

Abstract
Background
Females demonstrate improved cardiac recovery after ischemia-reperfusion (I/R) injury compared to males. Attenuation of myocardial dysfunction with pre-ischemic estradiol suggests that estrogen may be an important mediator of this cardioprotection. However, it remains unclear whether post injury estradiol may have clinical potential in the treatment of acute myocardial infarction. We hypothesize that post-ischemic administration of 17β-estradiol will decrease myocardial I/R injury, and improve left ventricular cardiac function.
Materials and Methods
Adult male Sprague-Dawley rat hearts (n=20) were isolated, perfused with Krebs-Henseleit solution via Langendorff model, and subjected to 15 min of equilibration, 25 min of warm ischemia, and 40 min reperfusion. Experimental hearts received post-ischemic 17β-estradiol infusion, 1nM (n=4), 10 nM (n=4), 25 nM (n=4), or 50 nM (n=4), throughout reperfusion. Control hearts (n=4) were infused with perfusate vehicle.
Results
Post-ischemic recovery of left ventricular developed pressure was significantly greater with 1nM (51.6 ± 7.4%) and 10 nM estradiol (47.7 ± 8.6%) than with vehicle (37.8 ± 9.7%) at end reperfusion. There was also greater recovery of the end diastolic pressure with 1 nM (47.8 ± 4.0 mmHg) and 10 nM estradiol (54.0 ± 4.0) compared to vehicle (75.3 ± 7.5). Further, 1nM and 10 nM estrogen preserved coronary flow after ischemia and decreased coronary effluent LDH compared to controls. Estrogen at 25 nM and 50 nM did not provide additional benefit in terms of functional recovery. Estrogen at all concentrations increased ERK phosphorylation.
Conclusions
Post-ischemic infusion of 17β-estradiol protects myocardial function and viability. The attractive potential for the clinical application of post-ischemic estrogen therapy warrants further study to elucidate the mechanistic pathways and differences between males and females.
Keywords: sex hormones, estrogen, myocardial infarction, recovery
INTRODUCTION
Cardiovascular disease is the leading cause of mortality in the United States for men and women. Reperfusion therapy, the treatment of choice for myocardial infarction, is accompanied by the deleterious effects of ischemia / reperfusion (I/R) injury (1, 2). Gender specific differences have been noted in I/R injury. Clinically, females seem to have relative cardiac protection from acute infarctions compared to males (3). Similarly, in animal studies, proestrus females demonstrate increased myocardial protection from I/R injury (4, 5), and improved systolic and diastolic function after ischemia (6) compared to males. Female animals also exhibit decreased left ventriclular remodeling after ischemic injury (7, 8), possibly due to decreased levels of inflammatory cytokines such as tumor necrosis factor α (TNF), interleukin (IL)-6, and IL-1β (9-12).
Estrogen is believed to play an important role in female cardioprotection from I/R injury. Chronic administration of estrogen provides protection from I/R injury in isolated hearts undergoing global ischemia and in hearts undergoing in vivo left anterior descending coronary artery (LAD) obstruction (13). Acute administration of estrogen before LAD occlusion also decreases cardiac myocyte injury and necrosis (14, 15), infarct size (16), and the incidence of ventricular arrhythmia (17). Estrogen may mediate these effects through estrogen receptor-alpha which increases extracellular signal-regulated protein kinase 1/2 (ERK) and decreases c-Jun NH(2)-terminal kinase (JNK) signaling (18). Estrogen receptor-alpha also appears important in decreasing calcium accumulation and increasing nitrite production, thereby minimizing ATP depletion and increasing myocardial perfusion (19). Intracellular estrogen signaling, by way of estrogen receptor alpha, is critical to female resistance to I/R injury.
Post-insult administration of estrogen appears to be cardioprotective in trauma-hemorrhage and sepsis models. Administration of 17β-estradiol fifteen minutes before the end of resuscitation following trauma-hemorrhage improves left ventricular and hepatocellular functioning (20, 21). Similarly, administration of estrogen following induction of sepsis decreases oxidative organ damage, improves liver function, and decreases pro-inflammatory cytokines (22, 23). However, it remains unknown whether post ischemic administration of estrogen confers myocardial protection in the isolated heart.
The administration of post-ischemic estrogen to confer cardioprotection has theoretical clinical appeal. We therefore hypothesize that post-ischemic administration of 17β-estradiol will decrease myocardial I/R damage, and improve left ventricular cardiac function in the isolated heart. The purposes of this study are to 1) assess myocardial function as measured by left ventricular developed pressure (LVDP), end diastolic pressure (EDP), the maximal positive and negative values of the first derivative of pressure (+dP/dt and -dP/dt), and coronary flow in male rats subjected to I/R and treated with 17β-estradiol; and 2) establish the most effective post-ischemic dose of 17β-estradiol via a dose-response curve.
MATERIALS AND METHODS
Animals
Age-matched (250–300 g, 9- to 10-week) normal male Sprague–Dawley rats (Harlan, Indianapolis, IN) were fed a standard diet and acclimated in a quiet quarantine room for 1 week before the experiments. The animal protocol was reviewed and approved by the Indiana Animal Care and Use Committee of Indiana University. All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 1985).
Experimental groups
All isolated rat hearts were subjected to the same I/R protocol: 15-min equilibration period, 25 min of global index ischemia (37°C), and 40 min of total reperfusion. Male rats were divided into five experimental groups: 1) vehicle infusion (n = 4); 2) 17β-estradiol infusion, 1 nM (n = 4); 3) 17β-estradiol infusion, 10 nM (n = 4); 4) 17β-estradiol infusion, 25 nM (n = 4); and 5) 17β-estradiol infusion, 50 nM (n = 4). As a reference, the circulating level of estrogen in a proestrus female rat is 0.2 nM. The 17β-estradiol solution was prepared with water soluble β-Estradiol (Sigma-Aldrich, St. Louis, MO) and Krebs-Henseleit solution. Vehicle and estradiol solutions were infused through a port above the aortic root throughout reperfusion (40 min). The rate of the 17β-estradiol infusion was titrated to the coronary flow in order to maintain the desired experimental concentration at all times.
Isolated heart preparation (Langendorff)
Rats were anesthetized (sodium pentobarbital, 60 mg/kg intraperitoneal) and heparinized (500 U intraperitoneal), and hearts were rapidly excised via median sternotomy and placed in 4°C Krebs–Henseleit solution. The aorta was cannulated and the heart was perfused (70 mmHg) with oxygenated (95% O2/5% CO2) Krebs–Henseleit solution (37 °C). A left atrial resection was performed prior to insertion of a water-filled latex balloon through the left atrium into the left ventricle. The preload volume (balloon volume) was held constant during the entire experiment to allow continuous recording of the LVDP and EDP. The balloon was adjusted to a mean EDP of 5 mmHg (range 4–8 mmHg) during the initial equilibration. Pacing wires were fixed to the right atrium and hearts were paced at approximately 6 Hz, 3 V, 2 ms (350 bpm) throughout perfusion. A three-way stopcock above the aortic root was used to create global ischemia, during which the heart was placed in a 37°C degassed organ bath. Coronary flow was measured by collecting pulmonary artery effluent. Data were continuously recorded using a PowerLab 8 preamplifier/digitizer (AD Instruments Inc., Milford, MA) and an Apple G4 PowerPC computer (Apple Computer Inc., Cupertino, CA). The +dP/dt and −dP/dt were calculated using PowerLab software. After reperfusion, the heart was removed from the apparatus, immediately sectioned, and snap frozen in liquid nitrogen.
Measurement of Myocardial Necrosis
Coronary effluent from various time points was collected. Lactage dehydrogenase (LDH) was determined using a commercially available cytotoxicity detection kit (Roche Applied Science, Indianapolis, IN).
Western blotting
Western blot analysis was performed to measure ERK proteins. Heart tissue was homogenized in cold buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-Glycerophosphate, 1 mM Na3VO4, 1 μg/ml Leupeptin, 1 mM PMSF, and centrifuged at 12000 rpm for 5 minutes. The protein extracts (30 μg/lane) were subjected to electrophoresis on a 12% tris-HCl gel from Bio-Rad and transferred to a nitrocellulose membrane, which was stained by Naphthol Blue-Black to confirm equal protein loading. The membranes were incubated in 5% dry milk for 1 hour and then incubated with the following primary antibodies: ERK antibody, phosphor-ERK (Thr202/Tyr204) antibody (Cell Signaling Technology, Beverly, MA), Subsequently, the membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG secondary antibody. Detection was performed using supersignal west pico stable peroxide solution (Pierce, Rockford, IL). Films were scanned using an Epson Perfection 3200 Scanner (Epson America, Long Beach, CA).
Presentation of data and statistical analysis
All reported values are mean ± S.E.M. Data were compared using two-way analysis of variance (ANOVA) with post-hoc Bonferroni test or Student’s t-test. A two-tailed probability value of less than 0.05 was considered statistically significant.
RESULTS
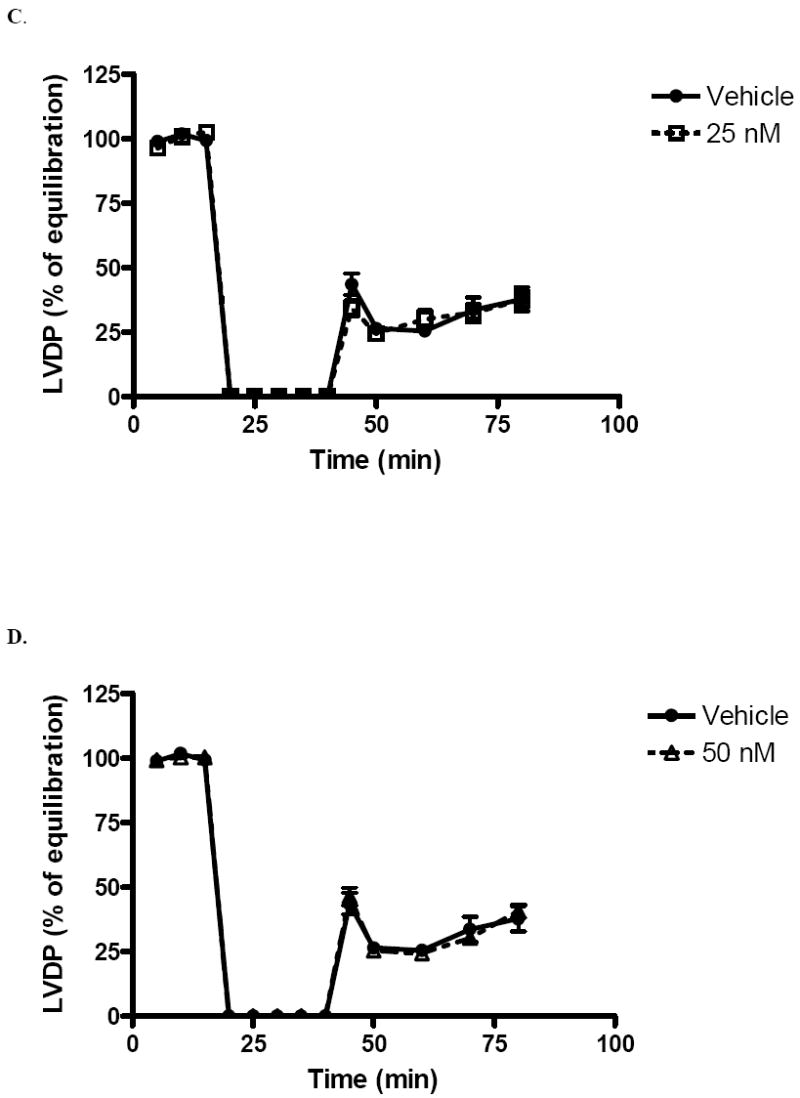
Postischemic recovery of LVDP (expressed as percentage of preischemic function) was significantly greater (p < .05) in the 1 nM group (51.6 ± 7.4%) and 10 nM group (47.7 ± 8.6%) than with vehicle (37.8 ± 9.7%) at end reperfusion (Fig 1A,B). However, there was no significant difference in postischemic recovery of LVDP in the 25 nM (37.5 ± 7.9%, Fig. 1C), or 50 nM group (40.7 ± 5.2%, Fig. 1D) compared to vehicle (37.8 ± 9.7%). Using the above data, a dose-response curve was generated (Fig. 2).
FIG. 1.
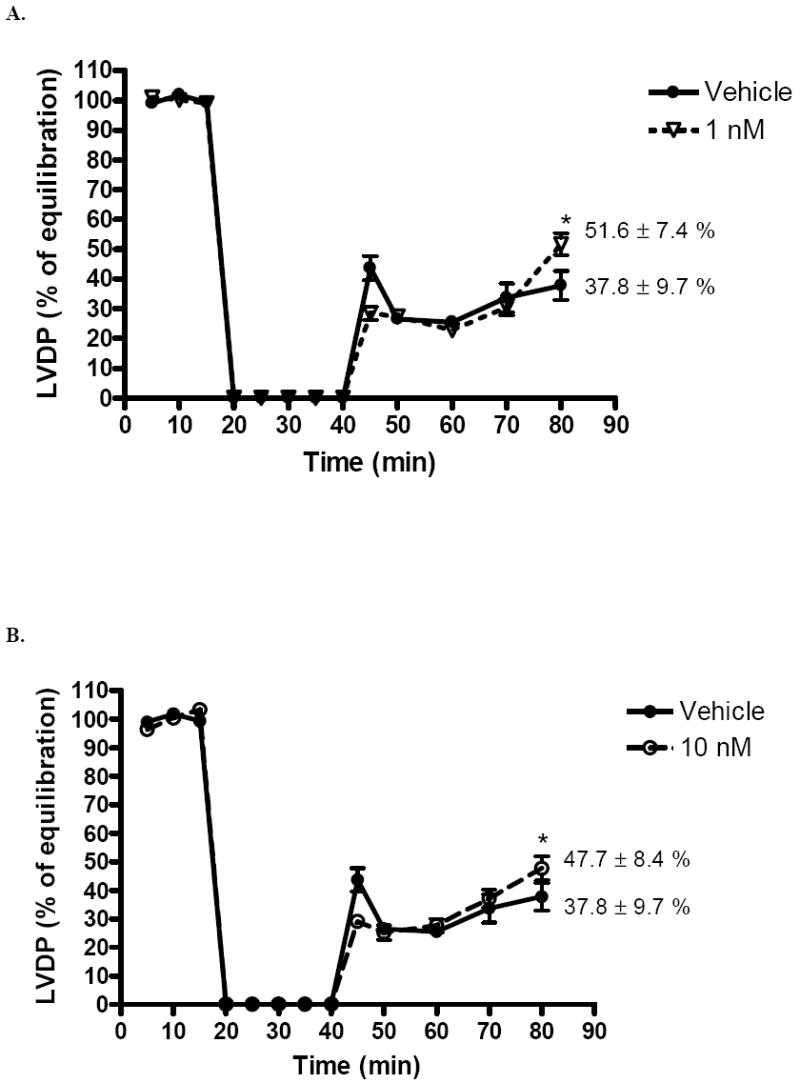
Changes in left ventricular developed pressure (LVDP % of equilibration) after I/R. (A) 1 nM compared to vehicle. (B) 10 nM compared to vehicle. (C) 25 nM compared to vehicle. (D) 50 nM compared to vehicle. Results are mean ± SEM, *p<0.05 vs vehicle.
FIG. 2.
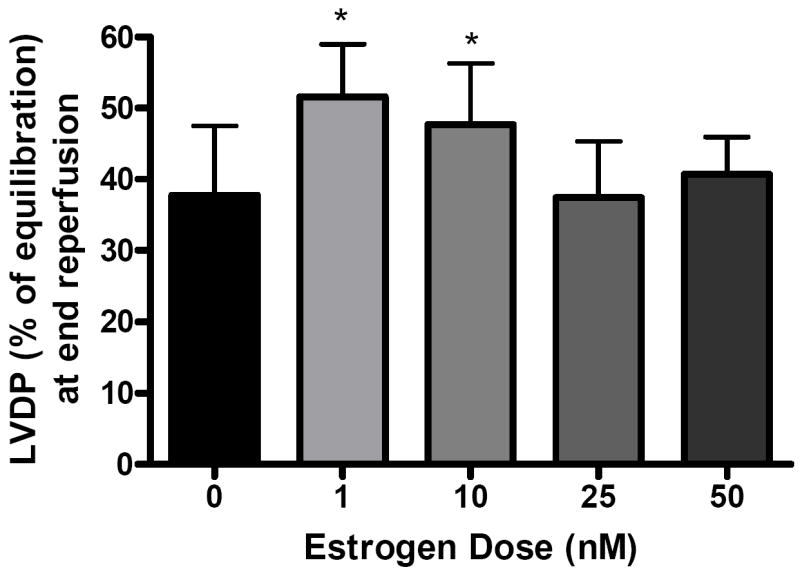
End reperfusion left ventricular developed pressure (LVDP % of equilibration) versus dose of estrogen infusion at 0 (vehicle), 1, 10, 25, and 50 nM. Results are mean ± SEM, *p<0.05 vs vehicle.
Left ventricular end diastolic pressure (mmHg) was elevated in response to I/R. However, male rats in the 1 nM (47.8 ± 4.0 mmHg) and 10 nM (54.0 ± 4.0 mmHg) group had significantly decreased EDP after forty minutes of reperfusion compared to those who received vehicle (75.3 ± 7.5, p < .001, Fig. 3A,B). The end reperfusion EDP of rats in the 25 nM and 50 nM groups was 72.8 ± 4.5 (Fig. 3C) and 70.0 ± 1.9 (Fig. 3D) respectively. Estrogen administration at these levels did not result in significantly different EDPs as compared to control. Maximum positive and negative dP/dt were impaired at the beginning of reperfusion. Significant differences were not noted between experimental groups and vehicle. While post-ischemic infusion of estrogen markedly reduces the EDP after I/R injury, it may not significantly improve contractility or compliance.
FIG. 3.
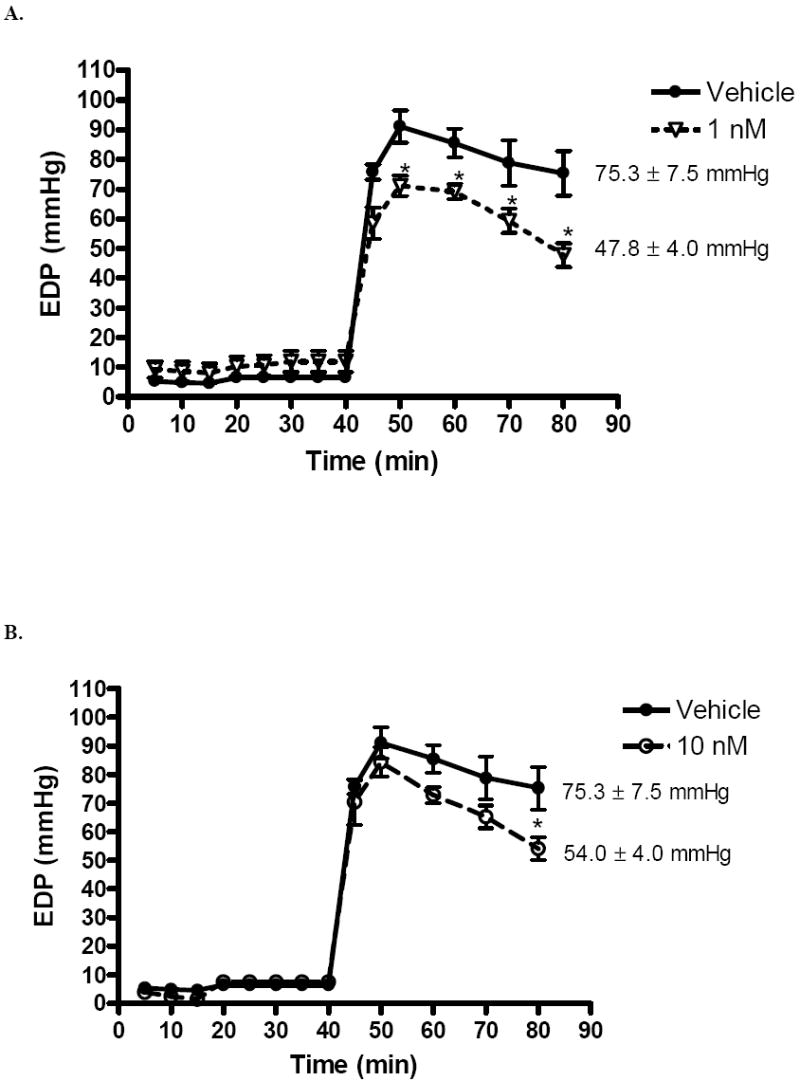
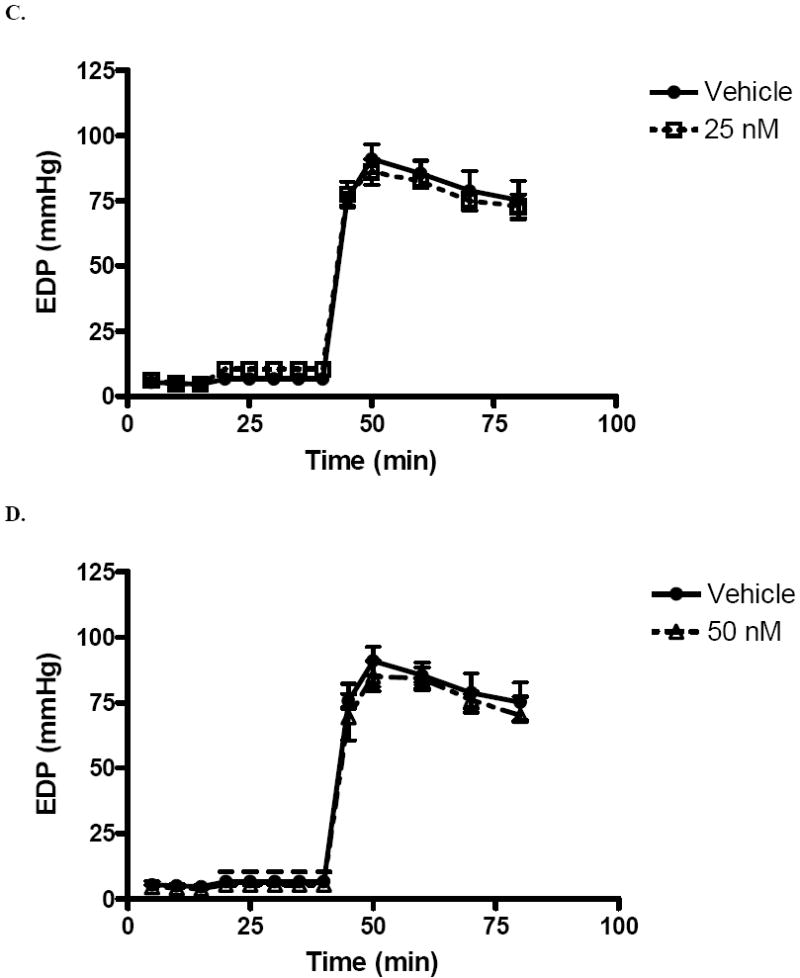
Changes in left ventricular end diastolic pressure (EDP) after I/R. (A) 1 nM compared to vehicle. (B) 10 nM compared to vehicle. (C) 25 nM compared to vehicle. (D) 50 nM compared to vehicle. Results are mean ± SEM, *p<0.05 vs vehicle.
Coronary flow was markedly decreased in the control (9.5 ± 1.0 to 7.5 ± 0.9 ml/min), 25 nM and 50 nM groups after ischemia, but remained stable in the 1 nM (9.6 ± 0.6 to 10.3 ± 1.0 ml/min) and 10 nM (9.8 ± 0.3 to 9.8 ± 1.1ml/min) group (Fig. 5B). Therefore, post-ischemic estrogen infusion at a concentration of 1 nM or 10 nM may help to maintain post-ischemic coronary flow, thereby promoting increased functional recovery.
FIG. 5.
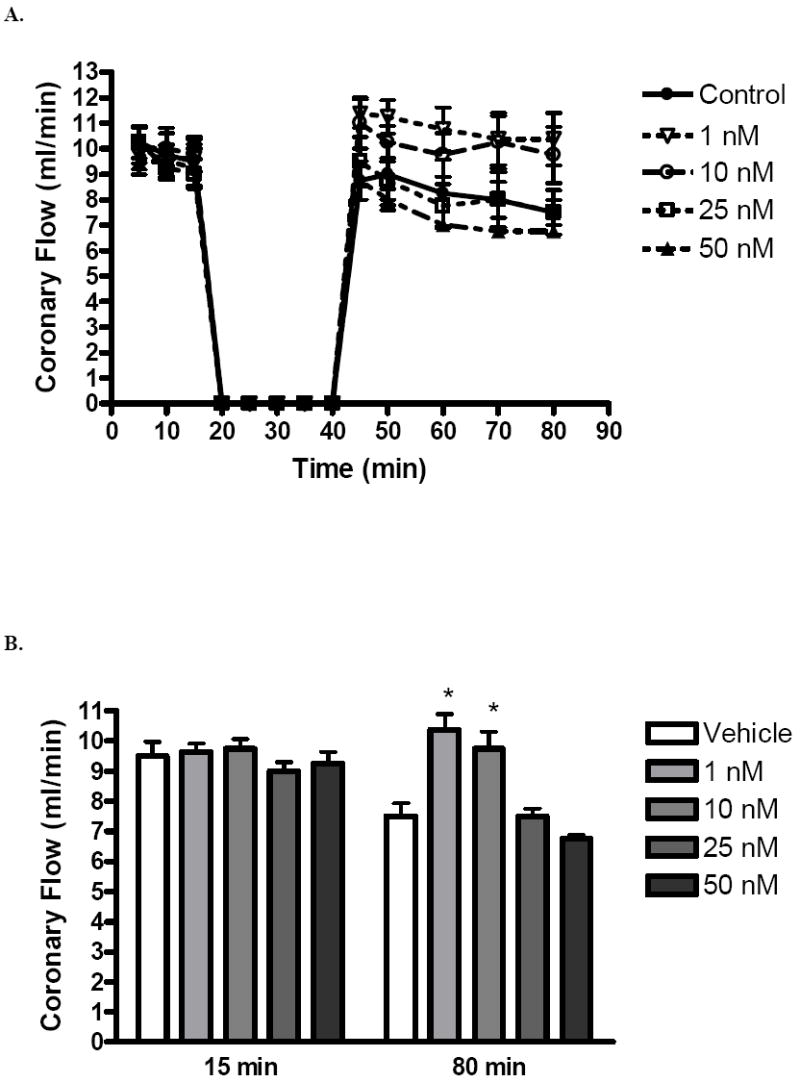
(A) Changes in coronary flow after I/R. (B) Coronary flow at 15 minutes (after equilibration and before ischemia) and 80 minutes (at the end of reperfusion). 1, 10, 25, and 50 nM groups compared to vehicle. Results are mean ± SEM, *p<0.05 vs vehicle.
Coronary effluent LDH was markedly increased after ischemia in all groups (Fig. 6A). However, LDH in 10 nM (1.9 ± 0.1 μU/ml) and 25 nM (2.4 ± 0.4 μU/ml) groups during early reperfusion were significantly lower than LDH in controls (Fig. 6B).
FIG. 6.
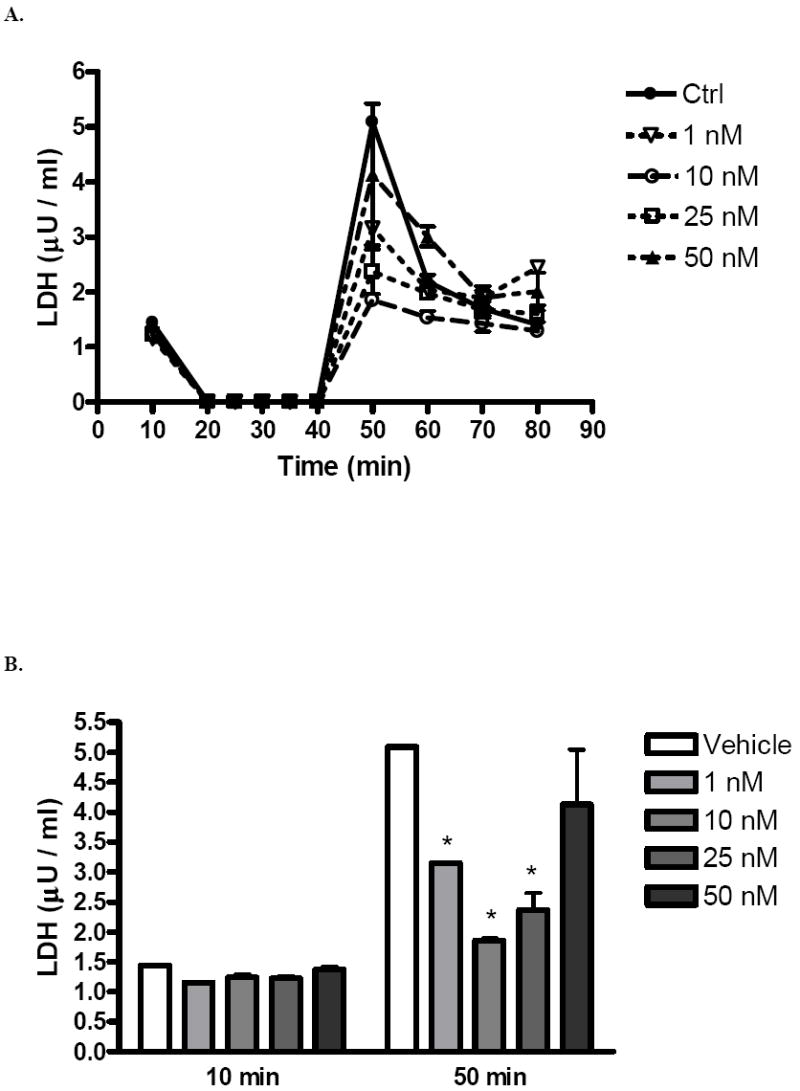
(A) Changes in coronary effluent LDH after I/R. (B) Coronary effluent LDH at 10 minutes (during equilibration, before ischemia) and 50 minutes (early reperfusion). 1, 10, 25, and 50 nM groups compared to vehicle. Results are mean ± SEM, *p<0.05 vs vehicle.
The myocardial activation of phosphorylated ERK and nonphosphorylated ERK were assessed by Western blot (Figure 7). The phosphorylated forms of ERK were increased in estradiol treated hearts compared tovehicle control hearts. However, no difference was found in the phosphorylated forms of ERK among different estradiol concentrations following I/R.
FIG 7.

I/R induced myocardial phosphorylation of p44/42 (ERK). The expression of activated ERK after I/R injury in control hearts and hearts exposed to estradiol at various concentrations. Shown are representative immunoblots. Row 1. Phosphorylated (activated) ERK; Row 2. Total ERK. All samples were on the same membrane.
DISCUSSION
The results of this study indicate that post-ischemic infusion of estrogen at a concentration of 1 nM or 10 nM (1) decreases myocardial injury and necrosis; (2) lowers EDP throughout reperfusion; (3) increases end reperfusion LVDP; and (4) preserves coronary flow. Post ischemic estradiol at all concentrations also increases phosphorylation of ERK. This study demonstrates that post-ischemic administration of estrogen may improve left ventricular functional recovery and viability.
Post ischemic functional recovery was marked by an improvement in the EDP, LVDP, and coronary flow in male rats. Elevations in end diastolic pressure, a marker of diastolic cardiac function, are typically seen with increasing severity of cardiac injury. This elevation is typically viewed as a reduction in the unstressed volume of the ventricle, which may be due to myocardial contracture or edema (24). In this study, EDP was elevated above equilibrium in all groups, indicating cardiac injury with the induced ischemia. However, estrogen at a dose of 1 nM or 10 nM significantly decreased the EDP during reperfusion as compared to control and other estrogen doses, indicating that this dose of estrogen protected hearts from ischemic injury. The protective effects of estrogen administration were also seen in other measures of systolic function. LVDP was improved at 40 minutes of reperfusion, although a significant difference was not observed at other time points during reperfusion. Interestingly, estrogen at all doses did not affect +dP/dT or −dP/dT between experimental groups. This discrepancy in measures of myocardial function suggests that estrogen may mediate its acute positive effects not by increasing contractility, but instead by increasing the end diastolic fluid volume of the heart.
Furthermore, coronary flow (25-27) was substantially decreased with ischemia in other experimental groups, but was maintained in the 1 nM and 10 nM group. This may be due to inhibition of voltage gated or receptor operated calcium channels. Estrogen has also been shown to promote coronary vasodilation (28). It is possible that a low dose of estrogen induces increased coronary vasodilation, improved cardiac perfusion, and overall increased functional recovery. However, it is important to note that other Langendorff models may differ in baseline coronary flow. Several reasons may exist which may account for differences in coronary flow such as Langendorff model (ADI vs others), perfusion pressure (70 vs 95 mmHg), and mode of perfusion (constant flow vs constant pressure). Thus, estradiol’s protective ability to preserve coronary flow may be greater or alternatively less depending on the magnitude of baseline coronary flow.
The acute cardioprotective effects of estrogen may occur through several mechanisms. Reactive oxygen species (ROS) are important mediators of myocardial dysfunction from I/R injury (29). Estrogen has been shown to decrease the production of oxygen radicals during I/R injury (30). Recent evidence shows that estrogen activates PI3K/Akt signaling. Activation of this pathway effectively decreases ROS generation, decreases pro-apoptotic p38 MAPK activation, and increases anti-apoptotic p38β MAPK signaling (31, 32). Estrogen also exerts an antioxidant effect by increasing reduced glutathione, a free radical scavenger in the myocardium (33, 34). Thus, one mechanism that estrogen may utilize in conferring cardioprotection from I/R injury is the reduction of oxidative damage.
In addition to decreasing oxidative damage, the post-ischemic protective effects of estrogen may be mediated through decreased proinflammatory cytokine signaling. This may be mediated in part by decreased TNF signaling through TNF receptor resistance (6, 13). Furthermore estrogen has previously been shown to upregulate the cardioprotective ERK MAPK pathway, while down-regulating JNK signaling. These pathways are also likely affected with post-ischemic administration of estrogen (18). Indeed, in this study, post ischemic estradiol infusion was associated with increased ERK phosphorylation compared to vehicle controls. Therefore, by minimizing proinflammitory cytokine signaling, estrogen effectively decreases cardiac dysfunction after I/R.
Estrogen also provides protection from I/R injury by decreasing apoptosis. Estrogen prevents the loss of cytochrome c from myocardial mitochondria, and decreases its accumulation in the cytosol. It reduces caspase-3 activation and decreases DNA strand breaks (35, 36). Estrogen also reduces cardiac apoptosis through a reduction in pro-apoptotic nuclear factor-κB (NF- κB), and also through increased PI3K/Akt signaling (37, 38). Interestingly, post ischemic estrogen compared to vehicle in this study also reduced myocardial necrosis as measured by coronary effluent LDH. By reducing cardiac cell death, estrogen increases the amount of viable myocardium after I/R injury, thereby facilitating increased cardiac recovery after I/R injury.
Our experiment, performed with leukocyte-free, buffer-perfused hearts demonstrates that post-ischemic administration of estrogen provides effective cardioprotection. Investigations have shown that estrogen modulates immune cells by decreasing systemic IL-6 release, MAPK activation, and Kupffer cell cytokine production (21, 39) after trauma-hemorrhage. Thus, in the future, in vitro studies on the heart may be helpful to test the possibility of additional cardioprotection with post-ischemic estrogen administration mediated through immune cell modulation.
The results of this study show that a post-ischemic infusion of 17β-estradiol at a concentration of 1 nM or 10 nM decreases myocardial dysfunction in male rats after I/R injury. It adds support to the use of estrogen as a therapeutic intervention for acute myocardial ischemia. Indeed, post-ischemic administration is even more clinically relevant and has attractive therapeutic potential. Further study in this area is imperative to further delineate the mechanisms of estrogen protection and to study the gender differences associated with post-ischemic estrogen administration.
FIG. 4.
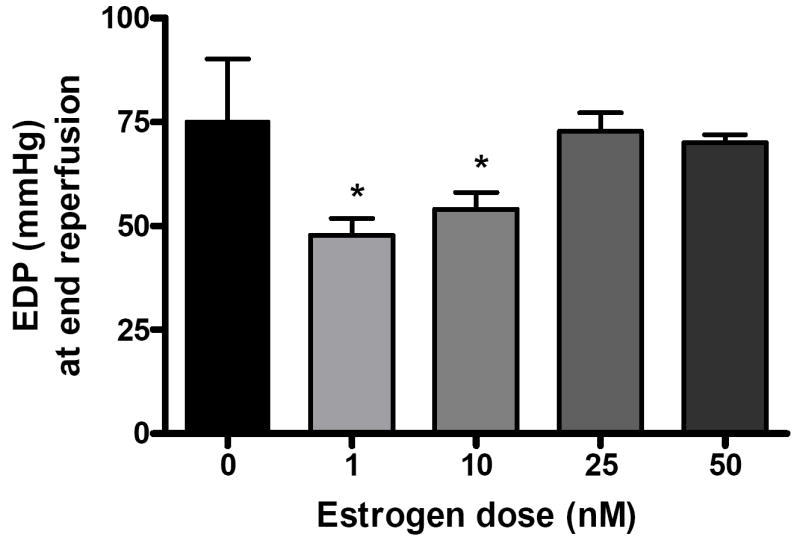
End reperfusion end diastolic pressure (EDP mmHg) versus dose of estrogen infusion at 0 (vehicle), 1, 10, 25, and 50 nM. Results are mean ± SEM, *p<0.05 vs vehicle.
Acknowledgments
This work was supported in part by NIH R01GM070628, NIH R01HL085595, NIH K99/R00 HL0876077-01, NIH NRSA, AHA Grant in aid, and AHA Post-doctoral Fellowship 0526008Z. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR015481-01 from the National Center for Research Resources, National Institutes of Health.
Footnotes
DISCLOSURES None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Matsumura K, Jeremy RW, Schaper J, Becker LC. Progression of myocardial necrosis during reperfusion of ischemic myocardium. Circulation. 1998;97:795–804. doi: 10.1161/01.cir.97.8.795. [DOI] [PubMed] [Google Scholar]
- 2.Meldrum DR. Tumor necrosis factor in the heart. Am J Physiol. 1998;274:R577–595. doi: 10.1152/ajpregu.1998.274.3.R577. [DOI] [PubMed] [Google Scholar]
- 3.Vaccarino V, Krumholz HM, Berkman LF, Horwitz RI. Sex differences in mortality after myocardial infarction. Is there evidence for an increased risk for women? Circulation. 1995;91:1861–1871. doi: 10.1161/01.cir.91.6.1861. [DOI] [PubMed] [Google Scholar]
- 4.Kher A, Wang M, Tsai BM, Pitcher JM, Greenbaum ES, Nagy RD, Patel KM, Wairiuko GM, Markel TA, Meldrum DR. Sex differences in the myocardial inflammatory response to acute injury. Shock. 2005;23:1–10. doi: 10.1097/01.shk.0000148055.12387.15. [DOI] [PubMed] [Google Scholar]
- 5.Yang S, Choudhry MA, Hsieh YC, Hu S, Rue LW, 3rd, Bland KI, Chaudry IH. Estrus cycle: influence on cardiac function following trauma-hemorrhage. Am J Physiol Heart Circ Physiol. 2006;291:H2807–2815. doi: 10.1152/ajpheart.00195.2006. [DOI] [PubMed] [Google Scholar]
- 6.Wang M, Tsai BM, Crisostomo PR, Meldrum DR. Tumor necrosis factor receptor 1 signaling resistance in the female myocardium during ischemia. Circulation. 2006;114:I282–289. doi: 10.1161/CIRCULATIONAHA.105.001164. [DOI] [PubMed] [Google Scholar]
- 7.Ramani R, Mathier M, Wang P, Gibson G, Togel S, Dawson J, Bauer A, Alber S, Watkins SC, McTiernan CF, Feldman AM. Inhibition of tumor necrosis factor receptor-1-mediated pathways has beneficial effects in a murine model of postischemic remodeling. Am J Physiol Heart Circ Physiol. 2004;287:H1369–1377. doi: 10.1152/ajpheart.00641.2003. [DOI] [PubMed] [Google Scholar]
- 8.Cavasin MA, Tao Z, Menon S, Yang XP. Gender differences in cardiac function during early remodeling after acute myocardial infarction in mice. Life Sci. 2004;75:2181–2192. doi: 10.1016/j.lfs.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 9.Cain BS, Meldrum DR, Dinarello CA, Meng X, Joo KS, Banerjee A, Harken AH. Tumor necrosis factor-alpha and interleukin-1beta synergistically depress human myocardial function. Crit Care Med. 1999;27:1309–1318. doi: 10.1097/00003246-199907000-00018. [DOI] [PubMed] [Google Scholar]
- 10.Cain BS, Meldrum DR, Meng X, Dinarello CA, Shames BD, Banerjee A, Harken AH. p38 MAPK inhibition decreases TNF-alpha production and enhances postischemic human myocardial function. The Journal of surgical research. 1999;83:7–12. doi: 10.1006/jsre.1998.5548. [DOI] [PubMed] [Google Scholar]
- 11.Meldrum DR, Dinarello CA, Shames BD, Cleveland JC, Jr, Cain BS, Banerjee A, Meng X, Harken AH. Ischemic preconditioning decreases postischemic myocardial tumor necrosis factor-alpha production. Potential ultimate effector mechanism of preconditioning. Circulation. 1998;98:II214–218. discussion II218-219. [PubMed] [Google Scholar]
- 12.Wang M, Baker L, Tsai BM, Meldrum KK, Meldrum DR. Sex differences in the myocardial inflammatory response to ischemia-reperfusion injury. Am J Physiol Endocrinol Metab. 2005;288:E321–326. doi: 10.1152/ajpendo.00278.2004. [DOI] [PubMed] [Google Scholar]
- 13.Xu Y, Arenas IA, Armstrong SJ, Plahta WC, Xu H, Davidge ST. Estrogen improves cardiac recovery after ischemia/reperfusion by decreasing tumor necrosis factor-alpha. Cardiovasc Res. 2006;69:836–844. doi: 10.1016/j.cardiores.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 14.Delyani JA, Murohara T, Nossuli TO, Lefer AM. Protection from myocardial reperfusion injury by acute administration of 17 beta-estradiol. J Mol Cell Cardiol. 1996;28:1001–1008. doi: 10.1006/jmcc.1996.0093. [DOI] [PubMed] [Google Scholar]
- 15.Hale SL, Birnbaum Y, Kloner RA. beta-Estradiol, but not alpha-estradiol, reduced myocardial necrosis in rabbits after ischemia and reperfusion. American heart journal. 1996;132:258–262. doi: 10.1016/s0002-8703(96)90419-6. [DOI] [PubMed] [Google Scholar]
- 16.Booth EA, Marchesi M, Kilbourne EJ, Lucchesi BR. 17Beta-estradiol as a receptor-mediated cardioprotective agent. The Journal of pharmacology and experimental therapeutics. 2003;307:395–401. doi: 10.1124/jpet.103.054205. [DOI] [PubMed] [Google Scholar]
- 17.Node K, Kitakaze M, Kosaka H, Minamino T, Funaya H, Hori M. Amelioration of ischemia- and reperfusion-induced myocardial injury by 17beta-estradiol: role of nitric oxide and calcium-activated potassium channels. Circulation. 1997;96:1953–1963. doi: 10.1161/01.cir.96.6.1953. [DOI] [PubMed] [Google Scholar]
- 18.Wang M, Crisostomo P, Wairiuko GM, Meldrum DR. Estrogen receptor-alpha mediates acute myocardial protection in females. Am J Physiol Heart Circ Physiol. 2006;290:H2204–2209. doi: 10.1152/ajpheart.01219.2005. [DOI] [PubMed] [Google Scholar]
- 19.Zhai P, Eurell TE, Cooke PS, Lubahn DB, Gross DR. Myocardial ischemia-reperfusion injury in estrogen receptor-alpha knockout and wild-type mice. Am J Physiol Heart Circ Physiol. 2000;278:H1640–1647. doi: 10.1152/ajpheart.2000.278.5.H1640. [DOI] [PubMed] [Google Scholar]
- 20.Mizushima Y, Wang P, Jarrar D, Cioffi WG, Bland KI, Chaudry IH. Estradiol administration after trauma-hemorrhage improves cardiovascular and hepatocellular functions in male animals. Ann Surg. 2000;232:673–679. doi: 10.1097/00000658-200011000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki T, Shimizu T, Yu HP, Hsieh YC, Choudhry MA, Bland KI, Chaudry IH. 17 beta-estradiol administration following trauma-hemorrhage prevents the increase in Kupffer cell cytokine production and MAPK activation predominately via estrogen receptor-alpha. Surgery. 2006;140:141–148. doi: 10.1016/j.surg.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 22.Sener G, Arbak S, Kurtaran P, Gedik N, Yegen BC. Estrogen protects the liver and intestines against sepsis-induced injury in rats. The Journal of surgical research. 2005;128:70–78. doi: 10.1016/j.jss.2005.02.019. [DOI] [PubMed] [Google Scholar]
- 23.Erikoglu M, Sahin M, Ozer S, Avunduk MC. Effects of gender on the severity of sepsis. Surgery today. 2005;35:467–472. doi: 10.1007/s00595-004-2958-3. [DOI] [PubMed] [Google Scholar]
- 24.Schaff HV, Gott VL, Goldman RA, Frederiksen JW, Flaherty JT. Mechanism of elevated left ventricular end-diastolic pressure after ischemic arrest and reperfusion. Am J Physiol. 1981;240:H300–307. doi: 10.1152/ajpheart.1981.240.2.H300. [DOI] [PubMed] [Google Scholar]
- 25.Feng NC, Satoh H, Urushida T, Katoh H, Terada H, Watanabe Y, Hayashi H. A selective inhibitor of Na+/Ca2+ exchanger, SEA0400, preserves cardiac function and high-energy phosphates against ischemia/reperfusion injury. J Cardiovasc Pharmacol. 2006;47:263–270. doi: 10.1097/01.fjc.0000202561.69291.ac. [DOI] [PubMed] [Google Scholar]
- 26.Lu C, Minatoguchi S, Arai M, Wang N, Chen XH, Bao N, Kawamura I, Yasuda S, Kobayashi H, Wu DJ, Takemura G, Fujiwara H. Nicorandil improves post-ischemic myocardial dysfunction in association with opening the mitochondrial K(ATP) channels and decreasing hydroxyl radicals in isolated rat hearts. Circ J. 2006;70:1650–1654. doi: 10.1253/circj.70.1650. [DOI] [PubMed] [Google Scholar]
- 27.Wang M, Sankula R, Tsai BM, Meldrum KK, Turrentine M, March KL, Brown JW, Dinarello CA, Meldrum DR. P38 MAPK mediates myocardial proinflammatory cytokine production and endotoxin-induced contractile suppression. Shock. 2004;21:170–174. doi: 10.1097/01.shk.0000110623.20647.aa. [DOI] [PubMed] [Google Scholar]
- 28.Moritz A, Radtke OA, Gust R, Glusa E, Pertz HH. Characterisation of the relaxant response to raloxifene in porcine coronary arteries. European journal of pharmacology. 2006;545:153–160. doi: 10.1016/j.ejphar.2006.06.049. [DOI] [PubMed] [Google Scholar]
- 29.Zhao ZQ. Oxidative stress-elicited myocardial apoptosis during reperfusion. Current opinion in pharmacology. 2004;4:159–165. doi: 10.1016/j.coph.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 30.McHugh NA, Merrill GF, Powell SR. Estrogen diminishes postischemic hydroxyl radical production. Am J Physiol. 1998;274:H1950–1954. doi: 10.1152/ajpheart.1998.274.6.H1950. [DOI] [PubMed] [Google Scholar]
- 31.Kim JK, Pedram A, Razandi M, Levin ER. Estrogen prevents cardiomyocyte apoptosis through inhibition of reactive oxygen species and differential regulation of p38 kinase isoforms. J Biol Chem. 2006;281:6760–6767. doi: 10.1074/jbc.M511024200. [DOI] [PubMed] [Google Scholar]
- 32.Camper-Kirby D, Welch S, Walker A, Shiraishi I, Setchell KD, Schaefer E, Kajstura J, Anversa P, Sussman MA. Myocardial Akt activation and gender: increased nuclear activity in females versus males. Circulation research. 2001;88:1020–1027. doi: 10.1161/hh1001.090858. [DOI] [PubMed] [Google Scholar]
- 33.Kim YD, Farhat MY, Myers AK, Kouretas P, DeGroot KW, Pacquing A, Ramwell PW, Suyderhoud JP, Lees DE. 17-Beta estradiol regulation of myocardial glutathione and its role in protection against myocardial stunning in dogs. J Cardiovasc Pharmacol. 1998;32:457–465. doi: 10.1097/00005344-199809000-00017. [DOI] [PubMed] [Google Scholar]
- 34.Urata Y, Ihara Y, Murata H, Goto S, Koji T, Yodoi J, Inoue S, Kondo T. 17Beta-estradiol protects against oxidative stress-induced cell death through the glutathione/glutaredoxin-dependent redox regulation of Akt in myocardiac H9c2 cells. J Biol Chem. 2006;281:13092–13102. doi: 10.1074/jbc.M601984200. [DOI] [PubMed] [Google Scholar]
- 35.Morkuniene R, Arandarcikaite O, Borutaite V. Estradiol prevents release of cytochrome c from mitochondria and inhibits ischemia-induced apoptosis in perfused heart. Experimental gerontology. 2006;41:704–708. doi: 10.1016/j.exger.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh YC, Yu HP, Suzuki T, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Upregulation of mitochondrial respiratory complex IV by estrogen receptor-beta is critical for inhibiting mitochondrial apoptotic signaling and restoring cardiac functions following trauma-hemorrhage. J Mol Cell Cardiol. 2006;41:511–521. doi: 10.1016/j.yjmcc.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 37.Pelzer T, Schumann M, Neumann M, deJager T, Stimpel M, Serfling E, Neyses L. 17beta-estradiol prevents programmed cell death in cardiac myocytes. Biochemical and biophysical research communications. 2000;268:192–200. doi: 10.1006/bbrc.2000.2073. [DOI] [PubMed] [Google Scholar]
- 38.Pelzer T, Neumann M, de Jager T, Jazbutyte V, Neyses L. Estrogen effects in the myocardium: inhibition of NF-kappaB DNA binding by estrogen receptor-alpha and -beta. Biochemical and biophysical research communications. 2001;286:1153–1157. doi: 10.1006/bbrc.2001.5519. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki T, Shimizu T, Yu HP, Hsieh YC, Choudhry MA, Schwacha MG, Chaudry IH. Tissue-Compartment-Specific Role of Estrogen Receptor Subtypes in Immune Cell Cytokine Production Following Trauma-Hemorrhage. J Appl Physiol. 2006 doi: 10.1152/japplphysiol.00964.2006. [DOI] [PubMed] [Google Scholar]