

Abstract
NMDA receptors bidirectionally modulate extracellular signal-regulated kinase (ERK) through the coupling of synaptic NMDA receptors to an ERK activation pathway that is opposed by a dominant ERK shut-off pathway thought to be coupled to extrasynaptic NMDA receptors. In the present study, synaptic NMDA receptor activation of ERK in cortical cultures was partially inhibited by the highly selective NR2B antagonist Ro25-6981 (Ro) and the less selective NR2A antagonist NVP-AAM077 (NVP). When Ro and NVP were added together, inhibition appeared additive and equal to that observed with the NMDA open-channel blocker MK-801. Consistent with a selective coupling of extrasynaptic NMDA receptors to the dominant ERK shut-off pathway, pre-block of synaptic NMDA receptors with MK-801 did not alter the inhibitory effect of bath applied NMDA on ERK activity. Lastly, in contrast to a complete block of synaptic NMDA receptor activation of ERK by extrasynaptic NMDA receptors, activation of extrasynaptic NMDA receptors had no effect upon ERK activation by brain-derived neurotrophic factor. These results suggest that the synaptic NMDA receptor ERK activation pathway is coupled to both NR2A and NR2B containing receptors, and that the extrasynaptic NMDA receptor ERK inhibitory pathway is not a non-selective global ERK shut-off.
Keywords: NMDA receptors, extracellular signal-regulated kinase, brain-derived neurotrophic factor, extrasynaptic, NR2A, NR2B
NMDA receptors play an important role in various forms of plasticity in the brain (Perez-Otano and Ehlers, 2005), and an important effector system implicated in NMDA receptor-associated plasticity is the Ras-ERK signaling cascade (Balazs, 2006, Wang et al., 2007). While it is well established that calcium influx through NMDA receptors can activate ERKs (Finkbeiner and Greenberg, 1996, Kornhauser and Greenberg, 1997), a curious observation in our previous study was that under basal conditions in which there is ongoing activity-dependent NMDA receptor-mediated ERK activation, bath application of NMDA did not further increase ERK activation but instead reduced it (Chandler et al., 2001). This lead us to propose that NMDA receptors bidirectionally modulate ERK activity via separate but competing stimulatory and inhibitory pathways. While we were able to largely define the components of the stimulatory pathway, the nature of the inhibitory pathway was not clear and it remained a mystery as to how activation of NMDA receptors could simultaneously activate opposing processes. Hardingham and colleagues (Hardingham et al., 2002) subsequently provided important insight into this process when they reported that synaptic NMDA receptors couple to a pro-survival CREB activation pathway whereas extrasynaptic NMDA receptors couple to an opposing pro-apoptotic pathway that exerts a dominant inhibitory action over CREB activation coupled to synaptic NMDA receptors. This seminal observation fit nicely with our bidirectional model and led us to propose a similar coupling of synaptic and extrasynaptic receptors for regulating ERK signaling (Sutton and Chandler, 2002). Recently, this suggestion was confirmed by convincing experimental evidence demonstrating coupling of extrasynaptic NMDA receptors to a dominant ERK shut-off pathway (Chandler and Luong, 2004, Ivanov et al., 2006).
The current study sought to address two important issues related to NMDA receptor modulation of ERK signaling. First, previous studies have shown important differences between NR2A- versus NR2B-containing NMDA receptors, including difference in scaffolding and signaling process to which they couple (van Zundert et al., 2004). Such differences may contribute to differences in plasticity associated with receptor subtypes. In relation to regulation of ERK signaling, it was reported that NR2B but not NR2A subunits associate with RasGRF1 leading to the suggestion that NMDA receptor activation of ERK signaling is selectively coupled to NR2B-containing NMDA receptors (Krapivinsky et al., 2003). However, this is in contrast to a subsequent report suggesting that NR2B-containing NMDA receptors selectively couple to inhibition of the ERK signaling pathway (Kim et al., 2005). Thus, we investigated this issue using subunit selective antagonists.
The second question addressed by the current study relates to the selectivity of the ERK shut-off pathway for the synaptic NMDA receptor activation pathway. While extrasynaptic NMDA receptors clearly exert a dominant inhibitory action over synaptic NMDA receptor activation of ERK, it is not known whether ERK activation through other processes is also subject to modulation by extrasynaptic NMDA receptors. We addressed this question by examining the effects of selective activation of extrasynaptic NMDA receptors on brain-derived neurotrophic factor (BDNF) activation of ERK. BDNF has emerged as a critical neuronal messenger in the brain not only because of its well-known pro-survival actions, but also because of its more recent identification as a mediator of experience-dependent plasticity (Lu, 2003, Binder and Scharfman, 2004). Interestingly, NMDA receptors and BDNF have important interactive actions as modulators of plasticity and survival. For example, it has been demonstrated that NMDA receptors play an important role in transcriptional regulation of BDNF formation.
Activation of synaptic NMDA receptors enhances BDNF transcription (via activation of CREB), while activation of extrasynaptic NMDA receptors inhibits BDNF transcription (via inhibition of CREB) (Vanhoutte and Bading, 2003). Whether this inhibitory action of extrasynaptic NMDA receptors at the level of the BDNF gene extends to a direct regulation of BDNF coupling to ERK activation is not known.
EXPERIMENTAL PROCEDURES
Reagents
All compounds were purchased from Sigma-Aldrich except for tetrodotoxin (TTX) (Calbiochem, San Diego, CA), Ro25-6981 (Tocris, Ellisville, MO), and NVP-AAM077 (generous gift from Novartis Labortories, Basel, Switzerland).
Preparation and Treatment of Cortical Neuronal Cultures
Postnatal day 1 rat pups from an established breeding colony of Sprague Dawley® rats (Harlan, Indianapolis, IN) were used to prepare cortical neuronal cultures as previously described (Chandler et al., 1997). In brief, brains were removed and placed in isotonic saline (pH 7.4) containing 100 units/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B. Pia matter and blood vessels were removed prior to finely mincing cortex with fine surgical scissors. Tissue was then dissociated using isotonic salt solution (pH 7.4) containing 0.25% trypsin (w/v) and incubated for 10 min at 37°C in a shaking water bath, followed by a 5 min incubation with 160 μg of DNase 1 prior to trituration (10 times). The cell suspension was placed in 50 ml of Dulbecco’s Modified Eagle’s Medium (DMEM) (Gibco, Grand Island, NY)/plasma-derived horse serum (PDHS) (Sigma-Aldrich, St. Louis, MO) and centrifuged for 10 min at 1000 × g. Cells were plated at a density of 3 × 106 cells in poly-L-lysine-precoated 35 mm culture dishes. Cultures were incubated in DMEM/PDHS for 3 days at 37°C in 7.5% CO2/92.5% air before replacing medium with fresh DMEM/PDHS containing 10 μM b-cytosine arabinoside. After 2 days treatment with b-cytosine arabinoside, the medium was replaced with DMEM/PDHS and cultures were grown for 14–16 days prior to their use.
To examine changes in phospho-ERK, cultures were washed twice with 1 ml of 25 mM HEPES buffer containing 140 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 10 μM glycine, 15 mM glucose (pH 7.4). After a 10 min acclimation period in the HEPES buffer, cultures were then subjected to various treatments as indicated in the results sections. In experiments examining the selective activation of extrasynaptic NMDA receptors by bath application of NMDA, synaptic NMDA receptors were first blocked with MK-801 in the presence of 50 μM bicuculline (Lu et al., 2001, Tovar and Westbrook, 2002). After 10 min of incubation, the MK-801-containing buffer was replaced with MK-801-free buffer, and the unblocked extrasynaptic receptors activated by bath application of NMDA.
Following treatment of the cultures, the cells were scraped into 100 μl of ice-cold homogenization buffer (50 mM Tris-HCl, 50 mM NaCl, 10 mM EGTA, 5 mM EDTA; 2 mM sodium pyrophosphate, 1 mM activated sodium orthovanadate, 0.2 mM AEBSF, 1 μg/ml aprotinin, 1 mM benzamide, 10 μg/ml leupeptin, 10 μg/ml pepstatin, pH 7.5). Isolated cells from three dishes were combined, probe sonicated for ~ 5 sec, and centrifuged at 15,000 × g for 30 min at 4°C. The resulting supernatant was removed and an aliquot was taken for determination of protein concentration by the bicinchoninic acid assay (Pierce Biotechnology, Inc., Rockford, IL). The remaining supernatant was stored at −80 °C until determination of ERK activity.
Gel Electrophoresis and Immunoblotting
An aliquot of each sample was diluted with an equal volume of 2× sample buffer yielding final concentrations of 50 mM Tris-HCl, 4% glycerol (w/v), 4% SDS, 1% 2-mercaptoethanol and bromophenol blue, pH 6.7. Samples were boiled for 5 min, and 20 μg of sample was separated on a 10% SDS-polyacrylamide gel using the buffer system of Laemmli and transferred to Millipore Immobilon-P PVDF membranes (Bedford, MA). After transfer, blots were washed with phosphate-buffered saline containing 0.05% Tween 20 (PBST) and then blocked with PBST containing 5% nonfat dried milk (NFDM) for 1 hr at room temperature with agitation. Levels of active ERK where assessed by immunoblot analysis of phospho-ERK 1/2. After blocking, the membranes were then incubated overnight at 4 °C with anti-phospho-ERK 1/2 (Cell Signaling Technology, Beverly, MA) diluted 1:1000 in PBST. The membranes were then washed in PBST prior to 1 hr incubation at 4 ºC with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody diluted 1:2000 in PBST containing 5% NFDM. Membranes received a final wash in PBST and the antigen-antibody complex was detected by enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL). Film autoradiograms were quantified by computer-assisted densitometry. Statistical analysis of raw optical density values was performed using Prism version 4.0c (GraphPad Software, Inc).
Whole-cell Patch-clamp Electrophysiology
Whole-cell patch-clamp recordings of spontaneous excitatory post-synaptic currents (sEPSC) measured across 30 sec intervals were carried out using an Axopatch 200B amplifier (Axon Instruments, Molecular Devices Corporation, Union City, CA). Currents were digitized at 5 kHz using an InstruTech (Port Washington, NY) ITC-16 interface and collected on a Macintosh (Apple Computers, Cupertino, CA) G4 computer running Axograph 4.9.2 graphics platform (Molecular Devices Corporation). Cells were bathed continuously with extracellular recording solution (pH 7.4, osmolarity 325 mOsm) containing the following (in mM): NaCl (140), KCl (5.4), CaCl2 (1.8), glucose (15), HEPES (5), glycine (0.01), and bicuculline (0.05). Patch pipettes (4–6 mOhm resistance) were pulled from borosilicate glass (0.86 mm ID; Warner Instruments, Hamden, CT) filled with internal recording solution (pH 7.2 osmolarity 280–290 mOsm) containing the following (in mM): K-gluconate (130), KCl (10) MgCl2 (2), EGTA (1), HEPES (10), K2ATP (2), GTP (0.3), and lidocaine N-ethyl bromide (5). Neurons selected for recordings were voltage-clamped at −65 mV. Pipette access resistance was monitored throughout each recording and only cells that demonstrated consistent access resistance (<15% change from baseline) were included in analyses.
Recordings were measured before, during and after washout of MK-801 in 30 sec epochs to determine the effect of treatment on sEPSC. The area of each sEPSC (in pA/sec) was determined using AxoGraph 4.9 software (Molecular Devices Corp) and the average sEPSC area was calculated across the 30 sec epoch. For measurement of NMDA-induced whole-cell currents, a multi-barreled Warner Perfusion Fast-Step system (Warner Instrument Corporation, Hamden, CT; ~8 ms switching time; 2 psi) was used to apply agonist for 5 sec by switching from the control barrel to the barrel containing NMDA and then back. Agonist-induced currents were recorded before and periodically after MK-801 bath application and the last 0.5 sec of steady-state amplitude was measured using AxoGraph 4.9 software. Data were analyzed using SPSS 12.0 software (SPSS Inc., Chicago, IL).
RESULTS
Both NR2A and NR2B receptor subtypes couple synaptic NMDA receptors to ERK activation
The role of NR2A versus NR2B subunits in NMDA receptor modulation of ERK signaling is controversial. To address the question of whether there is subunit selectivity in synaptic NMDA receptor activation of ERK in primary cortical cultures, we examined the effects of subunit antagonists upon activity-dependent NMDA receptor activation of ERK. Ro25-6981 (Ro) is a well-characterized antagonist that, at a concentration (e.g. 0.5 μM) that completely blocks NR2B/NR1 containing receptors, has no effect on NR2A/NR1 receptors (Bartlett et al., 2007). NVP-AAM077 (NVP) is an NMDA antagonist that, at a concentration (e.g. 0.5 μM) that completely blocks NR2A/NR1 receptors, produces a partial inhibition of NR2B/NR1 receptors (Bartlett et al., 2007). A 1-hr incubation of cortical neuronal cultures with either TTX (1 μM) to inhibit synaptic activity or MK-801 to block NMDA receptors, greatly reduced levels of phospho-ERK 1/2 (hereafter referred to as active ERK) by 83% and 75%, respectively (Fig 1A and B). This observation is in agreement with previous reports that the high basal levels of activated ERK in cortical neuronal cultures are primarily linked to activity-dependent activation of NMDA receptors (Chandler et al., 2001, Sutton and Chandler, 2002, Kim et al., 2005). In the mature high-density cortical cultures used in this study, removal of Mg2+ from the incubation buffer results in an increase in phospho-ERK levels and an increase in network activity (data not shown). In addition, removal of tonic inhibition by addition of bicuculline does not further increase phospho-ERK levels in Mg2+-free conditions (data not shown). The maximal synaptic NMDA receptor activation of ERK upon removal of Mg2+ most likely relates to the high levels of network driven synaptic activity that is characteristic of high-density cortical cultures (Wagenaar et al., 2005, Rolston et al., 2007). To examine the involvement of NR2A and NR2B subunits, cultures were incubated for 1-hr with Ro (0.5 μM) or NVP (0.5 μM). Separately, Ro and NVP partially reduced activate ERK by 42% and 33%, respectively, but in combination completely reduced levels of active ERK similar to that observed with MK-801. Together, these studies suggest that both NR2A- and NR2B-containing NMDA receptors couple to activation of ERK at the synapse.
Figure 1.
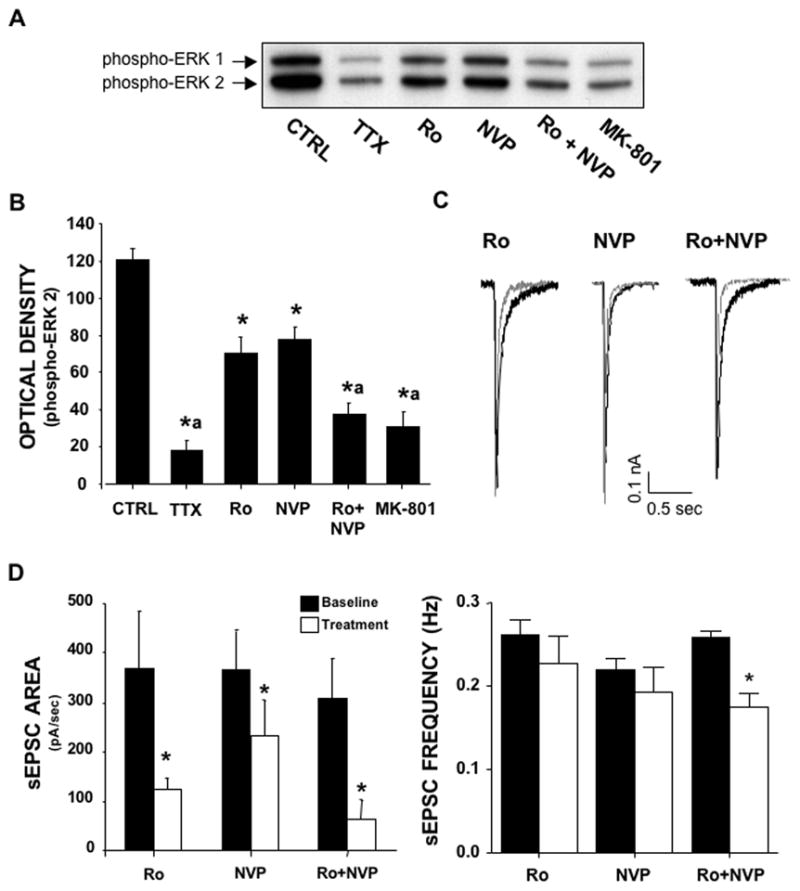
Contribution of NR2A- and NR2B-containing NMDA receptors to activity-dependent ERK activation. (A & B) Bath application (60 min) of the NR2B antagonist Ro (0.5 μM) or the NR2A antagonist NVP (0.5 μM) significantly reduced activity-dependent phospho-ERK levels. In comparison, treatment with TTX (1 μM), MK-801 (10 μM), or a combination of Ro and NVP markedly reduced phospho-ERK. (C & D) Bath application (20 min) of Ro or NVP reduced the area, but not the frequency of sEPSC. The combination of NVP and Ro largely attenuated the average area of sEPSC and produced a slight, yet significant, reduction in the frequency of sEPSC. Superimposed traces represent sEPSC before (black) and after (grey) exposure to antagonists alone or in combination.* p < .05 vs. CTRL; ª p < .05 vs. Ro or NVP; ANOVA with SNK post-hoc; n = 5–6.
The next experiments examined the effects of Ro and NVP on sEPSC that underlie activity-dependent activation of ERK. As shown in figure 1C and D, a 20 min exposure to Ro (0.5 μM) decreased the NMDA component of sEPSC as reflected by a large reduction in the average area of the current. A similar 20 min exposure to NVP (0.5 μM) also reduced the NMDA component of the sEPSC but to a lesser extent than did R0, while the combination of Ro and NVP completely eliminated the NMDA component of the sEPSC. Although Ro reduction in the area of the sEPSC was greater than that produced by NVP, caution should be exercised in regards to interpreting this as indicating that NR2B-containing receptors contribute more to the sEPSC than do NR2A-containing receptors. This is because NR2B-containing receptors have much slower kinetics than do NR2A-containing receptors. The remaining current was blocked by addition of NBQX (data not shown) consistent with it being the AMPA component of the sEPSC. Since activity-dependent NMDA receptor coupling to ERK activation depends upon spontaneous activity, it is possible that the reduction in the levels of active ERK by Ro or NVP does not reflect a direct coupling of the receptors to ERK activation, but instead an indirect effect via a reduction in the frequency of the sEPSC. However, neither inhibitor alone altered the frequency of the sEPSC (but there was a significant reduction when added in combination). Thus, it appears unlikely that a reduction in the frequency of spontaneous activity contributes to the inhibitory effect of either Ro or NVP.
Extrasynaptic NMDA receptors couple to a dominant ERK shut-off pathway
The next set of experiments examined extrasynaptic NMDA receptor modulation of ERK activity without the potential confound of synaptic NMDA receptor modulation of ERK. To isolate extrasynaptic receptors, we utilized the previously described MK-801 trapping technique to selectively inactivate synaptic NMDA receptors (Lu et al., 2001, Tovar and Westbrook, 2002). This procedure takes advantage of the fact that MK-801 is a use-dependent open channel blocker with a very slow off rate. When cultures that are spontaneously active are incubated with MK-801 by bath application, only NMDA receptors that are activated by spontaneous glutamate release (e.g. synaptic receptors) will be blocked by MK-801. In contrast, the inactive (e.g. extrasynaptic) NMDA receptors remain unblocked because they have not been activated. Following washout of MK-801 from the bath, the extrasynaptic NMDA receptors can then be selectively activated by bath application of NMDA.
Under certain experimental conditions, a time-dependent recovery of synaptic NMDA receptors has been reported following the MK-801 blocking procedure due to the lateral translocation into the synapse of unblocked extrasynaptic NMDA receptors (Tovar and Westbrook, 2002). As recovery of synaptic NMDA receptors could confound the use of this procedure in our in our studies, we again used whole-cell patch clamp to measure the block and potential recovery of synaptic NMDA currents. As shown in figure 2A, bath application of MK-801 (10 μM) resulted in a rapid decline in the NMDA component of the sEPSC that showed no recovery 30 min after washout. This observation is consistent with a recent report demonstrating a reduction in the NMDA component of miniature EPSCs following MK-801 trapping in cortical cultures (Liu et al., 2007). The effect of MK-801 was likely a reduction of the slow NMDA component of the sEPSC given that the remaining component of the sEPSC was completely eliminated by the local application of 50 μM NBQX (data not shown). In addition, the frequency of the sEPSC was not changed by MK-801 trapping (Fig 2B). Finally, the focal application of NMDA (50 μM) confirmed that while there was a large reduction in the whole-cell NMDA current following MK-801 trapping, there remained a substantial amount of current from unblocked extrasynaptic NMDA receptors (Fig 2C). This is consistent with the observation that preblocking all NMDA receptors with MK-801 by coincubation with NMDA completely eliminated all subsequent NMDA-activated current (data not shown). Together, these observations confirm that under the experimental conditions employed, bath application of MK-801 results in a selective block of synaptic receptors and no recovery of the synaptic NMDA currents 30 min after washout of MK-801.
Figure 2.

Persistent block of synaptic NMDA receptors using the MK-801 trapping procedure. (A) MK-801 (10 μM) exposure in the presence of 50 μM bicuculline for 10 min markedly reduced the area of sEPSC that persisted up to 30 min after MK-801 washout. Traces represent sEPSC before and during MK-801 exposure, as well as after washout of MK-801. (B) The frequency of sEPSC was not changed during MK-801 exposure or after washout. (C) The amplitude of steady-state whole-cell current induced by a 5 sec pulse of NMDA (50 μM) continued to be significantly attenuated 30 min washout of MK-801. The remaining current presumably reflects the presence of unblocked extrasynaptic NMDA receptors. Traces represent whole-cell NMDA currents before (black) and after (grey) MK-801 washout. * p < .05 vs. CTRL; ANOVA with SNK post-hoc; n = 6–9.
After establishing the validity of the MK-801 trapping technique in our experimental system, we next turned our attention to examining extrasynaptic modulation of ERK signaling. The first set of experiments compared the NMDA dose-response for inhibition of ERK activity in control cultures and after blocking synaptic NMDA receptors. As shown in figure 3A, the protocol involved an initial 30 min incubation with bicuculline to remove any GABAergic inhibition of network activity, thus ensuring maximal synaptic activation of ERK. After an additional 10 min incubation with and without MK-801, the culture medium was replaced with fresh medium with and without NMDA. As shown in figure 3B and 3C, bath application of NMDA reduced the levels of active ERK at all concentrations of NMDA tested. Furthermore, the NMDA dose-response curve was virtually identical regardless of whether the synaptic NMDA receptor ERK activation pathway was active or had been inactivated by the MK-801 trapping procedure. These results are consistent with the selective coupling of extrasynaptic NMDA receptors to the dominant ERK shut-off pathway.
Figure 3.

Bath application of NMDA reduces activity-dependent levels of phospho-ERK even in the presence of blocked synaptic NMDA receptors. (A) Treatment and time course of experimental protocol. (B) Representative immunoblots of phospho-ERK. (C) Corresponding graph of densitometry analysis of band intensity of phospho-ERK 2. In cultures pre-treated with 50 μM bicuculline, 30 min NMDA (25 – 100 μM) exposure significantly reduced phsopho-ERK levels. When synaptic receptors were blocked with 10 μM MK-801, a nearly identical reduction in phospho-ERK was observed following NMDA treatment. * p < .05 vs. CTRL; ** p < .01 vs. CTRL ; ANOVA with SNK post-hoc; n = 5.
The dominant extrasynaptic NMDA receptor ERK shut-off pathway is not a global mechanism for inhibition of ERK signaling
The next series of experiments addressed the question of whether the dominant extrasynaptic ERK inhibitory pathway exerts a similar inhibitory action over non-NMDA receptor-mediated activation of ERK as it does over the synaptic pathway. BDNF is a well characterized growth factor that has pro-survival actions and has been implicated in synaptic neuroplasticity (Lu, 2003, Binder and Scharfman, 2004), and many of the actions of BDNF occur through ERK signaling (Kaplan and Miller, 2000, Huang and Reichardt, 2003). To examine the effect of activation of extrasynaptic NMDA receptors on BDNF activation of ERK, synaptic NMDA receptors were initially blocked using the MK-801 trapping procedure (figure 4A). This was followed by a 60 min exposure to 1 μM TTX (to reduce basal levels of active ERK) and a subsequent 30 min exposure to NMDA (50 μM) with and without BDNF (5 ng/ml). As shown in figure 4B and C, BDNF alone caused a robust increase in the levels of active ERK consistent with its well-known ERK activation properties. Surprisingly, NMDA had no effect upon BDNF activation of ERK when added in combination. This is in stark contrast to the dominant inhibitory action of extrasynaptic NMDA receptors over synaptic NMDA receptor activation of ERK.
Figure 4.
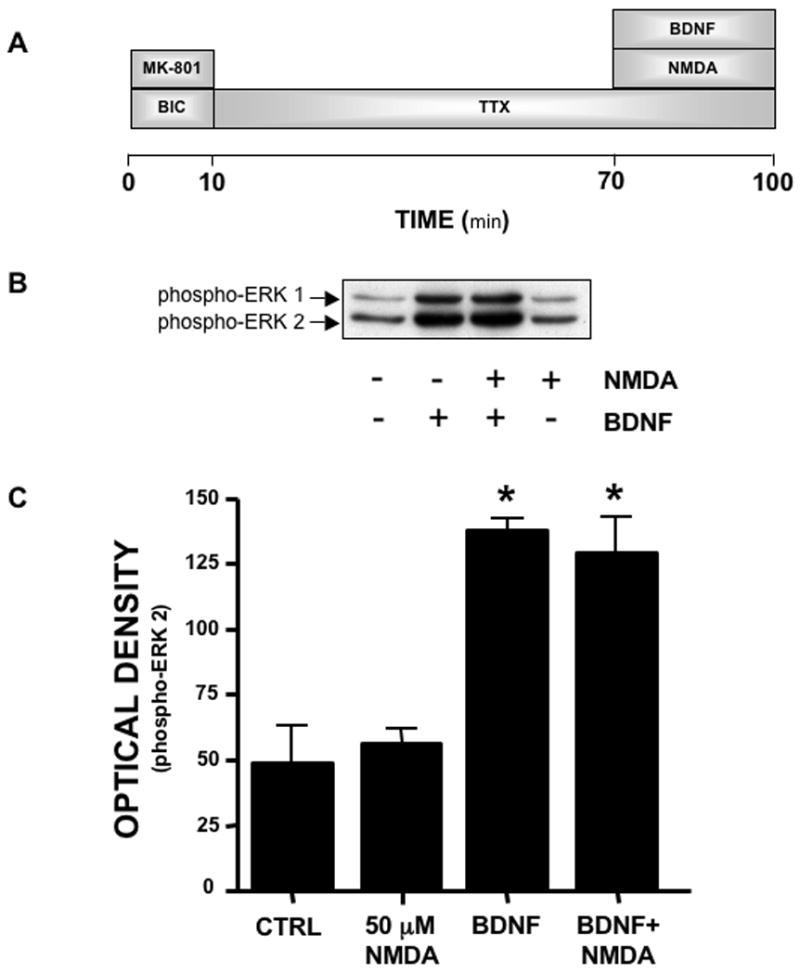
Activation of extrasynaptic NMDA receptors does not block BDNF activation of ERK. (A) Experimental protocol. (B) Representative immunoblots of phospho-ERK. (C) Analysis of band intensity of phospho-ERK 2. Following selective block of synaptic NMDA receptors with 10 μM MK-801, BDNF (5 ng/ml) exposure resulted in a significant increase in phospho-ERK. When 50 μM NMDA was co-exposed with BDNF, the level of phospho-ERK did not differ from that observed in cultures treated with BDNF alone. * p < .01 vs. CTRL (ANOVA with SNK post-hoc). n = 4.
A potential confound of the above experiments demonstrating a lack of effect of NMDA on BDNF activation of ERK may be differences in the temporal properties of the two processes. In particular, it is possible that the extrasynaptic NMDA receptor inhibitory pathway requires activation or priming prior to BDNF activation of ERK to have an inhibitory action. To examine this, we employed the same experimental treatment protocol used in figure 4, with the exception that addition of BDNF was delayed for 10 min after the addition of NMDA. Again, BDNF alone cause a large increase in the levels of active ERK that was only slightly reduced by NMDA. However, subtraction of the respective control values revealed that even this small reduction of the BDNF response by NMDA was most likely attributable to a reduction in the underlying basal levels of active ERK. Regardless, these results are again in stark contrast to the dominant inhibitory action of extrasynaptic NMDA receptors on the synaptic NMDA receptor ERK activation pathway.
DISCUSSION
Previous studies have shown that NMDA receptors bidirectionally modulate ERK activity by coupling to opposing stimulatory and inhibitory pathways (Chandler et al., 2001, Kim et al., 2005, Ivanov et al., 2006). The results of the present study in primary cortical cultures are in agreement with the suggestion that the opposing actions of NMDA receptors on ERK activity are a function of their spatial segregation within the dendritic membrane (Sutton and Chandler, 2002, Ivanov et al., 2006). We observed that the ERK activation pathway is selectively coupled to synaptic NMDA receptors while the dominant ERK inhibition pathway is selectively coupled to extrasynaptic NMDA receptors. In addition, we found no evidence in cortical cultures of a differential coupling of NR2A versus NR2B subunits to the activation and inhibition pathways. To the contrary, both NR2A and NR2B-containing NMDA receptors mediated synaptic activation of ERK. Finally, we found that BDNF activation of ERK is not subject to inhibitory modulation by extrasynaptic NMDA receptors but instead functions autonomously. This suggests that the dominant extrasynaptic NMDA receptor shut-off pathway is not a global mechanism for inactivation of ERK.
Although complex and potentially brain region specific, previous studies have largely defined many of the molecular components involved in NMDA receptor coupling to activation of the Ras-ERK signaling cascade (Chandler et al., 2001, Balazs, 2006, Wang et al., 2007). These components include a Src-like non-receptor kinase, phosphatidylinositide 3-kinase, calcium/calmodulin-dependent protein kinase II, and free Gbg subunits. In addition, guanine nucleotide exchange factors (GEFs) that activate Ras, and GTPase activating proteins (GAPs) that inhibit Ras, also play an important role in NMDA activation of Ras-ERK signaling (Thomas and Huganir, 2004, Balazs, 2006). Of note is the observation that the Ras-GEF RasGRF1 physically associates with NMDA receptors to facilitate activation of Ras-ERK signaling in hippocampus (Krapivinsky et al., 2003). This association was found to be specific for the NR2B subunit as there was no association of RasGRF1 with NR2A subunits, leading to the suggestion that only NR2B-containing NMDA receptors couple to activation of ERK signaling. Chen and colleagues (Chen et al., 2007) used the subunit antagonists Ro and NVP, and C-terminal NR2A and NR2B subunit selective mutants that interfere with the ability of the subunits to interact with intracellular signaling molecules. In agreement with the finding of Krapivinsky et al (2003), Chen et al (2007) found that NR2B but not NR2A receptors couple to activity-dependent ERK activation in hippocampus. However, in apparent contradiction, Kim and colleagues (2005) observed in hippocampus that NR2B but not NR2A receptors couple to inhibition rather than activation of ERK. This appeared to occur through the selective association of NR2B subunits with the Ras-GAP SynGAP. These authors also suggested that NMDA receptor activation of ERK signaling was selectively coupled to the NR2A subunit. Lastly, Li et al. (Li et al., 2006), also working in hippocampus and using Ro and NVP, reported that NR2A-containing receptors stimulate ERK via Ras-GRF2. While the results of the present study confirm that extrasynaptic NMDA receptors couple to the dominant inhibitory pathway, we found no evidence that synaptic NMDA receptors can couple to inhibition of ERK signaling or that extrasynaptic NMDA receptors can couple to activation of ERK signaling in cortical cultures. Instead, our results strongly support a bidirectional model of NMDA receptor coupling to ERK that is based upon the differential compartmentalization of the stimulatory pathway with synaptic NMDA receptors and the dominant inhibitory pathway with extrasynaptic NMDA receptors, and not upon a differential NR2A/B subunit coupling. The reason for the apparent discrepancy between our results and those of Chen et al. (2007) in terms of the coupling of NR2A receptors to activity-dependent ERK activation is not clear, but may relate to factors such as brain regional differences (cortex versus hippocampus) and/or differences in culture conditions.
Our conclusion that both NR2A and NR2B subunits couple to ERK activation is based upon the observation that separately, the NR2A antagonist NVP and the NR2B antagonist Ro only partially inhibit synaptic NMDA receptor activation of ERK, whereas in combination they completely blocked NMDA activation of ERK. It could be argued that the relatively low selectivity of NVP for NR2A over NR2B containing receptors confounds this interpretation. However, when considered with the results obtained with Ro alone (e.g. there was only partial inhibition of ERK activation at a concentration that nearly completely block NR2B/NR1 currents) the most likely explanation is that both NR2A and NR2B containing receptors contribute to synaptic NMDA receptor activation of ERK signaling in cortex.
The subunit composition of synaptic and extrasynaptic NMDA receptors is controversial and likely varies between brain region and even between neurons within a particular region. Studies in cell culture have demonstrated that early in development, both synaptic and extrasynaptic NMDA receptors are primarily NR2B-containing (van Zundert et al., 2004, Kohr, 2006). This is followed by a development-dependent addition of NR2A subunits that contribute to synaptic currents (Flint et al., 1997, Stocca and Vicini, 1998). This subunit-substitution hypothesis supported the idea that in the developed brain, NR2A and NR2B-containing NMDA receptors segregate almost exclusively into synaptic and extrasynaptic compartments, respectively. This segregation model gained additional significance in light of studies reporting functional differences in synaptic versus extrasynaptic receptors. However, more recent studies have cast doubt upon an absolute segregation of NR2A- and NR2B-containing NMDA receptors into synaptic and extrasynaptic compartments. For example, one study in mature hippocampal cultures reported that ~55% of the synaptic and ~49% of the extrasynaptic NMDA current was blocked by the NR2B subunit antagonist ifenprodil, respectively (Thomas et al., 2006). Similarly, a recent study in acutely dissected hippocampal slices reported synaptic and extrasynaptic NMDA receptors contain approximately equal amounts of NR2A and NR2B subunits (Harris and Pettit, 2007). Another recent study carried out in mature primary cortical cultures reported that ~34% of the synaptic NMDA current was NR2B subunit dependent whereas ~ 26% of the extrasynaptic current was NR2A subunit dependent (Liu et al., 2007). In the current study, our observations in mature cortical cultures using NR2A and NR2B subunit antagonist are generally consistent with the above findings. We observed that both NR2A and NR2B-containing receptors contribute substantially to the synaptic EPSC and to activity-dependent activation of ERK. However, we did not examine the effects of subunit antagonists upon extrasynaptic NMDA currents or upon the shut-off pathway, and we thus cannot make any conclusions concerning the NR2 subunit composition of the extrasynaptic NMDA receptor ERK shut-off pathway.
BDNF is a neurotrophic factor that plays an important role in neuronal survival, plasticity, and addiction, and many of the actions of BDNF occur through TrkB receptor coupling to the Ras-ERK signaling cascade (Kaplan and Miller, 2000, Huang and Reichardt, 2003). An increasingly popular view is that both NMDA receptors and BDNF modulate experience-dependent synaptic plasticity (Lu, 2003, Nagappan and Lu, 2005). Furthermore, evidence indicates important cross-talk between BDNF and NMDA receptors in modulation of synaptic plasticity (Yamada and Nabeshima, 2004). For example, BDNF synthesis and release is regulated by NMDA receptor activity, and BDNF acutely potentiates NMDA receptor-dependent LTP. Since extrasynaptic NMDA receptors activate a dominant ERK shut-off mechanism, and since there is a large pool of dendritic extrasynaptic TrkB receptors that presumably couple to ERK activation (Swanwick et al., 2004), we hypothesized that activation of extrasynaptic receptors would also inhibit BDNF activation of ERK. Surprisingly, this was found not to be the case in primary cortical cultures as selective activation of extrasynaptic NMDA receptors did not alter BDNF activation of ERK. This is in contrast to the reported ability of NMDA receptors to bidirectionally control BDNF gene expression (via synaptic NMDA receptor activation of CREB that enhances BDNF expression that is opposed by extrasynaptic NMDA receptor CREB shut-off pathway that inhibits BDNF expression) (Vanhoutte and Bading, 2003). Synaptic NMDA receptors were also recently shown to induce a coordinated upregulation of pro-survival genes and downregulation of pro-death genes (Zhang et al, 2007). In contrast, extrasynaptic NMDA receptors did not activate this neuroprotective program, but instead induced a small number of pro-death genes. An interesting future direction will be to characterize the interaction of synaptic and extrasynaptic NMDA receptors with BDNF in regulating the expression of pro-survival and pro-death gene programs.
In summary, the current study demonstrates that bidirectional NMDA receptor regulation of ERK activity occurs through the compartmentalization of distinct signaling pathways with spatially distinct populations of NMDA receptors. This is consistent with a model in which both synaptic NR2A and NR2B NMDA receptors couple to activation of ERK, while extrasynaptic NMDA receptors couple to the dominant ERK shut-off pathway. Furthermore, the extrasynaptic NMDA receptor pathway is not a global and non-specific mechanism for shutting-off ERK activity. These observations have important implications for the potential development of therapeutic strategies aimed at activating pro-survival pathways that oppose pathological activation of extrasynaptic NMDA receptors. In addition, sub-pathological activation of extrasynaptic NMDA receptors may play a critical role in modulating synaptic efficacy during periods of glutamate spill-over by providing a feedback mechanism that selectively attenuates synapse-specific ERK signaling coupled to synaptic NMDA receptors.
Figure 5.

Activation of extrasynaptic NMDA receptors prior to BNDF exposure does not block BDNF activation of ERK. (A) Treatment and time course of experimental protocol. In these experiments, BDNF exposure was delayed 10 min following extrasynaptic NMDA receptor activation. (B) Representative immunoblots of phospho-ERK. (C) Graph of densitometry analysis of band intensity of phospho-ERK 2. BDNF greatly increased phospho-ERK levels above that observed in control cultures. In cultures where extrasynaptic NMDA receptors were activated 10 min prior to BDNF exposure, phospho-ERK levels remained elevated above that observed in NMDA-treated cultures. (Δ) Represents BDNF-induced for each condition (Δ BDNF was calculated by subtracting the control value form the BDNF value; Δ BDNF+NMDA was calculated by subtracting the NMDA value from the BDNF+NMDA value. * p < .01 vs. CTRL; ANOVA with SNK post-hoc; n = 3.
Acknowledgments
This work was supported by NIH grant AA010983. P.J.M. is supported by National Research Service Award AA016450.
Abbreviations
- BDNF
brain-derived neurotrophic factor
- DMEM
Dulbecco’s Modified Eagle’s Medium
- ERK
extracellular signal-regulated kinase
- NVP
NVP-AAM077
- PBST
phosphate-buffered saline containing Tween 20
- PDHS
plasma-derived horse serum
- Ro
Ro25-6981
- sEPSC
spontaneous excitatory post-synaptic currents
- TTX
tetrodotoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balazs R. Trophic effect of glutamate. Curr Top Med Chem. 2006;6:961–968. doi: 10.2174/156802606777323700. [DOI] [PubMed] [Google Scholar]
- Bartlett TE, Bannister NJ, Collett VJ, Dargan SL, Massey PV, Bortolotto ZA, Fitzjohn SM, Bashir ZI, Collingridge GL, Lodge D. Differential roles of NR2A and NR2B-containing NMDA receptors in LTP and LTD in the CA1 region of two-week old rat hippocampus. Neuropharmacology. 2007;52:60–70. doi: 10.1016/j.neuropharm.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Binder DK, Scharfman HE. Brain-derived neurotrophic factor. Growth Factors. 2004;22:123–131. doi: 10.1080/08977190410001723308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandler LJ, Luong NT. Proceedings of the Society for Neuroscience, 2004 Abstract Viewer/Itinerary Planner. San Diego, CA. Washington, DC: Society for Neuroscience, Program No. 963.13; 2004. Extrasynaptic NMDA receptors stimulate an ERK shut-off pathway that is selective for the synaptic ERK activation pathway. [Google Scholar]
- Chandler LJ, Sutton G, Dorairaj NR, Norwood D. N-methyl D-aspartate receptor-mediated bidirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. J Biol Chem. 2001;276:2627–2636. doi: 10.1074/jbc.M003390200. [DOI] [PubMed] [Google Scholar]
- Chandler LJ, Sutton G, Norwood D, Sumners C, Crews FT. Chronic ethanol increases N-methyl-D-aspartate-stimulated nitric oxide formation but not receptor density in cultured cortical neurons. Mol Pharmacol. 1997;51:733–740. doi: 10.1124/mol.51.5.733. [DOI] [PubMed] [Google Scholar]
- Chen Q, He S, Hu XL, Zhou J, Zheng J, Zhang S, Zhang C, Duan WH, Xiong ZQ. Differential roles of NR2A- and NR2B-containing NMDA receptors in activity-dependent Brain-Derived Neurotrophic Factor gene regulation and limbic epileptogenesis. J Neurosci. 2007;27:542–552. doi: 10.1523/JNEUROSCI.3607-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Greenberg ME. Ca(2+)-dependent routes to Ras: mechanisms for neuronal survival, differentiation, and plasticity? Neuron. 1996;16:233–236. doi: 10.1016/s0896-6273(00)80040-9. [DOI] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- Harris AZ, Pettit DL. Extrasynaptic And Synaptic Nmdars Form Stable And Uniform Pools In Hippocampal Slices. J Physiol. 2007 doi: 10.1113/jphysiol.2007.137679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I. Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J Physiol. 2006;572:789–798. doi: 10.1113/jphysiol.2006.105510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kohr G. NMDA receptor function: subunit composition versus spatial distribution. Cell Tissue Res. 2006;326:439–446. doi: 10.1007/s00441-006-0273-6. [DOI] [PubMed] [Google Scholar]
- Kornhauser JM, Greenberg ME. A kinase to remember: dual roles for MAP kinase in long-term memory. Neuron. 1997;18:839–842. doi: 10.1016/s0896-6273(00)80322-0. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Manasian Y, Ivanov A, Tyzio R, Pellegrino C, Ben-Ari Y, Clapham DE, Medina I. The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron. 2003;40:775–784. doi: 10.1016/s0896-6273(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Li S, Tian X, Hartley DM, Feig LA. Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J Neurosci. 2006;26:1721–1729. doi: 10.1523/JNEUROSCI.3990-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wong TP, Aarts M, Rooyakkers A, Liu L, Lai TW, Wu DC, Lu J, Tymianski M, Craig AM, Wang YT. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Lu B. Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci. 2005;28:464–471. doi: 10.1016/j.tins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Perez-Otano I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 2005;28:229–238. doi: 10.1016/j.tins.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Rolston JD, Wagenaar DA, Potter SM. Precisely timed spatiotemporal patterns of neural activity in dissociated cortical cultures. Neuroscience. 2007;148:294–303. doi: 10.1016/j.neuroscience.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocca G, Vicini S. Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol. 1998;507(Pt 1):13–24. doi: 10.1111/j.1469-7793.1998.013bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton G, Chandler LJ. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. J Neurochem. 2002;82:1097–1105. doi: 10.1046/j.1471-4159.2002.01031.x. [DOI] [PubMed] [Google Scholar]
- Swanwick CC, Harrison MB, Kapur J. Synaptic and extrasynaptic localization of brain-derived neurotrophic factor and the tyrosine kinase B receptor in cultured hippocampal neurons. J Comp Neurol. 2004;478:405–417. doi: 10.1002/cne.20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CG, Miller AJ, Westbrook GL. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;95:1727–1734. doi: 10.1152/jn.00771.2005. [DOI] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- van Zundert B, Yoshii A, Constantine-Paton M. Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci. 2004;27:428–437. doi: 10.1016/j.tins.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Vanhoutte P, Bading H. Opposing roles of synaptic and extrasynaptic NMDA receptors in neuronal calcium signalling and BDNF gene regulation. Curr Opin Neurobiol. 2003;13:366–371. doi: 10.1016/s0959-4388(03)00073-4. [DOI] [PubMed] [Google Scholar]
- Wagenaar DA, Madhavan R, Pine J, Potter SM. Controlling bursting in cortical cultures with closed-loop multi-electrode stimulation. J Neurosci. 2005;25:680–688. doi: 10.1523/JNEUROSCI.4209-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JQ, Fibuch EE, Mao L. Regulation of mitogen-activated protein kinases by glutamate receptors. J Neurochem. 2007;100:1–11. doi: 10.1111/j.1471-4159.2006.04208.x. [DOI] [PubMed] [Google Scholar]
- Yamada K, Nabeshima T. Interaction of BDNF/TrkB signaling with NMDA receptor in learning and memory. Drug News Perspect. 2004;17:435–438. doi: 10.1358/dnp.2004.17.7.863702. [DOI] [PubMed] [Google Scholar]