

Abstract
The human interferon-induced protein kinase PKR is a key component of innate immunity, a process in which it senses pathogenic RNA. PKR consists of an N-terminal dsRNA-binding domain (dsRBD) and a C-terminal kinase domain. Upon binding long (>33 base pairs) stretches of pathogenic dsRNA, PKR undergoes autophosphorylation, which activates it to phosphorylate eIF2α, leading to inhibition of translation initiation. Many cellular and viral transcripts contain nucleoside modifications, and these could affect PKR activation. For example, a 5′-triphosphate confers the ability of relatively unstructured transcripts to activate PKR. Effects of internal RNA modifications on PKR activation have not been reported. Herein, PKR activation by ssRNA and dsRNA containing internal nucleobase, sugar, and phosphodiester modifications is analyzed. We find that for 5′-triphosphate-containing ssRNA, most base and sugar modifications abrogate activation, although 2′-fluoro-modified ssRNA does not, indicative of a critical role for hydrogen bonding at the ribose sugar. In the case of dsRNA, a more limited set of nucleoside modifications affect PKR activation. Watson–Crick base-pairing is required for activation, and some minor groove modifications abrogate activation while major groove modifications have little effect. Surprisingly, GU wobble pairs also largely abrogate dsRNA-mediated activation when present at modest levels. Modifications to dsRNA that abrogate activation have no significant effect on dsRBD binding, allowing such RNAs to act as inhibitors and suggesting a nonequivalence of binding ability and activation. Overall, the findings indicate that nucleoside modifications and wobble pairing may serve to discriminate self-RNA and pathogenic RNA in innate immunity.
Keywords: modified RNA, PKR activation, RNA folding, translation
INTRODUCTION
PKR is an interferon-inducible protein kinase and a key component of the innate immunity antiviral pathway (Williams 1999; Dever et al. 2007). Long stretches of double-stranded RNA (dsRNA) (>33 base pairs [bp]) activate PKR to undergo autophosphorylation and phosphorylate its cellular substrate, translation initiation factor eIF2α at serine 51 (Manche et al. 1992; Zheng and Bevilacqua 2004; Garcia et al. 2006). This process inhibits translation initiation, thus halting protein synthesis. In addition to its role as a translational regulator, PKR also affects signal transduction. For example, transcription factor, nuclear factor-κB (NF-κB), which is activated by a variety of stimuli, can be regulated by PKR-mediated signaling (Kumar et al. 1994).
PKR consists of a dsRNA-binding domain (dsRBD), which comprises two tandem copies of the dsRNA-binding motif (dsRBM), and a catalytic kinase domain (Fig. 1A). The dsRBD interacts with dsRNA in a non–sequence-specific fashion and does not bind appreciably to dsDNA or RNA–DNA hybrids (Hunter et al. 1975; Bevilacqua and Cech 1996). In addition to dsRNA, certain highly structured single-stranded viral and cellular RNAs with imperfect loops, bulges, and single-strand tails can activate PKR (Davis and Watson 1996; Bevilacqua et al. 1998; Ben-Asouli et al. 2002; Bommer et al. 2002; Zheng and Bevilacqua 2004). We recently demonstrated that PKR can also be activated by relatively unstructured ssRNA transcripts if a 5′-triphosphate is present, wherein the 5′-triphosphate likely serves as a pathogen-associated molecular pattern (PAMP) (Nallagatla et al. 2007). PKR can also be activated by non-RNA molecules including heparin, dextran sulfate, and poly-L-glutamate (Hovanessian and Galabru 1987; Fasciano et al. 2005). Despite identification of these diverse PKR activators, a clear understanding of the RNA molecular patterns that affect PKR activation has not been achieved.
FIGURE 1.

Effect of ssRNA-47 modifications on PKR activation. (A) Schematic of PKR protein. PKR is 551 residues and has two tandem N-terminal dsRNA-binding motifs (dsRBM1 and dsRBM2) that comprise its dsRNA-binding domain (dsRBD) and multiple C-terminal kinase subdomains that confer catalytic activity. P20 is the protein that goes from 1 to 184, and so contains the dsRBD. (B) Secondary structural model of ssRNA-47. This structure was derived from structure-mapping experiments in conjunction with energy minimization from mfold v3.2 as described in Nallagatla et al. (2007). (C) Modified nucleosides studied. (D) Activation assays for modified ssRNAs (10% SDS-PAGE). RNA concentrations are 0.16, 0.31, 0.63, 1.25, 2.5, and 5 μM. A no-RNA-added lane is provided. Phosphorylation activities are presented below each gel lane. Phosphorylation activities were normalized to 0.1 μM dsRNA-79 and rounded off to the nearest integer. Unmodified ssRNA-47 was included as a control in each separate experiment.
Many viruses have evolved strategies to evade PKR activation, including alteration of RNA structure and sequence and the production of inhibitory viral RNAs and proteins (Langland et al. 2006; Garcia et al. 2007; Unterholzner and Bowie 2008). It remains unclear, however, whether cellular RNAs also encode information to avoid activating PKR. Such mechanisms seem plausible given that self-RNA must be discriminated against for innate immunity to be effective. One mechanism by which an RNA might avoid activating PKR is through nucleoside modification. This is reasonable given that most bacterial and some viral RNAs are less modified than cellular RNAs (Bokar and Rottman 1998; Margulis and Chapman 1998; Hornung et al. 2006).
Cellular RNAs often undergo post-transcriptional modification. To date, more than 50 unique naturally occurring modified nucleosides have been identified in eukaryotes (Limbach et al. 1994; Rozenski et al. 1999). It has recently been shown that RNAs having natural modifications have a profound effect on the innate immunity functions of Toll-like receptors (TLRs) (Kariko et al. 2005; Kariko and Weissman 2007; Sioud et al. 2007; Unterholzner and Bowie 2008) and the retinoic-acid-inducible protein I (RIG-I) (Hornung et al. 2006). Effects of natural RNA modifications on PKR activation have not been described, however. Herein, we perform a systematic analysis of the effects of naturally occurring nucleobase, sugar, and phosphodiester modifications on activation of PKR by ssRNA and dsRNA. Our results indicate that RNA loses its ability to activate PKR upon undergoing certain nucleoside modifications, as well as in the presence of GU wobble pairings.
RESULTS
Effect of nucleoside modifications on activation of PKR by single-stranded RNA
We recently found that short stem–loop-containing ssRNAs activate PKR in a 5′-triphosphate-dependent manner (Nallagatla et al. 2007). The shortest model ssRNA that activates PKR in this fashion is ssRNA-47, which is a 47-nucleotide (nt) transcript with two experimentally verified short stem-loops (Fig. 1B). The effects of nucleoside modifications in ssRNA-47 on PKR activation were evaluated by incorporating modified nucleosides during T7 transcription.
We fully substituted the 12 uridines in ssRNA-47 with 2-thiouridine (s2U), 4-thiouridine (s4U), pseudouridine (ψ), 5-methyluridine (m5U), 5-iodouridine (I5U), 2′-fluorouridine (2′-FU), 2′-deoxyuridine (2′-dU), or α-phosphorothiouridine (PS-U). The structures of these modified bases are provided in Figure 1C, and the positions of the modification relative to the grooves and the Watson–Crick base-pairing face are tabulated in Table 1. In additional experiments, the 15 adenosines were substituted with N-6-methyladenosine (m6A), while the five guanosines were replaced with α-phosphorothioguanosine (PS-G). These modified nucleosides are commercially available as triphosphates, which are suitable for in vitro transcription reactions (see Materials and Methods). We tested the ability of modified ssRNA-47 transcripts to activate PKR in the presence of a 5′-triphosphate since it is required for activation (Nallagatla et al. 2007). Activation was judged by incubating PKR with the appropriate RNA in the presence of [γ-32P]ATP and assaying for a 32P-labeled protein at the appropriate position on a 10%-SDS polyacrylamide gel (Fig. 1D).
TABLE 1.
Summary of effects of RNA modifications on PKR activation

Many of the nucleoside modifications strongly abrogated activation of PKR (Fig. 1D; Table 1). Of the modified RNAs tested, s2U-, s4U-, Ψ-, m5U-, I5U-, m6A-, and 2′-dU-containing ssRNA-47 transcripts had significantly impaired ability to activate PKR. For example, when compared at an RNA concentration of 2.5 μM, s2U-, Ψ-, m6A-, and s4U-containing ssRNA-47 showed ∼150-, 30-, 30-, and sevenfold reduced activation of PKR, respectively, as compared to unmodified ssRNA-47. The modified nucleosides Ψ, m5U, I5U, m6A, and 2′-dU do not have altered Watson–Crick base-pairing faces (Fig. 1C); thus, perturbation of base-pairing within the short stem–loops in ssRNA-47 is unlikely (Fig. 1B).
However, not all modifications led to loss of activation. In particular, PS-U-, PS-G-, and 2′-FU-containing ssRNA-47 transcripts largely retained their ability to activate PKR (Fig. 1D). The 2′-FU transcript retains the ability to accept hydrogen bonds from PKR, which likely accounts for its high activity. The PS-U and PS-G transcripts have activity within twofold of the unmodified transcript, suggestive of a minor role for phosphates in recognition of ssRNA.
Next, we tested the effect of representative inactivating nucleoside modifications on binding of ssRNA to a catalytically inactive variant of PKR, K296R. We chose to use the inactive K296R variant to avoid having to dephosphorylate PKR after purification; previous studies indicate that K296R binds RNA with similar properties as wild-type PKR (Katze et al. 1991). Filter binding experiments revealed no appreciable effect of nucleoside modifications on K296R binding (Supplemental Fig. S1). Indeed, ssRNA-47 containing s4U and m5U modifications bound somewhat tighter to K296R (up to 2.2-fold) than unmodified ssRNA-47, while Ψ-containing RNA bound just 2.4-fold weaker. Apparently, nucleoside modifications that abrogate 5′-triphosphate-dependent activation of PKR do not abrogate binding, consistent with the presence of PKR-interacting features in these transcripts.
Effect of nucleoside modifications on activation of PKR by double-stranded RNA
Next, we tested effects on PKR activation of the same modifications in a double-stranded version of ssRNA-47, termed “dsRNA-47” (Fig. 2A). In this dsRNA construct, the unmodified complementary transcript to ssRNA-47 was CIP-treated and annealed with modified ssRNA-47 transcript (note that the 5′-triphosphate of the complementary strand to ssRNA-47 was removed since this strand is in slight excess of the top strand [see Materials and Methods] and might otherwise activate PKR directly). Modified dsRNA-47 duplexes were tested for their ability to activate PKR (Fig. 2B; Table 1). Activation was abrogated by s2U-, s4U-, and 2′-dU-containing dsRNAs, with ∼20-fold reduced activity when compared to unmodified dsRNA at the maximal PKR-activating concentration of 0.125 μM dsRNA. Duplexes containing Ψ and I5U, on the other hand, showed intermediate activation with ∼3-fold reduced activity. Finally, m5U-, PS-U-, m6A- and 2′-FU-containing dsRNAs retained their ability to activate PKR at near wild-type levels. We tested loss of activation upon s2U substitution in the longer and unrelated 79-bp dsRNA as well (Zheng and Bevilacqua 2004). Greatly diminished activation of PKR was still observed upon incorporation of s2U into the top strand (Fig. 2C), suggesting that s2U effects are independent of dsRNA sequence and length.
FIGURE 2.

Effect of dsRNA-47 modifications on PKR activation. (A) Sequence of dsRNA-47. Only the top strand was modified. (B,C) Activation assays for modified dsRNA-47 (10% SDS-PAGE) (B) and s2U-modified dsRNA-79 (C). For both panels, the radiolabeled top strand was modified and annealed with a slight excess of unmodified and unlabeled CIP-treated bottom strand (see Materials and Methods). The dsRNA concentrations are 0.016, 0.032, 0.062, 0.125, 0.25, and 0.5 μM. A no-RNA-added lane is provided. Phosphorylation activities are normalized to 0.1 μM dsRNA-79 and rounded off to the nearest integer. Unmodified dsRNA-47 was included as a control in each separate experiment.
Effect of A–s4U, G–s4U, and GU base-pairings on activation of PKR by double-stranded RNA
Substitution of s4U into dsRNA resulted in 20-fold lower levels of PKR activation (Fig. 2B). This may arise because of disruption in dsRNA structure, as s4U modifies the Watson–Crick base-pairing face of U (Fig. 3A), although any effects on structure have been shown to give minimal effects on duplex stability (Testa et al. 1999). A GU wobble pair, on the other hand, can still form upon introduction of the s4U modification (Fig. 3A). Indeed, Testa and co-workers demonstrated that G–s4U wobble pairs have significantly enhanced stability relative to unmodified GU wobbles (Testa et al. 1999). We therefore prepared dsRNAs with a variable number of A–s4U and G–s4U wobble pairs, making A-to-G changes in the complementary, unmodified bottom strand of ssRNA-47 as necessary (Fig. 3B). The duplex termed “dsRNA1” is the same as dsRNA-47, while “dsRNA2” and “dsRNA3” have 10 and seven A-to-G changes in the bottom strand, respectively (Fig. 3B) (note that, upon modification, a duplex is renamed with the modification at the end of the name; for example, dsRNA1 transcribed with s4UTP rather than UTP is “dsRNA1–s4U”).
FIGURE 3.

Effect of A–s4U, G–s4U, and GU pairs on PKR activation by dsRNA-47. (A) Structures of A–s4U, G–s4U, and GU base pairs. (B) dsRNA-47 variants having 12 s4U modifications in the top strand and variable number of opposing A or G bases in the bottom strand. The A–s4U pairs are boxed, and the G–s4U pairs are shaded with the G in the GU wobbles shown by lower-case letters. The s4U-modified dsRNA1 duplex, “dsRNA1–s4U,” has a bottom strand with all As opposite the 12 s4U substitutions. (The unmodified version of dsRNA1 is the same sequence used in Fig. 2.) dsRNA2–s4U is a sequence variant in which the bottom strand has two As and 10 Gs opposite the 12 s4U substitutions, while dsRNA3–s4U is a sequence variant in which the bottom strand has five As and seven Gs opposite the 12 s4U substitutions. The percent modification or GU wobble pairing is provided in panels C and D. (C) PKR activation assays by s4U-modified dsRNAs (10% SDS-PAGE). s4U-modified versions of dsRNA1, dsRNA2, and dsRNA3 as well as an unmodified dsRNA1 were tested for PKR activation. The dsRNA concentrations are 0.016, 0.032, 0.062, 0.125, 0.25, and 0.5 μM. (D) PKR activation assay by dsRNA1 in which top strand has no modification and bottom strand has no A-to-G changes, and dsRNA2 in which top strand has no modifications and bottom strand has 10 A-to-G changes. The dsRNA concentrations are 0.032, 0.062, 0.125, 0.25, and 0.5 μM. A no-RNA-added lane is provided. Phosphorylation activities are normalized to 0.1 μM dsRNA-79 and rounded off to the nearest integer.
We transcribed the top strand in the presence of s4UTP and no UTP and tested these modified dsRNAs for PKR activation. Activation assays revealed that none of the modified duplexes activated PKR significantly compared to unmodified dsRNA1 (Fig. 3C). In the case of dsRNA1–s4U, this observation suggests that a 4-thio substitution in the major groove interferes with PKR interaction. In the case of dsRNA2–s4U and dsRNA3–s4U, which contain different levels of A–s4U and G–s4U pairing (Fig. 3B), either the 4-thio in the major groove and/or the GU wobbles interfere with activation. To test the latter possibility, we conducted activation assays with unmodified dsRNA2, which contains 12 GU pairs distributed throughout the 47-bp duplex (Fig. 3D). Surprisingly, this duplex did not support activation either, indicating that certain types and levels of wobble base pairs interfere with activation of PKR by dsRNA. Thus, both a 4-thio group and wobble pairing appear to be inhibitory toward PKR activation by dsRNA.
Activation of PKR by varying the number of modified nucleosides in dsRNA
The experiments described so far involved dsRNA with approximately one-fourth of the top-strand bases modified. One question is how fewer nucleoside modifications modulate PKR activation. We therefore decreased the number of modifications in dsRNA-47 and tested PKR activation. The s2U modification was chosen for this study since this base strongly interferes with activation by dsRNA-47 (Fig. 2B,C) but does not disrupt Watson–Crick AU base-pairing (Fig. 4A). We prepared dsRNA-47s with either 12 (26%), six (13%), or three (6%) s2U substitutions (Fig. 4B). As expected, as the number of modifications in the RNA decreases, the level of abrogation of PKR activation decreases. Incorporation of 6% and 13% A–s2U pairing reduced PKR activation up to 20% and 62%, respectively (Fig. 4C). Thus, even a modest level of substitution of a dsRNA sequence with s2U leads to an appreciable loss of activation, with a near complete loss of activation occurring upon 25% substitution.
FIGURE 4.

Effect of different levels of A–s2U base-pairing on PKR activation by dsRNA-47. (A) Structures of A–U and A–s2U base pairs. (B) dsRNA-47 variants having variable numbers of s2U modifications in the top strand. dsRNA1–s2U has 12 s2U substitutions (26% modification) in the top strand. (The unmodified version of dsRNA1 is the same sequence used in Fig. 2.) dsRNA4–s2U is a sequence variant in which the top strand has six s2U substitutions (13% modification), while dsRNA5–s2U has 3 s2U substitutions (6% modification) in the top strand. (C) PKR activation assays by these RNAs (10% SDS-PAGE). Unmodified and s2U-modified versions of dsRNA1, dsRNA4, and dsRNA5 were tested for PKR activation. The dsRNA concentrations are 0.016, 0.032, 0.062, 0.125, 0.25, and 0.5 μM. A no-RNA-added lane is provided. Phosphorylation activities are normalized to 0.1 μM dsRNA-79 and rounded off to the nearest integer.
Next, we tested the ability of representative modified dsRNA-47 to bind protein. Native mobility-shift experiments with the dsRBD of PKR (P20) (Fig. 1A) were carried out as previously described (Bevilacqua and Cech 1996). No appreciable difference in binding of modified and unmodified dsRNAs was observed (Fig. 5). All RNAs gave about five band shifts, as expected from earlier studies on dsRNAs of this length (Manche et al. 1992), and the concentration of P20 required to shift nearly all of the free RNA was the same for all duplexes within a factor of 2. Thus, much like with ssRNA (Supplemental Fig. S1), the ability of a modified duplex to abrogate activation of PKR is independent of its ability to bind.
FIGURE 5.

Binding of unmodified and modified dsRNA-47 to P20 by mobility-shift assays. Trace amounts of radiolabeled modified dsRNAs were mixed with P20 and analyzed by 10% native PAGE (79:1 crosslink). Modified (mod.) top strand (p*TS) oligonucleotide was 5′-32P-labeled and pre-annealed to excess unlabeled and unmodified (unmod.) bottom strand (BS) oligonucleotide. Formation of duplex was confirmed by a microshift of the top strand upon addition of bottom strand. (Compare first and second lanes in each RNA set.) PS-U/PS-A represents dsRNA-47 in which top strand has PS-U modification and bottom strand has PS-A modification. Protein concentrations used are 0.005, 0.01, 0.02, 0.04, 0.08, 0.16, 0.32, 0.63, 1.25, 2.5, and 5 μM.
Inhibition of PKR autophosphorylation by modified single- and double-stranded RNAs
RNAs can have three different properties with respect to PKR autophosphorylation: activation, inhibition, or no effect. This classification can be further delimited since all known dsRNA and ssRNA activators of PKR serve as inhibitors at high enough concentrations (Zheng and Bevilacqua 2004; Nallagatla et al. 2007), presumably because high RNA concentrations titrate PKR dimers out to monomers, which abrogates activation. We therefore refer to inhibitors that do not activate PKR at any concentration as “intrinsic inhibitors.” It has been previously reported that 24- and 33-bp RNAs are intrinsic inhibitors, presumably because they are too short to dimerize PKR (Manche et al. 1992; Zheng and Bevilacqua 2004). Since some of the modified ssRNA and dsRNAs did not activate PKR, we sought to address whether they can act as intrinsic inhibitors.
We first tested whether modified ssRNA transcripts could inhibit activation of PKR by 5′-triphosphate ssRNA-47 (Fig. 6A). Equal concentrations (5 μM) of unmodified and modified ssRNA-47s were mixed prior to PKR activation assays. Modified ssRNA-47 transcripts containing s2U, 2′-dU, s4U, m6A, and m5U showed only modest levels of inhibition (23%, 22%, 13%, 16%, and 7%, respectively) while Ψ had no observable effect (Fig. 6A).
FIGURE 6.

Inhibition of PKR activation by modified ssRNA-47 and dsRNA-47. (A) Inhibition of PKR activation by modified ssRNA-47s. To each lane containing 5 μM of unmodified activating ssRNA-47, an equal concentration of modified ssRNA was added, and PKR activation was assayed (10% SDS-PAGE). Phosphorylation activities are normalized to 0.1 μM dsRNA-79 and rounded off to the nearest integer. (B) Inhibition of PKR activation by modified dsRNA-47s. To each lane containing 0.125 μM of unmodified activating dsRNA-47, 0.125 or 0.5 μM modified dsRNA was added, and PKR activation was assayed (10% SDS-PAGE). Phosphorylation activities are normalized to 0.1 μM dsRNA-79 and rounded off to the nearest integer.
Next, we tested whether modified dsRNA duplexes could inhibit PKR activation by dsRNA-47 (Fig. 6B). Unmodified and modified dsRNA-47 duplexes were mixed at equal concentrations (0.125 μM) or with a fourfold excess of the modified duplex (0.5 μM). In contrast to ssRNAs, modified dsRNAs served as strong inhibitors of PKR. An equal concentration of modified dsRNA led to a 50% or greater reduction in PKR activation, while a fourfold excess of modified dsRNA inhibited PKR activation almost completely (Fig. 6B). Given that modified dsRNA-47 duplexes can bind avidly to the dsRBD (Fig. 5), they presumably inhibit activation of PKR by blocking the binding of activating RNAs.
DISCUSSION
PKR is an essential molecular sensor in the innate immunity antiviral pathway (Williams 1999; Dever et al. 2007). Activation of PKR by long stretches of dsRNA, which can have viral origins, has been clearly demonstrated in vitro (Hunter et al. 1975; Manche et al. 1992). Recent findings indicate that relatively unstructured RNAs can also activate PKR if they begin with a triphosphate (Nallagatla et al. 2007). Since most self-RNAs have a m7G cap or a monophosphate at their 5′-ends while many bacterial and viral transcripts begin with a triphosphate, a 5′-triphosphate may serve as a pathogen-associated molecular pattern (PAMP) (Hornung et al. 2006; Pichlmair et al. 2006; Nallagatla et al. 2007). In addition to a unique 5′-end, self-RNAs also have an abundance of modified nucleosides relative to pathogenic RNA (Limbach et al. 1994; Rozenski et al. 1999). We therefore examined effects of internal nucleoside modifications on PKR activation in the context of both ssRNA and dsRNA.
We recently reported that PKR is activated by short stem-loop RNAs in a 5′-triphosphate dependent manner (Nallagatla et al. 2007). Herein, we performed transcription reactions for ssRNA and dsRNA in which a naturally occurring NTP, typically UTP, was completely substituted with a modified NTP. Our results reveal that incorporation of minor groove-modified nucleosides (s2U and 2′-dU), major groove-modified nucleosides (s4U, ψ, m5U, I5U, and m6A), and Watson–Crick base-pairing-modified nucleosides (s2U and s4U) (Fig. 1; Table 1) abrogate activation of PKR in a model ssRNA with two short stem-loops. Strikingly, ssRNA-47 with the minor groove-modifying nucleoside, 2′-FU, retains the ability to activate PKR, while modification of the phosphodiester group to a phosphorothioate in PS-U and PS-G leads to within twofold lower activation (Fig. 1D).
The opposing effect of 2′-FU and 2′-dU substitutions in ssRNA-47 strongly supports the importance of hydrogen bonding between PKR and the ribose portion of ssRNA. In fact, the 2′-FU transcript maximally activated PKR at a greater than fourfold lower concentration than unmodified ssRNA-47 (Fig. 1D). The importance of 2′-OH hydrogen bonding for activation of PKR by 5′-triphosphate-containing ssRNA-47 is consistent with earlier observations that a DNA version of ssRNA-47 does not activate PKR (Nallagatla et al. 2007). The only slightly lower activation of PKR by PS-U and PS-G is suggestive of loss of relatively few contacts between PKR and the phosphodiester backbone of ssRNA-47.
The molecular basis for abrogation of PKR activation by most other nucleoside modifications in ssRNA-47 is unclear. However, filter binding experiments on representative nonactivating ssRNA-47 transcripts revealed that binding to K296R is relatively unaffected (Supplemental Fig. S1). Thus, there appears to be a nonequivalence of ssRNA binding to PKR and activation of PKR. One possibility is that dimerization of PKR is altered or disrupted by many ssRNA modifications.
Finally, given that nucleoside modifications did not strongly affect binding of ssRNA-47 to PKR, it might seem surprising that modified ssRNAs are not potent inhibitors of the ssRNA-mediated activation of PKR (Fig. 6A). However, binding of ssRNA is relatively weak, with apparent K d values in the 2–10 μM range (Supplemental Fig. S1), consistent with the weak inhibition observed at 5 μM concentrations (Fig. 6A) (note that higher concentrations of the modified ssRNA-47 competitor were not used due to limitations in production of these transcripts). In summary, functional groups on the major groove, minor groove, and Watson–Crick base-pairing faces of the nucleobases, as well as the ribose sugar itself, abrogate the 5′-triphosphate-dependent activation of PKR by ssRNA. These observations suggest that PKR interacts with 5′-triphosphate-containing ssRNA in an intimate manner at both the 5′-end and internal regions of the transcript.
Activation of PKR by modified dsRNA-47 duplexes, in which ssRNA-47 was allowed to base pair with its complement, was also measured. On the one hand, modifications to the minor groove and Watson–Crick base-pairing face led to similar effects on PKR activation as in the ssRNA-47 backgrounds. For example, the minor groove changes of s2U and 2′-dU abrogated activation, while 2′-FU duplexes retained activation in both dsRNA-47 and ssRNA-47 (Fig. 2B; Table 1). In addition, changes to the Watson–Crick base-pairing face in s4U and s2U (s2U also alters the minor groove) abrogated activation in both RNA structural backgrounds.
Modifications to the major groove, however, modulated activation of PKR by ssRNA and dsRNA in strikingly different fashions. Whereas major groove modifications abrogated the 5′-triphosphate-dependent activation of PKR by ssRNA (Fig. 1D), such changes had little effect on activation of PKR by dsRNA (Fig. 2B). For example, m5U- and m6A-containing duplexes had either full or enhanced ability to activate PKR, while ψ- and I5U-containing duplexes had only partially compromised activation ability (Fig. 2B). Overall, we conclude that nucleoside modifications modulate the activation of PKR in an RNA structure-specific fashion. This conclusion supports the notion that activation of PKR by ssRNA and dsRNA occurs by inherently different mechanisms. The previous observation that a 5′-triphosphate contributes to activation by ssRNA but not dsRNA is also consistent with this conclusion (Nallagatla et al. 2007).
The effects of nucleoside modifications on PKR activation by dsRNA are also largely consistent with a previously reported crystal structure of dsRNA complexed with the dsRBM from a Xenopus laevis protein (Ryter and Schultz 1998) (note that there is presently no structure of the dsRBD from PKR in complex with dsRNA). This crystal structure revealed that RNA–protein interactions are largely in the minor groove with water-bridged contacts to the 2′-hydroxyls of dsRNA and just a few water-bridged hydrogen bonds to phosphates across the major groove. This structure thus explains how a dsRBM can interact with dsRNA in a nonsequence-specific fashion but still discriminate against dsDNA and RNA–DNA hybrids (Hunter et al. 1975; Bevilacqua and Cech 1996). In agreement with the importance of hydrogen bonding with the 2′-hydroxyl, dsRNA substituted with 2′-dU does not activate PKR, while dsRNA substituted with 2′-FU does (Fig. 2B). Since 2′-FU can serve as a hydrogen bond acceptor, these observations are consistent with a direct or water-bridged interaction between the 2′-hydroxyl of dsRNA and PKR.
The crystal structure also shows that the dsRNA is largely A-form in the complex with the dsRBM (Ryter and Schultz 1998). Thus, it is reasonable to expect modifications to the Watson–Crick base-pairing face, which would perturb the A-form geometry, to abrogate activation. In agreement, the s4U change, which interferes with hydrogen bonding in AU base pairs (Fig. 3A), abrogates activation (Fig. 2B). Loss of activation upon incorporation of s2U opposite A, on the other hand, may be due to interference in the minor groove, since s2U has an unperturbed ability to hydrogen bond with A (Fig. 4A). Sulfur has very different hydration patterns in DNA than oxygen (White et al. 1996), and therefore s2U substitutions might also alter the spine of hydration around the dsRNA. Such an effect would be consistent with disruption of some of the many observed water-mediated contacts between the dsRBD and dsRNA (Ryter and Schultz 1998).
The observation that most nucleosides with major groove modifications have little effect on activation of PKR by dsRNA (Fig. 2B) is consistent with few major groove interactions in the dsRBM–dsRNA crystal structure (Ryter and Schultz 1998). Moreover, lack of any substantial effect of the PS-U substitution in dsRNA (Fig. 2B) is consistent with the relatively few interactions of the dsRBM to the phosphodiester backbone of dsRNA inferred from both biochemical (Bevilacqua and Cech 1996) and structural studies (Ryter and Schultz 1998). Finally, we note that partially compromised activation of PKR upon substitution of U with ψ in dsRNA-47 might be because the imino proton at the 5-position of ψ alters the structure or hydration patterns of the major groove (Auffinger and Westhof 1997).
In principle, abrogation of PKR activation by minor groove- and Watson–Crick-modified dsRNA could be due to inability of modified dsRNA to bind protein. However, native mobility-shift analysis on representative nonactivating dsRNA-47 duplexes revealed dsRBD binding affinity and stoichiometry similar to dsRNA-47 (Fig. 5). Consistent with avid binding of modified dsRNA-47 duplexes to the dsRBD, all modified duplexes were strong competitors of PKR activation (Fig. 6B). Therefore, as with ssRNA, there appears to be a nonequivalence of dsRNA binding to PKR and activation of PKR. One possibility is that nucleoside modifications alter dimerization of PKR, which is a critical step for activation.
Finally, we consider the observation that GU wobble pairing leads to abrogation of PKR activation. Incorporation of 10 GU wobble pairs into the 47-bp dsRNA led to a major loss of ability to activate PKR (Fig. 3D). The major groove of the A-form helix, which is normally deep and inaccessible, is made wide and shallow upon introduction of GU wobble pairs (Weeks and Crothers 1993; Cate and Doudna 1996; Masquida and Westhof 2000). Thus alteration of the A-form geometry upon GU wobbling may perturb dsRBM interaction, as inferred from the dsRNA–dsRBM crystal structure being largely A-form (Ryter and Schultz 1998).
It has been shown that other key components of innate immunity, in particular TLRs and RIG-I, are regulated by nucleoside modification as well (Kariko et al. 2005; Hornung et al. 2006; Kariko and Weissman 2007; Sioud et al. 2007). TLR7 and TLR8 are ssRNA-sensing proteins, whereas TLR3 and RIG-I sense dsRNA. In addition, RIG-I has been shown to sense the 5′-triphosphate group of RNA. Modifications such as m5C, m5U, ψ, m6A, and s2U abrogated activation of TLR7 and TLR8 by a 1571-nt structured RNA, whereas only s2U and m6A abrogated TLR3 activation. In addition, s2U, ψ, and 2′-O-Me modifications in 24-nt 5′-triphosphate ssRNA abrogated RIG-I activation. In general, these findings are similar to results observed herein with PKR and modified ssRNA and dsRNA. Nonetheless, it is unlikely that the molecular basis for the effects is identical in PKR since the TLRs and RIG-I do not have dsRBMs. Finally, it is important to note that TLR3, TLR7, and TLR8 are located in the endosomal membrane, while RIG-I and PKR are located in the cytosol (Schlee et al. 2006; Yoneyama and Fujita 2007). Thus, there may be a synergy between endosomal and cytosolic viral recognition processes.
Recent observations indicate that small interfering RNAs (siRNA) containing s2U, ψ, s4U, Br5U, and locked nucleic acid (LNA) modifications display similar silencing activities as unmodified siRNAs (Chiu and Rana 2002; Sipa et al. 2007). We found that substitution of just 13% s2U abrogates 62% of PKR activation (Fig. 4C), a feature that may be useful in siRNA design (Schlee et al. 2006; Aagaard and Rossi 2007; Unterholzner and Bowie 2008). On a related note, RNAs with 2′-F ribose and phosphorothioates may be attractive candidates as immunostimulatory RNAs (isRNA) for treating cancer or chronic viral infections (Schlee et al. 2006; Lan et al. 2007), since these nucleic acids activate PKR as well as or better than unmodified RNA and have enhanced stability in vivo (Layzer et al. 2004; Allerson et al. 2005).
CONCLUSION
PKR serves important roles in innate immunity in which it senses general features of pathogens and offers the host protection. The major regulator of PKR is RNA, and while long stretches of dsRNA activate PKR in vitro, in vivo other RNAs may be responsible for activation. Largely unstructured RNAs have been shown to activate PKR in a 5′-triphosphate-dependent fashion in vitro and in a cellular context (Nallagatla et al. 2007). Herein, we demonstrated that a large number of modified nucleosides abrogate this activation in both ssRNA and dsRNA contexts, although they do so in an RNA structure-specific fashion. Since the majority of self-RNA has some level of modification, the modified nucleosides may serve, at least in part, to confer lack of activation of PKR by self. Moreover, since most bacterial and some viral RNA is unmodified, this mechanism may allow selective sensing of pathogens. Where possible, these effects could be explained by structural data. Overall, these observations suggest RNA modifications can serve as part of a code that allows the innate immune system to discriminate against self-RNA. Intriguingly, non-Watson–Crick wobble pairing also negatively affects activation in some instances, suggesting that self-RNA can be distinguished not only by covalent modification of the nucleosides but by altered secondary structures as well. A better understanding of the effects of non-Watson–Crick secondary structure on PKR activation will require additional studies. The present study paves a way to deduce new mechanisms of inhibiting PKR and may aid in the identification of novel RNA therapeutic agents.
MATERIALS AND METHODS
Protein preparation
PKR, K296R, and P20 proteins have an N-terminal (His)6 tag that has been shown to not interfere with binding to and activation by RNA (Bevilacqua and Cech 1996). P20 was cloned into the T7 expression vector pET-14b (Novagen), while PKR and K296R were cloned into pET-28a (Novagen) and transformed into Escherichia coli BL21(DE3)Rosetta cells (Novagen) and purified on a Ni2+-NTA column as previously reported (Bevilacqua and Cech 1996; Bevilacqua et al. 1998). After purification, PKR was obtained in phosphorylated form, which was dephosphorylated with λ-PPase before being subjected to activation assays (Zheng and Bevilacqua 2004). Protein concentrations were determined spectrophotometrically at 280 nm (Gill and von Hippel 1989).
RNA sequences
A number of modified or unmodified ssRNA and dsRNAs were used in this study. These were prepared by T7 transcription reactions (Milligan and Uhlenbeck 1989). dsRNA-79 and dsRNA-47 are 79- and 47-bp dsRNAs without base-pairing imperfections derived from pUC-19 and the 3′-UTR of lin-41, respectively, and were prepared by annealing top- and bottom-strand RNAs (Nallagatla et al. 2007). ssRNA-47 was derived from the 3′-UTR of lin-41, which has been shown to be especially unstructured (Nallagatla et al. 2007). The sequences of RNAs used in this study are provided in the figures.
Preparation, purification, CIP treatment, and radiolabeling of RNA
Unmodified and s2U-, s4U-, ψ-, m5U-, m5C-, I5U-, m6A-, PS-U-, and PS-G-containing RNAs were prepared by T7 transcription reaction (Ambion) or the following protocol: 1 μg of linearized or hemi-duplex DNA with T7 promoter sequence was incubated with 0.5 μg of T7 RNA polymerase in a buffer containing 40 mM Tris (pH 8), 20 mM Mg(OAc)2, 40 mM DTT, 2 mM spermidine, and 7 mM of each NTP for 3 h at 37°C. Inorganic pyrophosphate was removed by spinning the sample, and the reaction was stopped by adding 95% formamide loading buffer. RNAs were purified by denaturing polyacrylamide gel electrophoresis (PAGE). For each RNA, the corresponding modified nucleoside triphosphate was substituted in the transcription reactions. The transcript was identified by UV shadowing, excised from gel, and eluted overnight in TEN250 (10 mM Tris [pH 7.5], 1 mM EDTA, and 250 mM NaCl) buffer at 4°C. The RNA was precipitated in ethanol, dissolved in TE (10 mM Tris [pH 7.5], 1 mM EDTA), and stored at −20°C. RNA concentrations were determined spectrophotometrically. In general, yields were lower with modified RNAs, which limited the range of RNA concentrations possible in inhibition studies.
To make 2′-FU-containing RNA, the DuraScribe T7 transcription (Epicentre Technologies Corp.,) kit was used according to the following protocol: 1 μg of linearized or hemi-duplex DNA with T7 promoter sequence was incubated in the manufacturer's transcription buffer with 5 mM of each NTP in which UTP was replaced with 2′-FUTP, 10 mM DTT, and the manufacturer's enzyme mix for 6 h at 37°C. The reaction was then quenched by adding 95% formamide loading buffer. The transcript was purified as described above.
To make 2′-dU-containing RNA, T7 R&DNA modified polymerase (Epicentre) was used according to the following protocol: 1 μg of linearized or hemi-duplex DNA with T7 promoter sequence was incubated in the manufacturer's transcription buffer with 1 mM of each NTP in which UTP was replaced with 2′-dUTP (Sigma), 10 mM DTT, and 25 units of T7 R&DNA polymerase (Epicentre). After mixing on ice, the reaction was carried out overnight at 37°C. The transcript was purified as described above.
Calf intestinal alkaline phosphatase (CIP) treatment of RNA and formation of dsRNA were performed as described (Bevilacqua and Cech 1996; Zheng and Bevilacqua 2004; Nallagatla et al. 2007). The bottom strand was treated with CIP and in 1.1-fold excess over the top strand. For 5′-end labeling, RNAs were treated with CIP, purified, reacted with T4 polynucleotide kinase (T4 PNK) and [γ-32P]ATP, repurified by gel electrophoresis, excised from the gel, eluted overnight in TEN250, ethanol precipitated, and resuspended in TE, as previously described (Bevilacqua and Cech 1996; Zheng and Bevilacqua 2004; Nallagatla et al. 2007).
PKR activation assays
Modified and unmodified RNAs were tested for their ability to activate or inhibit PKR autophosphorylation. Activation assays were carried out largely as previously described (Zheng and Bevilacqua 2004; Nallagatla et al. 2007). Briefly, purified PKR was dephosphorylated with λ-PPase (New England Biolabs), quenched by addition of freshly made sodium orthovanadate, and incubated with [γ-32P]ATP for 10 min at 30°C. The time of 10 min was chosen because this is in the plateau region of phosphorylation versus time plots (Nallagatla et al. 2007). Reactions were quenched by adding SDS loading buffer, heated at 95°C for 5 min, and loaded on a 10% SDS-PAGE gel (Pierce). Gels were exposed to a storage PhosphorImager screen, and the labeled bands were quantified on a PhosphorImager (Molecular Dynamics). In all experiments, data were normalized to the counts in a 0.1 μM dsRNA-79 lane.
Binding assays
Binding of P20 to unmodified and modified dsRNA-47 was carried out by native gel-mobility shift assays as described (Bevilacqua and Cech 1996). Duplex RNA was 5′-32P-labeled and present in limiting concentration relative to protein concentration. Samples were prepared in standard binding buffer (BB: 25 mM HEPES [pH 7.5], 10 mM NaCl, 5% glycerol, 5 mM DTT, 0.1 mM EDTA and 0.1 mg/mL tRNA [Sigma-Aldrich]). tRNA was included to prevent sticking of the complex in the wells of the gel. Binding reactions were loaded onto a 10% (79:1 acrylamide/bis) running native gel. The gel and the running buffer contained 0.5× TBE (50 mM Tris base, 41.5 mM boric acid, and 1 mM EDTA [final pH 8.3]). Electrophoresis was performed at 300 V, at 16°C.
Binding of K296R to unmodified and modified ssRNA-47 was performed by filter binding in a 96-well dot blot apparatus essentially as described (Wong and Lohman 1993; Bevilacqua et al. 1998). Nitrocellulose (0.45 μm, Schleicher & Schuell from Sigma Aldrich) and Hybond N+ (0.45 μm) were equilibrated in BB for 30 min at room temperature. Wells were washed with 100 μL of BB, after which four reactions were filtered. Wells were immediately washed again with 100 μL of ice-cold BB. Dissociation constants for unmodified and modified ssRNA-47 RNAs were determined by quantifying the fraction (θ) of radiolabeled (nM) RNA bound with a PhosphorImager (Molecular Dynamics) and fitting by nonlinear least squares to Equation 1:
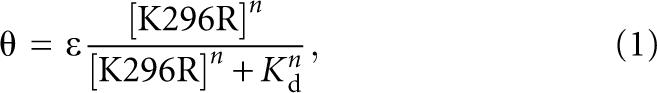 |
where θ is the fraction of RNA bound, ε is the observed maximum fraction bound, K d is the dissociation constant, and n is the Hill coefficient.
SUPPLEMENTAL DATA
Supplemental material can be found at http://research.chem.psu.edu/pcbgroup/Pages/pubs.html.
ACKNOWLEDGMENTS
This work was supported by NIH Grant R01-58709. We thank Laurie Heinicke, Nate Siegfried, and Rebecca Toroney for helpful comments on the manuscript.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.1007408.
REFERENCES
- Aagaard L., Rossi J.J. RNAi therapeutics: Principles, prospects, and challenges. Adv. Drug Deliv. Rev. 2007;59:75–86. doi: 10.1016/j.addr.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allerson C.R., Sioufi N., Jarres R., Prakash T.P., Naik N., Berdeja A., Wanders L., Griffey R.H., Swayze E.E., Bhat B. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005;48:901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- Auffinger P., Westhof E. RNA hydration: Three nanoseconds of multiple molecular dynamics simulations of the solvated tRNAAsp anticodon hairpin. J. Mol. Biol. 1997;269:326–341. doi: 10.1006/jmbi.1997.1022. [DOI] [PubMed] [Google Scholar]
- Ben-Asouli Y., Banai Y., Pel-Or Y., Shir A., Kaempfer R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell. 2002;108:221–232. doi: 10.1016/s0092-8674(02)00616-5. [DOI] [PubMed] [Google Scholar]
- Bevilacqua P.C., Cech T.R. Minor-groove recognition of double-stranded RNA by the double-stranded RNA-binding domain from the RNA-activated protein kinase PKR. Biochemistry. 1996;35:9983–9994. doi: 10.1021/bi9607259. [DOI] [PubMed] [Google Scholar]
- Bevilacqua P.C., George C.X., Samuel C.E., Cech T.R. Binding of the protein kinase PKR to RNAs with secondary structure defects: Role of the tandem A–G mismatch and noncontiguous helixes. Biochemistry. 1998;37:6303–6316. doi: 10.1021/bi980113j. [DOI] [PubMed] [Google Scholar]
- Bokar J.A., Rottman F.M. Biosynthesis and functions of modified nucleosides in eukaryotic mRNA. In: Grosjean H., Benne R., editors. Modification and Editing of RNA. ASM Press; Washington, D.C: 1998. pp. 183–200. [Google Scholar]
- Bommer U.A., Borovjagin A.V., Greagg M.A., Jeffrey I.W., Russell P., Laing K.G., Lee M., Clemens M.J. The mRNA of the translationally controlled tumor protein P23/TCTP is a highly structured RNA, which activates the dsRNA-dependent protein kinase PKR. RNA. 2002;8:478–496. doi: 10.1017/s1355838202022586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cate J.H., Doudna J.A. Metal-binding sites in the major groove of a large ribozyme domain. Structure. 1996;4:1221–1229. doi: 10.1016/s0969-2126(96)00129-3. [DOI] [PubMed] [Google Scholar]
- Chiu Y.L., Rana T.M. RNAi in human cells: Basic structural and functional features of small interfering RNA. Mol. Cell. 2002;10:549–561. doi: 10.1016/s1097-2765(02)00652-4. [DOI] [PubMed] [Google Scholar]
- Davis S., Watson J.C. In vitro activation of the interferon-induced, double-stranded RNA-dependent protein kinase PKR by RNA from the 3′ untranslated regions of human α-tropomyosin. Proc. Natl. Acad. Sci. 1996;93:508–513. doi: 10.1073/pnas.93.1.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dever T.E., Dar A.C., Sicheri F. The eIF2a kinases. In: Mathews M.B., et al., editors. Translational Control in Biology and Medicine. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2007. pp. 319–344. [Google Scholar]
- Fasciano S., Hutchins B., Handy I., Patel R.C. Identification of the heparin-binding domains of the interferon-induced protein kinase, PKR. FEBS J. 2005;272:1425–1439. doi: 10.1111/j.1742-4658.2005.04575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M.A., Gil J., Ventoso I., Guerra S., Domingo E., Rivas C., Esteban M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006;70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M.A., Meurs E.F., Esteban M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie. 2007;89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Gill S.C., von Hippel P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989;182:319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K.K., Schlee M., et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Hovanessian A.G., Galabru J. The double-stranded RNA-dependent protein kinase is also activated by heparin. Eur. J. Biochem. 1987;167:467–473. doi: 10.1111/j.1432-1033.1987.tb13360.x. [DOI] [PubMed] [Google Scholar]
- Hunter T., Hunt T., Jackson R.J., Robertson H.D. The characteristics of inhibition of protein synthesis by double-stranded ribonucleic acid in reticulocyte lysates. J. Biol. Chem. 1975;250:409–417. [PubMed] [Google Scholar]
- Kariko K., Buckstein M., Ni H., Weissman D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Kariko K., Weissman D. Naturally occurring nucleoside modifications suppress the immunostimulatory activity of RNA: Implication for therapeutic RNA development. Curr. Opin. Drug Discov. Devel. 2007;10:523–532. [PubMed] [Google Scholar]
- Katze M.G., Wambach M., Wong M.-L., Garfinkel M., Meurs E., Chong K., Williams B.R.G., Hovanessian A.G., Barber G.N. Functional expression and RNA binding analysis of the interferon-induced, double-stranded RNA-activated, 68,000-M r protein kinase in a cell-free system. Mol. Cell. Biol. 1991;11:5497–5505. doi: 10.1128/mcb.11.11.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Haque J., Lacoste J., Hiscott J., Williams B.R.G. Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating IκB. Proc. Natl. Acad. Sci. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T., Kandimalla E.R., Yu D., Bhagat L., Li Y., Wang D., Zhu F., Tang J.X., Putta M.R., Cong Y., et al. Stabilized immune modulatory RNA compounds as agonists of Toll-like receptors 7 and 8. Proc. Natl. Acad. Sci. 2007;104:13750–13755. doi: 10.1073/pnas.0706059104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langland J.O., Cameron J.M., Heck M.C., Jancovich J.K., Jacobs B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–110. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Layzer J.M., McCaffrey A.P., Tanner A.K., Huang Z., Kay M.A., Sullenger B.A. In vivo activity of nuclease-resistant siRNAs. RNA. 2004;10:766–771. doi: 10.1261/rna.5239604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbach P.A., Crain P.F., McCloskey J.A. Summary: The modified nucleosides of RNA. Nucleic Acids Res. 1994;22:2183–2196. doi: 10.1093/nar/22.12.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manche L., Green S.R., Schmedt C., Mathews M.B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell. Biol. 1992;12:5238–5248. doi: 10.1128/mcb.12.11.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulis L., Chapman M.J. Endosymbioses: Cyclical and permanent in evolution. Trends Microbiol. 1998;6:342–345. doi: 10.1016/s0966-842x(98)01325-0. (discussion, 345–346). [DOI] [PubMed] [Google Scholar]
- Masquida B., Westhof E. On the wobble GoU and related pairs. RNA. 2000;6:9–15. doi: 10.1017/s1355838200992082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan J.F., Uhlenbeck O.C. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989;180:51–62. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- Nallagatla S.R., Hwang J., Toroney R., Zheng X., Cameron C.E., Bevilacqua P.C. 5′-Triphosphate-dependent activation of PKR by RNAs with short stem–loops. Science. 2007;318:1455–1458. doi: 10.1126/science.1147347. [DOI] [PubMed] [Google Scholar]
- Pichlmair A., Schulz O., Tan C.P., Naslund T.I., Liljestrom P., Weber F., Reis e Sousa C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 2006;314:997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Rozenski J., Crain P.F., McCloskey J.A. The RNA Modification Database: 1999 update. Nucleic Acids Res. 1999;27:196–197. doi: 10.1093/nar/27.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryter J.M., Schultz S.C. Molecular basis of double-stranded RNA-protein interactions: Structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 1998;17:7505–7513. doi: 10.1093/emboj/17.24.7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M., Hornung V., Hartmann G. siRNA and isRNA: Two edges of one sword. Mol. Ther. 2006;14:463–470. doi: 10.1016/j.ymthe.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Sioud M., Furset G., Cekaite L. Suppression of immunostimulatory siRNA-driven innate immune activation by 2′-modified RNAs. Biochem. Biophys. Res. Commun. 2007;361:122–126. doi: 10.1016/j.bbrc.2007.06.177. [DOI] [PubMed] [Google Scholar]
- Sipa K., Sochacka E., Kazmierczak-Baranska J., Maszewska M., Janicka M., Nowak G., Nawrot B. Effect of base modifications on structure, thermodynamic stability, and gene silencing activity of short interfering RNA. RNA. 2007;13:1301–1316. doi: 10.1261/rna.538907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa S.M., Disney M.D., Turner D.H., Kierzek R. Thermodynamics of RNA-RNA duplexes with 2- or 4-thiouridines: implications for antisense design and targeting a group I intron. Biochemistry. 1999;38:16655–16662. doi: 10.1021/bi991187d. [DOI] [PubMed] [Google Scholar]
- Unterholzner L., Bowie A.G. The interplay between viruses and innate immune signaling: Recent insights and therapeutic opportunities. Biochem. Pharmacol. 2008;75:589–602. doi: 10.1016/j.bcp.2007.07.043. [DOI] [PubMed] [Google Scholar]
- Weeks K.M., Crothers D.M. Major groove accessibility of RNA. Science. 1993;261:1574–1577. doi: 10.1126/science.7690496. [DOI] [PubMed] [Google Scholar]
- White A.P., Reeves K.K., Snyder E., Farrell J., Powell J.W., Mohan V., Griffey R.H. Hydration of single-stranded phosphodiester and phosphorothioate oligodeoxyribonucleotides. Nucleic Acids Res. 1996;24:3261–3266. doi: 10.1093/nar/24.16.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams B.R. PKR: A sentinel kinase for cellular stress. Oncogene. 1999;18:6112–6120. doi: 10.1038/sj.onc.1203127. [DOI] [PubMed] [Google Scholar]
- Wong I., Lohman T.M. A double-filter method for nitrocellulose-filter binding: Application to protein–nucleic acid interactions. Proc. Natl. Acad. Sci. 1993;90:5428–5432. doi: 10.1073/pnas.90.12.5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M., Fujita T. RIG-I family RNA helicases: Cytoplasmic sensor for antiviral innate immunity. Cytokine Growth Factor Rev. 2007;18:545–551. doi: 10.1016/j.cytogfr.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Zheng X., Bevilacqua P.C. Activation of the protein kinase PKR by short double-stranded RNAs with single-stranded tails. RNA. 2004;10:1934–1945. doi: 10.1261/rna.7150804. [DOI] [PMC free article] [PubMed] [Google Scholar]