol. 2006;208:261–269. doi: 10.1002/path.1906. [DOI] [PubMed] [Google Scholar]
- van den Berg TK, Breve JJ, Damoiseaux JG, Dopp EA, Kelm S, Crocker PR, Dijkstra CD, Kraal G. Sialoadhesin on macrophages: its identification as a lymphocyte adhesion molecule. J Exp Med. 1992;176:647–655. doi: 10.1084/jem.176.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis. 2004;38:1261–1265. doi: 10.1086/383317. [DOI] [PubMed] [Google Scholar]
- Worbs T, Mempel TR, Bolter J, von Andrian UH, Forster R. CCR7 ligands stimulate the intranodal motility of T lymphocytes in vivo. J Exp Med. 2007;204:489–495. doi: 10.1084/jem.20061706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.