

Abstract
Absorption and fluorescence emission spectra of the polycyclic aromatic hydrocarbons benzo[a]pyrene (BaP) and benzo[e]pyrene (BeP) in solution and adsorbed on silica have been obtained and compared to examine the spectroscopic effects of clustering. Molecular mechanics calculations with the UFF potential were done to optimize monomer, dimer and trimer geometries, and energy differences were determined by MP2/6-31G* calculations. Fluorescence emission spectra of adsorbed BeP and BaP display a red shift that progresses with increased loading, and the two differ in their photodegradation kinetics. The experimental and theoretical results are found to be consistent.
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are environmental pollutants found principally in airborne particles, but to some extent also in water, soil, sediments and food [1]. Several PAHs have been classified by the International Agency for Research on Cancer and the U. S. Environmental Protection Agency as probable human carcinogens [1, 2]. They have also been proposed as photoelectronic components, in which their extensive π-electron systems could act as charge-carriers [3, 4]. Because they are nonpolar, hydrophobic compounds with planar structures, PAH molecules tend to aggregate as stacked clusters [4]. Aggregation affects their chemical, photophysical, and biological properties and is therefore an important attribute [5, 6].
PAHs are formed primarily as products of incomplete combustion of organic fuels [7]. Because of their low volatility, PAHs readily adsorb onto airborne particles or condense to initiate particle formation [8]. Condensed PAHs released into the atmosphere are susceptible either to photochemical reaction if exposed to sunlight or to thermal reaction with other pollutants [7]. The microenvironment in which PAHs are adsorbed and their arrangement on particle surfaces affect their spectroscopic and chemical properties [9], and monomer-dimer differences in absorption and emission spectra of some of the smaller PAHs have been studied.
Anthracene forms ground-state pairs that exhibit a red shift in the absorption spectrum with respect to the monomer [10]. The fluorescence spectrum of ground-state anthracene dimer is also shifted to longer wavelengths than that of the monomer and presents some vibronic structure. Phenanthrene and chrysene adsorbed on silica gel at high loading, on the other hand, tend to form microcrystals [11], as evidenced by the absence of a red shift in the absorption spectrum, but with broadening and a red shift in the emission spectrum. The changes in the fluorescence spectra with increased loading are gradual, indicating that crystal growth is also gradual. Photooxidation of polyaromatics is also affected by cluster formation, and because it is a route to formation of toxic compounds, aggregation that affects the rate of reaction may affect their toxicological properties [12].
Polyaromatic hydrocarbons are large, nonpolar molecules that interact via van der Waals forces between their π–electron systems. Interaction potential surfaces are consequently shallow, and accurate theoretical calculation of PAH cluster geometries and interaction energies is difficult. Density functional theory (DFT) is the most common all-electron theoretical method applied to large molecules, but the current generation of functionals does not accurately represent van der Waals interactions [13]. Recently, however, the addition of dispersion terms to DFT energies has yielded promising results [14]. Interactions among the smaller PAH dimers, benzene, naphthalene and anthracene, have been examined with all-electron wavefunction-based methods with correlation correction such as MP2 or CCSD, while larger molecules are more commonly treated by force-field or semiempirical methods.
Benzene dimer has been extensively studied theoretically [15] and experimentally [16, 17]. Theoretical studies find two stable isomers, one having a T-shaped or perpendicular geometry and the other a stacked parallel structure in which the monomers are displaced from the eclipsed conformation [15]. The T-shaped isomer is the global minimum on the shallow potential surface. The leading multipole interaction, the quadrupole-quadrupole interaction, stabilizes both isomers but destabilizes the eclipsed form [5, 18]. In naphthalene dimer, and in the naphthalene-anthracene complex the T-shaped and stacked geometries are very close in energy [18]. In larger PAHs stacked, parallel displaced structures are expected to be the more stable form [19, 20].
A variety of semiempirical methods have been employed in studies of PAH clusters [15, 21]. Force-field molecular mechanical methods have been notably successful in describing PAH stacking [20–22]. These latter can be parametrized in accurate calculations on smaller molecules [14, 19]. The parameters of the Universal Force Field (UFF) [23, 24], which is employed in this study, have been derived from fundamental properties and empirical rules available for every element, with special emphasis on organics, for which a greater depth of data is available. The van der Waals interaction energy is represented as a Lennard-Jones potential. UFF and similar potentials have proven reliable in determining stacking geometries of polyaromatics [20, 22].
We here report on the results of a study undertaken to examine the effects of clustering on the absorption and emission spectra of benzo[e]pyrene (BeP) and benzo[a]pyrene (BaP) (Fig. 1) and on their photoreactivity. To better understand the relation between molecular cluster structure and observed absorption and fluorescence spectra the geometries of BeP and BaP monomers, dimers and trimers have been optimized in molecular mechanics theoretical calculations, and MP2/6-31G* single-point calculations performed at the optimum geometries.
1.
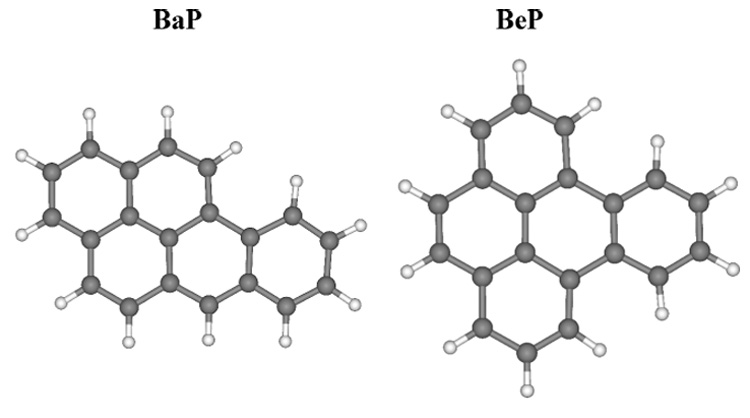
UFF optimized structures of BaP and BeP.
MP2/6-31G* single-point calculations at the UFF optimized geometries yield total energies and energies of the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals. Although the HOMO-LUMO energy difference does not in general yield accurate spectral transition energies, it has in fact long been used to correlate with spectral shifts and other physicochemical properties of PAHs [25, 26], and HOMO-LUMO differences do correlate satisfactorily with the shifts in λmax that accompany increases in the number of chromophores in PAHs, even to the extent of being able to distinguish among isomers [27].
Methods
Experimental
BeP and BaP (Sigma-Aldrich) were used as received; their purity was verified by HPLC to be at least 99%. Methanol, acetonitrile and hexane (Optima, Fisher Scientific) were used as solvents. The adsorbed samples were prepared by adding a measured volume of standard solution of PAH in hexane to a weighed amount of adsorbent (unactivated silica gel grade 923, 25Å pore size, Sigma-Aldrich). The solvent was evaporated using a rotary evaporator at 40°C. A Cary 3 UV-visible spectrophotometer was used to record the absorption spectra of the samples. Excitation and emission fluorescence spectra were measured on a Cary Eclipse (Varian, Inc.) spectrofluorometer. The excitation and emission wavelengths were set at 330 and 398 nm, respectively for BeP samples, and at 384 and 430 nm for BaP. For solid samples the fluorescence spectra were recorded in a front face arrangement. In photodegradation kinetics studies samples were irradiated with an Oriel 1000W Xe(Hg) lamp. A Corning 7–54 glass filter was used to isolate the wavelength range 250 – 350 nm. A water filter was placed between the lamp and the sample cell to prevent overheating by infrared radiation incident on the sample.
Computational
Structures of the monomers, dimers and trimers of BeP and BaP were optimized in molecular mechanics calculations using the Universal Force Field (UFF) [23, 24] potential as implemented in Gaussian 03 [28]. B3LYP/ and MP2/6-311G* optimizations of the monomer geometries were conducted to verify the UFF results, and the UFF geometries were found to be in close agreement with these. An MP2/6-31G* single-point calculation of each optimized structure was then done to obtain relative energies and HOMO-LUMO separations.
Results and Discussion
The absorption spectra of BeP and BaP in polar solvents and adsorbed on silica are similar, except that slight broadening of the bands is observed in the spectra of the adsorbed samples. The fluorescence excitation and emission spectra of BeP and BaP are similar for samples adsorbed at loadings lower than 3 × 10−7 moles PAH/gram of silica (Fig. 2 and 3) and in solutions in polar solvents. Some PAH-surface interaction broadening is seen in the adsorbed state. As surface loading increases a bathochromic shift is observed, and the shift progresses with increased loading. The maxima in the excitation and emission spectra of BaP of 3 × 10−6 mol/g are shifted 2 nm to longer wavelengths relative to maxima at 3 × 10−7 mol/g. In the case of BeP the shift between the maxima in samples of 5 × 10−7 mol/g and samples ten times more concentrated is 3 nm. This observation is consistent with the formation of aggregates on the silica caused by a surface pooling effect during sample preparation. As solvent is removed by evaporation, PAH molecules adsorb one over another, forming stacked clusters. UFF molecular mechanics optimizations of BeP and BaP monomers (Fig. 1), dimers and trimers (Fig. 4) were performed, and geometrical parameters are presented in Table 1. Columns of the Table from left to right, respectively, display the total MP2/6-31G* energies, the energy of each isomer relative to the global minimum, binding energies of the global minimum dimers and trimers with respect to the appropriate number of monomers, the HOMO-LUMO separation, the interplanar separation(s) of each isomer and the rotation angle relative to the top molecule of each monomer composing the dimers and trimers. A progression to lower energy is seen in the calculated HOMO-LUMO differences for both BeP and BaP (Table 1) corresponding to a progression from monomer to dimer to trimer, qualitatively reflecting the shifts seen in the fluorescence spectra, and thus reinforcing the interpretation that the red shift arises from clustering.
2.
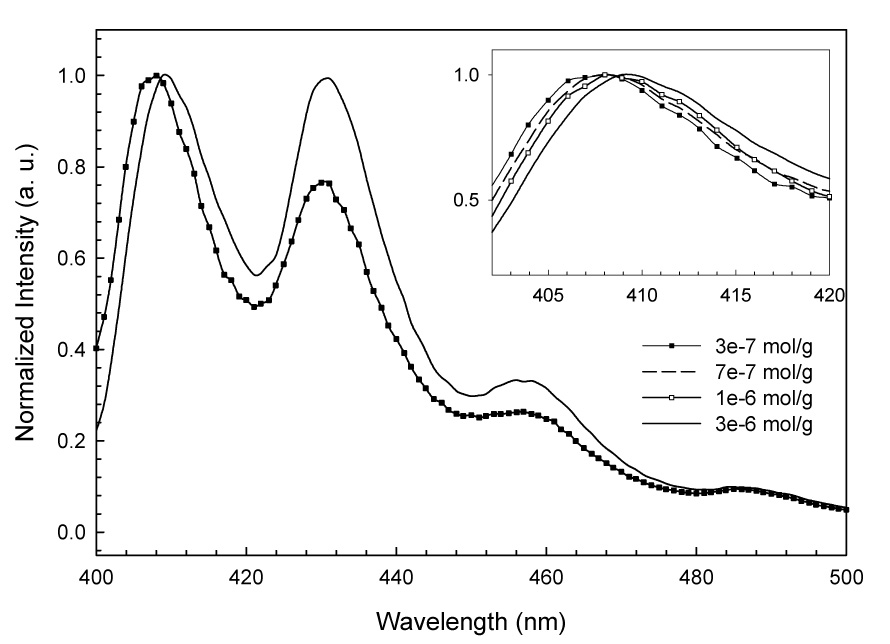
Bathochromic shift of the fluorescence emission spectra of BaP adsorbed on silica as loading increases. The inset shows the gradual red shift of the emission maxima at intermediate loadings.
3.
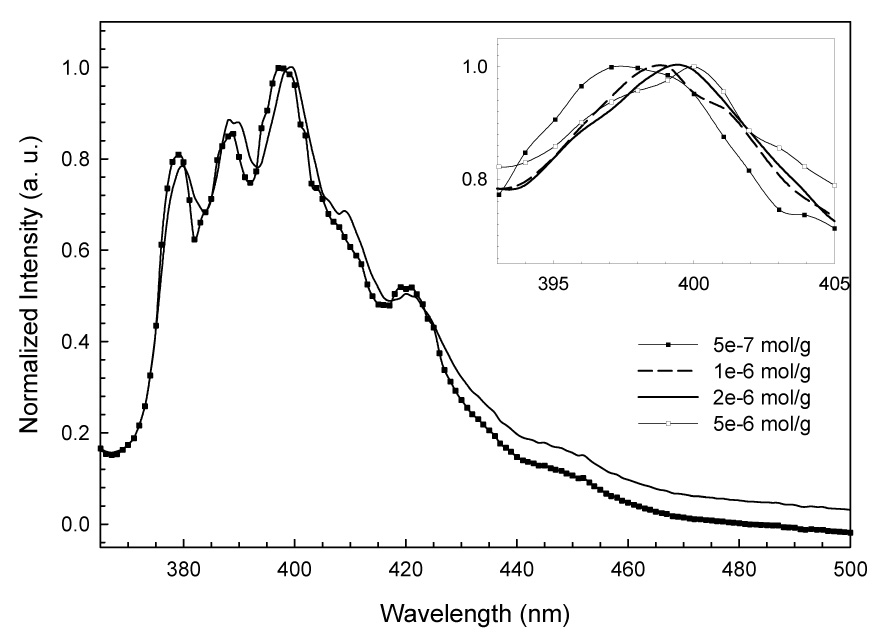
Bathochromic shift of the fluorescence emission spectra of BeP adsorbed on silica as loading increases. The inset shows the gradual red shift of the emission maxima at intermediate loadings.
4.
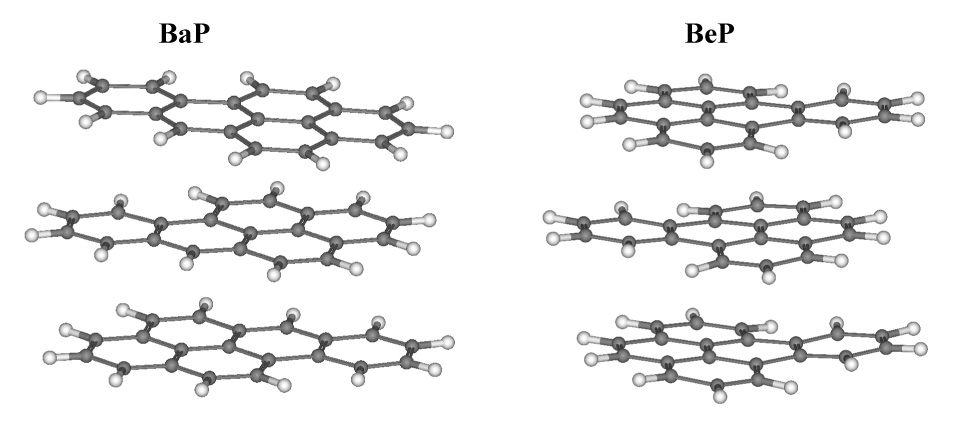
UFF optimized global minimum stacking structures of BaP and BeP trimers.
Table 1.
UFF optimized interplanar distances and rotation angles and MP2/6-31G* energies for BaP and BeP monomers, dimers and trimers.
| Isomer | EMP2/6-31G*(a. u.) | ΔErelative (kcal/mole) | ΔEbinding (kcal/mole) | ΔEHOMO-LUMO (a.u.) | Interplanar Distance (Å) | Rotation Angle (°) | |
|---|---|---|---|---|---|---|---|
| BaP | |||||||
| Monomer | −765.86209 | 0.2776 | |||||
| Dimer | A | −1531.75063 | 0.00 | 16.60 | 0.2615 | 3.46 | 179.6 |
| B | −1531.74841 | 1.40 | 0.2625 | 3.44 | 19.4 | ||
| C | −1531.74767 | 1.86 | 0.2625 | 3.44 | 339.0 | ||
| D | −1531.74112 | 5.97 | 0.2414 | 3.48 | 0.0 | ||
| Trimer | A | −2297.63785 | 0.00 | 32.36 | 0.2545 | 3.43/3.45 | 179.8/206.7 |
| B | −2297.63733 | 0.33 | 0.2533 | 3.43/3.45 | 179.2/158.7 | ||
| C | −2297.63516 | 1.68 | 0.2565 | 3.43/3.43 | 20.8/41.7 | ||
| D | −2297.63177 | 3.81 | 0.2550 | 3.45/3.45 | 179.3/360.0 | ||
| BeP | |||||||
| Monomer | −765.86553 | 0.3183 | |||||
| Dimer | A | −1531.75717 | 0.00 | 16.38 | 0.3164 | 3.43 | 180.0 |
| B | −1531.75697 | 0.12 | 0.3070 | 3.41 | 31.4 | ||
| C | −1531.75621 | 0.60 | 0.3032 | 3.46 | 146.5 | ||
| D | −1531.75517 | 1.25 | 0.3104 | 3.41 | 84.8 | ||
| E | −1531.75513 | 1.28 | 0.3090 | 3.44 | 151.1 | ||
| F | −1531.74695 | 6.41 | 0.2814 | 3.48 | 0.0 | ||
| Trimer | A | −2297.65083 | 0.00 | 34.03 | 0.3103 | 3.42/3.45 | 177.2/355.3 |
| B | −2297.65124 | 0.26 | 0.3036 | 3.40/3.40 | 27.4/56.3 |
The most stable conformations of BaP and BeP dimers and trimers are stacked, parallel and slightly displaced. The stacked, parallel structure is favored in most large PAHs [20]. Therefore the interplanar distances and rotation angles shown in Table 1 define the geometries relative to the global minimum energy structure. In the most stable BaP and BeP dimers (Table 1) the two molecules are rotated 180° with respect to each other and slightly displaced. In BaP trimer (Fig. 4) the topmost two molecules of the most stable isomer show a similar rotation, while the third is rotated by less than 30° with respect to the second. In BeP trimer the 180° rotation of the top two molecules is less stable than rotation through a smaller angle. This is the pattern seen in pyrene [20]. BeP also displays a slightly shorter intermolecular distance than BaP, indicating a more compact cluster structure.
The nature of cluster formation was experimentally tested by extracting the adsorbed samples with hexane. The hexane extracts analyzed spectrophotometrically indicate only monomer PAH present in solution, rather than the aggregates seen in the adsorbed samples. The aggregation/disaggregation process is thus confirmed to be completely reversible. This behavior is consistent with the view of PAH clusters being formed by weak interactions among the molecules. Adding water to the surface prior to adsorption of the PAH is also found to induce broadening and bathochromic shifts in the fluorescence spectra, even at loadings lower than 3 × 10−7 mol/g. When water molecules occupy the active sites of the silica surface, the hydrophobic BeP and BaP molecules shun the surface and adhere to each other. The effect is more pronounced with BeP than BaP, indicating either that BeP is more hydrophobic than BaP and/or that BeP clusters are more stable than those of BaP.
The MP2/6-31G* binding energies (Table 1) of the most stable isomers of BeP and BaP dimer are essentially identical, although the average BeP dimer is about 0.5 kcal/mole more stable than the average BaP dimer. The global minimum energy BeP trimer is more than 1.5 kcal/mol more stable than that of BaP with respect to three monomers. Therefore the calculated binding energies are consistent with the picture that BeP clusters are more stable than BaP.
PAH aggregation also affects the photodegradation kinetics of BeP and BaP. When samples of adsorbed BeP and BaP were irradiated with UV light, emission intensity decreased, indicating that photodegradation was underway. The photoproducts of the adsorbed PAHs have been previously reported. The principal products of BeP photodegradation are diones, diols and hydroxyl derivatives,9 and for BaP mainly diones.7 The photodegradation kinetics depends on surface coverage as can be noted from Figure 5. At low loading degradation of BaP and BeP follows first-order kinetics with apparent rate constants of 0.01 sec−1 for BeP and 0.03 sec−1 for BaP. For loadings of 1 × 10−6 mol/g or higher, kinetics is no longer first order, displaying rapid initial decay followed by a much slower second step. The initial decay kinetics are similar for low- and high-loading samples, but the slower parts of the degradation curves fit neither first nor second-order kinetics. This behavior suggests that monomeric PAH is being rapidly degraded by UV light, and that after the monomer is consumed, a slower reaction involving clusters takes place. The photoreactivity differences between the monomer and the clusters are more pronounced for BeP than for BaP, again in agreement with the slightly smaller trimer stabilization energies calculated for BaP than for BeP.
5.
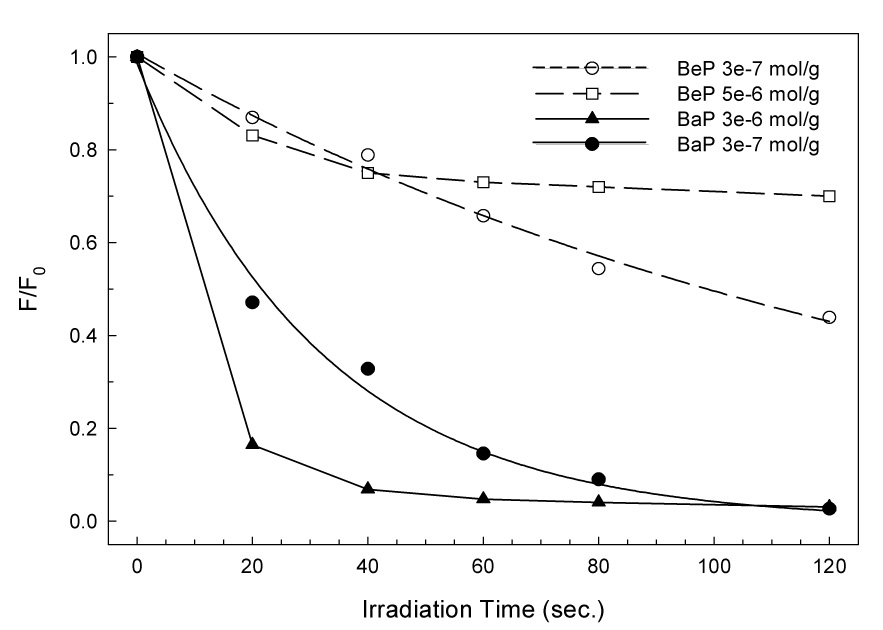
Photodegradation kinetics (relative fluorescence intensity vs. time) of BaP and BeP adsorbed on silica at high and low loadings.
BeP trimers are more structurally compact than BaP (Fig. 4). Attributing to BaP a looser aggregate structure and poorer stabilization of the trimer provides a good rationale for the experimental results that show less effect of BaP aggregation on reactivity and spectroscopic red shift in the adsorbed molecules.
Conclusions
Fluorescence spectra of BeP and BaP adsorbed on silica gel present a progressive red shift with increasing loading that is consistent with PAH cluster formation on the surface. The aggregation process of both PAHs is completely reversible, evidence consistent with weak π–π interaction between the stacked molecules. UFF molecular mechanics optimization of BaP and BeP dimers and trimers locate a number of energetically closely-spaced isomers. MP2/6-31G* single-point calculations reveal a narrowing of the HOMO-LUMO gap in proceeding from monomer to dimer to trimer in both molecules, consistent with the spectroscopically observed red shifts. The calculated dimer and trimer binding energies of BeP are slightly greater than those of BaP, consistent with observation that photodegradation kinetics of BeP differs from that of BaP and that BeP clusters are more stable. Although both the spectral shifts and calculated binding energies are small, the experimental results show a tendency that qualitatively correlates with the theoretical data.
Acknowledgements
The authors gratefully acknowledge support from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) through Grant P20 RR016470. The contents of this work are solely the responsibility of the authors and do not necessarily represent the official view of the NCRR or NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.IARC. Polynuclear Aromatic Hydrocarbons Part I: Chemical, Environmental and Experimental Data. Lyon: International Agency for Research on Cancer; 1983. [Google Scholar]
- 2.U. S. Department of Health and Human Services. P.H.S., ATSDR, Toxicological Profile for Polycyclic Aromatic Hydrocarbons (PAHs) Atlanta: 1995. [Google Scholar]
- 3.Shtein M, Mapel J, Benziger JB, Forrest SR. Appl. Phys Lett. 2002;81:268. [Google Scholar]
- 4.Kim KS, Tarakeshwar P, Lee JY. Chem Rev. 2000;100:4145. doi: 10.1021/cr990051i. [DOI] [PubMed] [Google Scholar]
- 5.Wasserfallen D, Kastler M, Pisula W, Hofer WA, Fogel Y, Wang Z, Müllen K. J. Am. Chem. Soc. 2006;128:1334. doi: 10.1021/ja056782j. [DOI] [PubMed] [Google Scholar]
- 6.Panne U, Knöller A, Kotzick R, Niessner R, Fresenius J. Anal. Chem. 2000;366:408. doi: 10.1007/s002160050083. [DOI] [PubMed] [Google Scholar]
- 7.Finlayson-Pitts BJ, Pitts JN. Atmospheric Chemistry. Wiley, New York: Fundamentals and Experimental Techniques; 1986. [Google Scholar]
- 8.Appel J, Bockhorn H, Frenklach M. Combustion and Flame. 2000;121:122. [Google Scholar]
- 9.Fioressi S, Arce R. Environ. Sci. Technol. 2005;39:3646. doi: 10.1021/es049192e. [DOI] [PubMed] [Google Scholar]
- 10.Dabestani R, Ellis KJ, Sigman ME. J. Photochem. Photobiol. 1995;86:231. [Google Scholar]
- 11.Dabestani R. Inter-Am. Photochem. Soc. Newsl. 1997;20:24. [Google Scholar]
- 12.Fouillet B, Chambon P, Chambon R, Castegnaro M, Weill N. Bull. Environ. Contam. and Toxicology. 1991;47:1. doi: 10.1007/BF01689445. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Pan W, Yang W. J. Chem. Phys. 1997;107:7921. [Google Scholar]
- 14.Grimme S, Antony J, Schwabe T, Mück-Lichtenfeld C. Org. Biomol. Chem. 2007;5:741. doi: 10.1039/b615319b. [DOI] [PubMed] [Google Scholar]
- 15.Špirko V, Engkvist O, Soldán P, Selzle HL, Schlag EW, Hobza P. J. Chem. Phys. 1999;111:572. [Google Scholar]
- 16.Arunan E, Gutowsky HS. J. Chem. Phys. 1993;98:4294. [Google Scholar]
- 17.Radloff W, Stert V, Freudenberg T, Hertel IV, Jouvet C, Dedonder-Lardeux C, Solgadi D. Chem. Phys. Lett. 1997;281:20. [Google Scholar]
- 18.Lee NK, Park S, Kim SK. J. Chem. Phys. 2002;116:7902. [Google Scholar]
- 19.Lee NK, Park S, Kim SK. J. Chem. Phys. 2005;122:031102. doi: 10.1063/1.1845432. [DOI] [PubMed] [Google Scholar]
- 20.Rapacioli M, Calvo F, Spiegelman F, Joblin C, Wales DJ. J. Phys. Chem. A. 2005;109:2487. doi: 10.1021/jp046745z. [DOI] [PubMed] [Google Scholar]
- 21.Borosky GL. J. Org. Chem. 1999;64:7738. [Google Scholar]
- 22.Coelho LAF, Marchut A, De Oliveira JV, Balbuena PB. Ind. Eng. Chem. Res. 2000;39:227. [Google Scholar]
- 23.Rappe AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM. J. Am. Chem.Soc. 1992;114:10024. [Google Scholar]
- 24.Casewit CJ, Colwell KS, Rappe AK. J. Am. Chem. Soc. 1992;114:10035. [Google Scholar]
- 25.Wohl AJ. Tetrahedron. 1968;24:6889. [Google Scholar]
- 26.Aihara J. J. Phys. Chem. A. 1999;103:7487. [Google Scholar]
- 27.Ruiz-Morales YJ. Phys. Chem. A. 2002;106:11283. [Google Scholar]
- 28.Frisch MJ, et al. Gaussian 03, Revision B.4, Gaussian, Inc. Pittsburgh, PA: 2003. [Google Scholar]