

Abstract
β-Rich self-assembly is a major structural class of polypeptides, but still little is known about their atomic structures and biophysical properties. Major impediments for structural and biophysical studies of peptide self-assemblies include their insolubility and heterogeneous composition. We have developed a model system, termed “peptide self-assembly mimic (PSAM)”, based on the single-layer β-sheet of Borrelia outer surface protein A (OspA). PSAM allows for the capture of a defined number of self-assembly-like peptide repeats within a water-soluble protein, making structural and energetics studies possible. In this work, we extend our PSAM approach to a highly hydrophobic peptide sequence. We show that a penta Ile peptide (Ile5), which is insoluble and forms β-rich self-assemblies in aqueous solution, can be captured within the PSAM scaffold in a form capable of self-assembly. The 1.1-Å crystal structure revealed that the Ile5 stretch forms a highly regular β-strand within this flat β-sheet. Self-assembly models built with multiple copies of the crystal structure of the Ile5 peptide segment showed no steric conflicts, indicating that this conformation represents an assembly-competent form. The PSAM retained high conformational stability, suggesting that the flat β-strand of the Ile5 stretch primed for self-assembly is a low-energy conformation of the Ile5 stretch and rationalizing its high propensity for self-assembly. The ability of the PSAM to “solubilize” an otherwise insoluble peptide stretch suggests the potential of the PSAM approach to the characterization of self-assembling peptides.
Keywords: amyloid fibril, single-layer β-sheet, solubilization, protein engineering, x-ray crystallography
Introduction
It has become evident that many polypeptides can be transformed into β-rich self-assemblies.1 β-Rich self-assembly is the core structure of the so-called cross-β amyloid fibrils that are associated with devastating human diseases. Discovery of diverse peptide sequences that can form self-assemblies has increased interest in peptide self-assembly as a means for creating nano-scale structures. Self-assembly of molecules is becoming a popular and powerful strategy to create nanomaterials, and because peptide building blocks with desired chemical composition can be readily synthesized, peptide self-assembly offers a technology platform for the production of diverse nanomaterials. Indeed, many types of self-assemblies have been produced using synthetic peptides.2; 3 Therefore an increased understanding of the factors governing peptide self-assembly will have broad impacts on material sciences, biology and medicine.
Despite their importance, still little is known about high-resolution structure and biophysical properties of peptide self-assemblies. Their insoluble nature and inherently heterogeneous stoichiometry make it extremely difficult to apply the standard biophysical techniques for water-soluble proteins. Solid-state NMR studies have elucidated the conformations of peptide units and modes of higher-order assemblies.4; 5; 6; 7 Although powerful, it is a laborious method, and it is difficult to obtain high-resolution structural information. Pioneering work by the Eisenberg group has determined the atomic structures of amyloidgenic peptides assembled into micro crystals, which has established the cross-β spine as a common structure of peptide fibrils.8; 9 Although the crystallization approach is also powerful, it is limited to structural determination and it seems that peptides are crystallized in a high-energy state.10 In addition, it may not be applicable to a broad range of amyloids beyond short peptides.
To overcome these fundamental difficulties associated with characterization of self-assembling peptides, several groups have employed transplantation of a peptide segment into a water-soluble, globular protein. In this approach, it is hoped that an engineered protein harboring the peptide segment remains soluble and insights into the structure and thermodynamics of self-assembly can be gained by characterizing the engineered protein with standard biophysical techniques such as x-ray crystallography. Perutz et al. introduced Gln repeats into a flexible loop of chymotrypsin inhibitor 2 (CI2).11 Although the engineered CI2 oligomerized, its crystal structure revealed that the oligomerization was due to domain swapping and the Gln repeats were disordered and not forming β-rich self-assembly.12 Similarly, Takano et al. replaced a C-terminal segment of ribonuclease HI with an Aβ fragment, which prevented aggregation of the Aβ fragment.13 The x-ray crystal structure of this fusion protein revealed that, although the Aβ segment had a β-sheet conformation, it did not form β-sheet-mediated interactions with other molecules, suggesting that the observed form lacked the capability of self-assembly. These examples illustrate the difficulty of capturing a peptide in a form primed for assembly into higher-order structures.
We have developed a protein engineering strategy to overcome the fundamental challenges in high-resolution characterization of peptide self-assemblies. Our approach, termed peptide self-assembly mimic (PSAM), captures a peptide segment of interest within the structural context of a flat β-sheet. A highly regular, flat β-sheet is a hallmark of β-rich self-assemblies. Thus, our approach is distinct from other transplantation attempts in that it grafts a peptide of interest in an environment close to that of self-assembly.
As the host for PSAM, we use the single-layer β-sheet (SLB) segment of outer surface protein A (OspA) from Borrelia Burgdorferi. The SLB exists as a segment linking two globular domains, which in turn serve as “end caps” for the SLB. Although it is exposed to the solvent and thus it does not contain a hydrophobic core, it is highly stable. Indeed, the presence of the globular domain would appear to inhibit the lamination of the β-sheet, regardless of its composition.14; 15 The single-layer architecture eliminates complications caused by long-range interactions through a hydrophobic core that are commonly present in a water-soluble β-sheet protein. We have demonstrated that a β-hairpin sequence from the SLB forms β-rich fibrils16 and the self-assembly of this β-hairpin segment can be captured within OspA in the form of extended SLB.17 The x-ray crystal structures of these PSAMs have enabled to establish structural linkage between the atomic structures of self-assembling peptides and the macroscopic morphology.14 The two globular domains sequester the “sticky” β-sheet edges, thus enabling the PSAM system to capture β-rich self-assembly within a water-soluble protein. Furthermore, unlike most β-sheets found in globular proteins,18 the SLB has a highly regular geometry and is flat, which may closely mimic the β-sheet conformation within actual peptide assemblies. Recent studies have shown that many peptide sequences self-assemble into an anti-parallel β-sheet and small sequence changes can alter the β-sheet topology between parallel and anti-parallel.9; 19 Therefore, although Our PSAM system using OspA can mimic only self-assemblies consisting of anti-parallel β-sheets, its detailed structural and energetic studies would provide significant knowledge relevant to the molecular mechanism of peptide-self assembly.
In our initial work to establish the PSAM strategy, we used a hydrophilic peptide sequence derived from the SLB itself. In this work, we wished to explore the potential of the PSAM to capture diverse self-assembling peptides. To this end, we introduced penta-Ile motif in the PSAM. We show that a penta-Ile peptide forms β-rich self-assemblies and it can be stably captured into the PASM. The high-resolution x-ray crystal structure of the peptide captured in the PSAM reveals molecular details of β-rich peptide self-assembly.
Results
An oligo-Ile peptide forms β-rich self-assemblies
It has been shown that certain poly-amino acids self-assemble into fibrils.20 Subsequently, amyloid aggregation propensities of amino acids have been derived.21; 22 Ile has the highest aggregation propensity among aliphatic amino acids on the scale of Pawer et al. 21 and it is also among the most aggregation-prone amino acids on the scale of Rousseau et al.22 Ganesh et al. have reported that short peptides containing three consecutive Ile form β-rich self-assembly.23 Therefore, we chose an oligo-Ile peptide for this study. An oligo-Ile peptide is very hydrophobic and distinct from the highly hydrophilic sequence of the OspA SLB, and thus it represents a challenging target for transplantation into the PSAM system.
A penta-Ile peptide (Ile5) was synthesized with solid phase peptide synthesis. The purity and molecular weight of the peptide were confirmed with LC-MS. Because the lyophilized peptide only sparingly dissolved in an aqueous buffer, we prepared peptide solutions in two methods. First, the peptide was suspended at a 2mg/ml concentration in an aqueous buffer with vigorous voltexing, and the supernatant of this suspension after centrifugation was used. Second, the peptide was first completely dissolved in 100% DMSO at 20 mg/ml and this solution was diluted tenfold into an aqueous buffer, resulting in a solution containing 10% DMSO.
Atomic force microscopy (AFM) characterization revealed needle-like objects in the peptide solution (Figure 1b and 1c). Their widths ranged from ∼20 to ∼100 nm and their lengths were a few μm. The needle-like objects were also found in the sample in 10% DMSO (Figure 1d), and the shape of the objects in the 10% DMSO sample was similar to that in the water sample, indicating that these objects can form from monomeric peptides in solution and they are not an artifact from incomplete solubilization of peptide powder. The suspension supernatant exhibited a far-UV circular dichroism (CD) spectrum characteristic of β-sheet (Figure 1a), indicating the peptide self-assembles had high β-sheet content. The AFM objects showed no twist commonly observed for peptide self-assemblies. Both samples show X-ray diffraction maxima corresponding to 4.6 Å and 10 Å spacings (Figure 1e, f), a hallmark of β-rich peptide assemblies.24. Commonly, the 4.6 Å spacing is interpreted as the spacing between β-strands within a β-sheet and the the 10 Å spacing as the spacing between laminated two β-sheets, which are the basis for the cross-β structure.24 Interestingly, the Ile5 self-assembly showed little Thioflavin-T (ThT) binding activity. It should be noted that not all peptide fibrils bind ThT binding.25 Taken together, their morphology, high β-sheet content and the X-ray diffraction patterns suggest that the oligo-Ile peptide forms a cross-β self-assembly, as seen for fibrils of other peptides.
Figure 1.

(a) Far UV CD spectrum of a supernatant of homo Ile peptide suspension in water. (b, c, and d) AFM scan images. 1 μm scale bars are indicated in the images. The brightness of features increases as a function of height. The CD spectrum is plotted with the ellipticity, not with the molar ellipticity, because we could not determine the concentration of the peptide solution. (b, c) The images from water suspension sample which is taken from different area of the same sample surface. (d) The image from 10% DMSO sample. (e, f) X-ray fiber diffraction patterns of the water (e) and 10% DMSO (f) samples. The diffraction maxima corresponding to 4.6 Å and 10 Å spacings are marked with the arrows.
Stable grafting of an oligo-Ile stretch into the PSAM
Having confirmed the ability of oligo-Ile peptide to self-assemble into a β-rich structure, we proceeded to graft the Ile5 sequence within the PSAM β-sheet. We hypothesized that, if the Ile5 peptide has a high propensity to self-assemble into flat β-sheets, replacing a flat β-strand with an Ile5 stretch should not reduce the stability of the PSAM. Thus, we grafted the Ile5 sequence into the central β-strand (strand 9) of the OspA SLB (Figure 2a). The SLB consists of three solvent-exposed strands. We chose strand 9 as the host because it is centrally located in the SLB and highly exposed to the solvent. These characteristics minimize interactions of residues in strand 9 with those in the globular domains. The five consecutive Ile residues replaced residues 120-124 (SSTEE), and we term the resulting mutant PSAM-Ile5.
Figure 2.

(a) The amino acid sequence of OspA SLB region. The line rectangles indicate residues in a β-strand of which side-chains face toward the reader and the shaded rectangles indicate those facing backward. The mutational residues are shown in rectangle. The backbone hydrogen bonds are shown as dashed lines (b) Elution profiles of the wild type (dashed line) and PSAM-Ile5 (solid line) from a size-exclusion column detected using absorbance at 280 nm. (c) Urea-induced unfolding of PSAM-Ile5. Normalized CD (circles) and fluorescence (cross) intensities for PSAM-Ile5 are plotted as a function of urea concentration. The profiles for the wild-type protein are also shown (CD, solid line; fluorescence, dashed line).
In spite of the introduction of highly hydrophobic segment at highly solvent-exposed positions, PSAM-Ile5 was expressed as a soluble protein in E. coli and it was predominantly monomeric in aqueous solution as judged with gel-filtration chromatography (Figure 2b). These results indicate that the PSAM scaffold effectively “solubilizes” the otherwise insoluble Ile5 peptide by sequestering within the SLB.
PSAM-Ile5 underwent a reversible unfolding reaction induced by urea (Figure 2c). The unfolding curve was interpreted in terms of a three-state model as for the wild type OspA.15; 26 In this three-state model, the first transition corresponds to unfolding of the C-terminal domain and a portion of the SLB, and the second to that of the rest of the SLB and the N-terminal domain. The overall stability of PSAM-Ile5 was nearly identical to that of wild-type (ΔΔGNU3M = 0.07 kcal/mol), but the first transition was destabilized (ΔΔGNI3M = -0.75 kcal/mol) and the second transition was stabilized (ΔΔGIU3M = 0.82 kcal/mol) (ΔGNU3M, ΔGNI3M and ΔGIU3M are the free energy difference at 3M urea between the native and unfolded states, that between the native and intermediate states and that between the intermediate and unfolded state, respectively; ΔΔGNU3M, ΔΔGNI3M and ΔΔGIU3M are differences in ΔGNU3M, ΔGNI3M and ΔGIU3M between the wild-type and mutant, respectively). These compensatory changes suggest that the Ile5 replacement has altered the nature of the intermediate state. Although the detailed effects of the strand replacement on the unfolding mechanism are not clear, these results demonstrate that PSAM accommodates the strand replacement without a significant stability loss.
High-resolution x-ray crystal structure of PSAM-Ile5
Our crystallization efforts of PSAM-Ile5 did not yield any single crystals. To facilitate crystallization, we introduced a set of surface mutations in the globular domains that have been highly effective in crystallizing OspA and its variants.14; 27 These mutations are distant from the grafted Ile5 segment and have previously shown to cause little structural change.27 The resulting surface engineered protein, termed PSAM-Ile5-sm1, readily crystallized, and its crystal structure was determined at a resolution of 1.10-Å (Figure 3a). The data collection and refinement statistics are summarized in Table 1. Despite the fact that Ile5 replacement in strand-9 completely disrupts the native inter-strand pairings (Figure 2a), the global fold of PSAM-Ile5-sm1 was nearly identical to the wild-type OspA containing the same set of surface mutations (Figure. 3b), with a root-mean-squared (RMS) deviation of 0.3 Å for the Cα atoms common between the wild-type and mutant structures. This structural resemblance is remarkable, because the SLB does have a level of structural plasticity. For example, a series of PSAMs with different numbers of peptide inserts exhibited a range of backbone conformations,14 and a single mutation at Phe126 located at the C-terminus of strand 9 (Figure 2a) significantly perturbs the backbone structure, increasing the Cα RMS deviation to 1.15 Å.15 Thus the little structural deviation due to the Ile5 replacement suggests that the PSAM SLB provides a structural environment that is compatible with a stable conformation of the Ile5 segment.
Figure 3.
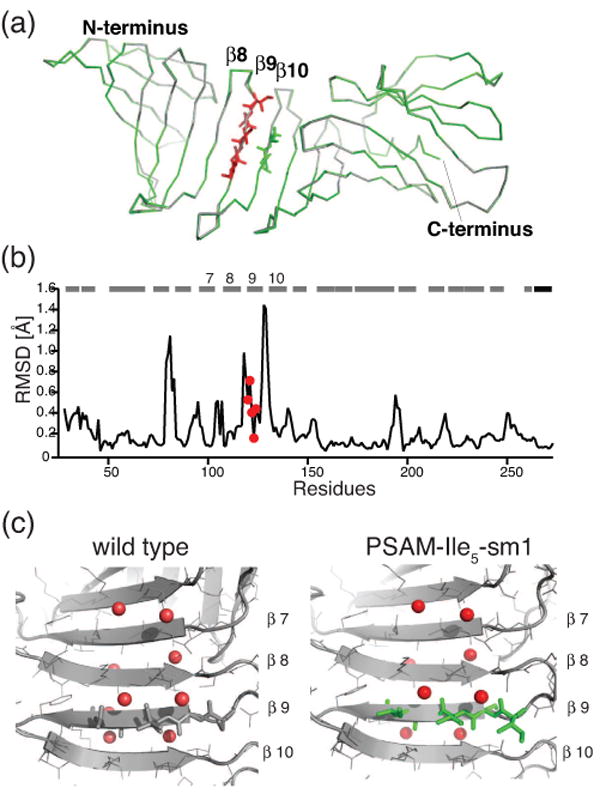
Crystal structure of PSAM-Ile5-sm1. (a) Superposition of PSAM-Ile5-sm1 (green) onto OspA-sm1 (gray). The mutated residues are shown in red sticks and Ile residues in strand-10 are also shown in green sticks. N- and C- terminus positions are indicated. (b) The root-mean-square deviations (RMSD) for the Cα atoms are plotted versus residue number. The positions of β-strands and the C-terminal helix are shown as bars. The mutation residues are indicated as a red circle. (c) Back-bone hydration of the SLB. The water molecules are shown as red spheres. The mutation residues are shown as green sticks.
Table 1.
Data collection and refinement statistics for PSAM-Ile5-sm1
| Data collection statistics | |
| Space group | P21 |
| Cell parameters | a=33.26 |
| b=54.71 | |
| c=66.50 | |
| β=99.91 | |
| Beamline | APS-23ID |
| Wavelength | 0.9795 Å |
| Resolution (Å) (highest resolution shell) a) | 50-1.1(1.14-1.10) |
| Completeness (%) | 97.8(86.2) |
| I/σ(I) | 19.25(2.88) |
| Rmerge b) | 0.053(0.318) |
| Average redundancy | 3.3(2.3) |
| Refinement statistics | |
| Resolution range (Å) | 20.0-1.1 |
| Reflections used (free) | 88420 (4669) |
| R factor c) | 0.155 |
| Rfree d) | 0.171 |
| RMS deviations | |
| Bonds (Å) | 0.008 |
| Angles (°) | 1.281 |
| No. protein residues | 246 |
| No. waters | 378 |
| Average B factor (Å2) | 9.82 |
| Ramachandran plot statistics | |
| Most favored (%) | 91.9 |
| Additionally allowed (%) | 8.1 |
| Generally allowed (%) | 0.0 |
Highest resolution shell is shown in parenthesis.
R-merge = ΣhklΣi| I(hkl)i - <I(hkl)> |/ΣhklΣi<I(hkl)i> over i observations of a reflection hkl.
R-factor = Σ | |F(obs)| - |F(calc)| |/Σ|F(obs)|.
Rfree is R with 5% of reflections sequestered before refinement.
In the crystal structure, the Ile5 segment takes on a regular β-sheet conformation with right-hand twist along the strand (Figure 4a). In addition to the Ile5 stretch, PSAM-Ile5 contains two cross-strand Ile-Ile pairs involving the Ile5 stretch and residues in strand 10 (Figures 3a and 4b). These are a non-hydrogen bonded (NHB) pair (Ile122-Ile137) and a hydrogen bonded (HB) pair (Ile123-Ile136). These cross-strand pairs provide an opportunity to see the side-chain conformations and interactions of Ile residues in the context of self-assembly, since these combinations are present in an anti-parallel β-sheet structure made of homo-Ile peptides. We found distinct modes of interactions in the HB and NHB pairs (Figure 4b). As pointed out previously,28 the Cγ atoms of the NHB Ile pair face each other with slight offset along with the strand direction. They are tightly packed and exclude solvent molecule between the side chains. In contrast, the Cγ atoms of the HB pair point to mutually opposite directions and creating voids aside the Cβ atoms (Figure 4c). These voids are filled with well-defined water molecules, thus rescuing the “weak points”.27; 29 This hydration pattern is conserved in the wild-type OspA-sm1 and PSAM-Ile5-sm1, suggesting this it is a general feature found on the surface of a flat β-sheet (Figure 3c). Together, in the Ile peptide self-assembly, adjacent Ile peptides are held through the backbone hydrogen bonds, the side chains of the NHB pairs would form tight interactions and those of the HB pair would form suboptimal interactions that may be compensated by the main chain hydration at the weak points.
Figure 4.

Construction of the β-assembly models. (a) Ile5 structure in PSAM-Ile5-sm1. (b) H-bond pair and non H-bond pair of Ile residues in the crystal structure. The mutation residues are shown as red sticks and Ile residues in strand-10 are shown as green sticks. Back-bone H-bonds are shown as blue dashed line. (c) The inter-strand Ile pairing in the crystal structure. Ile residues are shown as CPK representation. Left: non-hydrogen bonding pair (shown in red). Right: hydrogen bonding pair (shown in red). Water molecules positioned “weak points” are shown in cyan. (d) Models of single-layer β-sheet assembly. Anti-parallel β-sheets model is created based on the amyloid-forming peptide crystal structure.
Atomic models of the Ile5 self-assembly
To examine if the Ile5 structure in PSAM-Ile5-sm1 reflects the structure of an Ile5 peptide within the β-rich self-assemblies, we constructed an atomic model of a self-assembly from the x-ray crystal structure of the Ile5 segment. In this model, multiple copies of the peptide structure were placed side by side so that a continuous β-sheet is formed (Figure 4d). We used the backbone structures an insulin peptide that forms anti-parallel β-sheet assembly (PDBID: 2OMQ)9 as the template for placing the Ile5 structures. It is important to emphasize that the template was used only to define the spacing and that we did not alter the conformation of the Ile5 segment in this modeling.
The resulting β-sheet structure of the model has no steric clashes, indicating that the Ile5 conformation in PSAM-Ile5-sm1 can self-assemble without conformational adjustment. Thus, this model may represent the basic unit for the self-assembly of the Ile5 peptide. Although further lamination of the β-sheet units are required to account for the dimensions of the self-assembly seen in the AFM images, this model helps to connect the atomic structure and the macroscopic morphology of peptide self-assembly.
Discussion
We have shown that an Ile5 peptide forms a β-rich self-assembly and that it can be captured within a soluble PSAM in a form that is clearly compatible with self-assembly. To our knowledge, this is the first example of the high-resolution structure of a homo-oligomer of an amino acid in an assembly-competent form. PSAM-Ile5 was highly stable and it retained the flat β-sheet seen in other PSAMs, strongly suggesting that the highly regular, flat β-strand is an energetically favorable conformation of the Ile5 peptide.
Our results with PSAM-Ile5 are in a stark contrast to previous results of grafting studies of self-assembling peptides. Often, soluble host proteins are recruited into amyloids and/or aggregates.30; 31 In other cases where the grafting location was carefully chosen so that the protein solubility was maintained, the grafted peptide was found to be in a conformation incapable of self-assembly.12; 13 The main difference between our host and the others is that ours captures a peptide of interest within a well-defined, flat β-sheet while others capture a peptide within a flexible segment. Lamination of β-sheet does not occur, presumably because this is prevented by the globular domains. The flat β-sheet is the native conformation of the host SLB. Thus, the formation of a flat β-strand conformation of the grafted peptide does not promote a nonnative conformation of the PSAM host that might cause denaturation and/or misfolding. Because the flat β-sheet is a favorable conformation of self-assembling peptides, grafting of a peptide segment with a high self-assembly propensity stabilizes the native structure of the PSAM scaffold.
β-Sheet assembly requires the formation of a backbone H-bond network across β-strands. In general, an exposed β-sheet edge is poised to interact with another β-sheet edge, and in natural proteins multitudes of negative elements are installed at an exposed β-sheet edge to prevent aberrant inter-molecular interactions.18 Thus, it is unlikely that a grafted peptide segment finds a highly regular β-sheet edge in the host protein to which it can dock. Accordingly, it is unlikely that simple grafting of a self-assembling peptide unit in a natural host protein would promote the formation of a self-assembly-mimicking conformation (e.g. β-sheet) of the peptide. Furthermore, simple grafting provides no clear mechanisms for preventing uncontrolled self-assembly of a grafted peptide.
Our PSAM approach converts peptide self-assembly into protein folding, allowing for high-resolution characterization of structural features in peptide self-assemblies using well-established tools. While the PSAM scaffold promotes the β-sheet formation of the grafted sequence, the scaffold has enough structural plasticity so that the grafted peptide can adapt its own, favorable conformation. The work presented here demonstrates that the PSAM is applicable to diverse peptide sequences including extremely hydrophobic ones such as Ile5, suggesting sequences from natural fibril-forming proteins can also be captured in the PSAM. We speculate that, with the ability of the PSAM to accommodate larger segments of self-assembly mimics,14 this strategy will complement existing ones such as solid-state NMR spectroscopy and micro crystallography to shed new light to molecular structure and biophysical properties of peptide self-assemblies.
Materials and Methods
Peptide preparation
The Ile5 peptide was synthesized by solid phase peptide synthesis using standard FMOC chemistry on a Rink MBHA resin. Its N-terminus was unblocked and its C-terminus was amidated. The product was cleaved from the resin using TFA:triisopropylsilane:H2O = 95:2.5:2.5 (v:v:v) and dissolved in 50% acetic acid and purified using reversed-phase HPLC.
Protein preparation
Methods for site-directed mutagenesis, urea denaturation experiments and analysis of denaturation data have been described previously.26 The gene for the PSAM-Ile5 was subcloned in the equivalent region of the OspA surface mutant for crystallization (“OspAsm1”)27 using SpeI and PstI restriction enzyme sites, resulting twelve surface mutations in total. The mutant was expressed and purified as described before.27 Analytical size exclusion chromatography was performed using a Superdex 75 HR 10/30 column (GE healthcare) in 50mM Tris-HCl pH8 containing 200 mM NaCl at room temperature.
Circular dichroism spectroscopy
CD measurements were carried out using an Aviv 202 spectropolarimeter (Aviv Associates, Lakewood, NJ). The spectrum was taken at 25°C in a 1-mm path length cuvette with a 1-nm step size, a 3-nm bandwidth and a 1-second averaging time.
Atomic force microscopy
Samples were imaged after incubation for ∼1 month at room temperature. Samples were deposited onto a freshly cleaved mica surface and left on mica for 30 s and then rinsed with deionized water. The mica surface with the adsorbed peptide was then dried with nitrogen gas. The images shown were obtained by scanning the mica surface in air by AFM (Multimode, Digital Instruments, Santa Barbara, CA) with spring constant of 2 N/m and tip radius of curvature of <10 nm (AC240TS-C2; Olympus). AFM scans were taken at 512 × 512 pixels resolution and produced topographic images of the samples, in which the brightness of features were as follows: tapping frequency ∼70 kHz, RMS amplitude before engage 1 V, integral and proportional gains 0.2-0.6 and 0.3-1 respectively, set point 0.8V, and scanning speed 1 Hz.
X-ray fiber diffraction
The same batch of Ile5 samples that were used in the AFM experiments were placed in 1-mm diameter quartz capillary and vacuum dried. Diffraction data were collected at synchrotron radiation beamline 23ID (the Advanced Photon Source (APS) at the Argonne National Laboratory) and examined using the Mosflm software.32
Crystallization and structure determination
The crystals were grown in 30% PEG400, 0.1M Tris-HCl pH8.0 using the hanging drop vapor diffusion method. The x-ray diffraction data were collected at the APS 23-ID beamline. Crystal data, data collection statistics is summarized in Table 1. x-ray diffraction data were processed with HKL2000.33 The structure was determined by molecular replacement with the program MOLREP in CCP4.32 The OspA-sm1 structure (PDBID: 2G8C) was used as the search models. CNS1.1 34 and Refmac5 35 were used for the structural refinement, and final positional and anisotropic temperature factor refinement was performed using Refmac5. Model building was carried out using the Coot program.36 The coordinates have been deposited in the Protein Data Bank with an entry code 2I5V. The self-assembly model was built by superposing the five Cα positions of the Ile5 structure of PSAM-Ile5 onto the Ca positions of the crystal structure of the insulin peptide. Molecular graphics were generated using Pymol (http://www.pymol.org).
Acknowledgments
We are thankful to Dr. Justin Jureller of the University of Chicago NanoBio facility for his assistance with atomic force microscopy. This work was supported in part by NIH grant R01-GM72688 and NSF grant CMMI-0709079. MB was supported by NIH grant T90-DK070076 and the Paul K. Richter and Evalyn E. Cobb Richter Memorial Fund. Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. W-31-109-ENG-38. GM/CA CAT (APS 23-ID) has been funded in whole or in part with Federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Science (Y1-GM-1104).
Footnotes
Accession Code: The coordinates have been deposited in the Protein Data Bank with an entry code 2I5V
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dobson CM. Protein folding and disease: a view from the first Horizon Symposium. Nat Rev Drug Discov. 2003;2:154–60. doi: 10.1038/nrd1013. [DOI] [PubMed] [Google Scholar]
- 2.Rajagopal K, Schneider JP. Self-assembling peptides and proteins for nanotechnological applications. Curr Opin Struct Biol. 2004;14:480–6. doi: 10.1016/j.sbi.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Fairman R, Akerfeldt KS. Peptides as novel smart materials. Curr Opin Struct Biol. 2005;15:453–63. doi: 10.1016/j.sbi.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Jaroniec CP, MacPhee CE, Bajaj VS, McMahon MT, Dobson CM, Griffin RG. High-resolution molecular structure of a peptide in an amyloid fibril determined by magic angle spinning NMR spectroscopy. Proc Natl Acad Sci U S A. 2004;101:711–6. doi: 10.1073/pnas.0304849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer's amyloid-beta(1-42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–7. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer's beta-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tycko R. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr Opin Struct Biol. 2004;14:96–103. doi: 10.1016/j.sbi.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 8.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–8. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJ, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–7. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 10.Esposito L, Pedone C, Vitagliano L. Molecular dynamics analyses of cross-{beta}-spine steric zipper models: {beta}-Sheet twisting and aggregation. Proc Natl Acad Sci U S A. 2006 doi: 10.1073/pnas.0602345103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stott K, Blackburn JM, Butler PJ, Perutz M. Incorporation of glutamine repeats makes protein oligomerize: implications for neurodegenerative diseases. Proc Natl Acad Sci U S A. 1995;92:6509–13. doi: 10.1073/pnas.92.14.6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen YW, Stott K, Perutz MF. Crystal structure of a dimeric chymotrypsin inhibitor 2 mutant containing an inserted glutamine repeat. Proc Natl Acad Sci U S A. 1999;96:1257–61. doi: 10.1073/pnas.96.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Takano K, Endo S, Mukaiyama A, Chon H, Matsumura H, Koga Y, Kanaya S. Structure of amyloid beta fragments in aqueous environments. Febs J. 2006;273:150–8. doi: 10.1111/j.1742-4658.2005.05051.x. [DOI] [PubMed] [Google Scholar]
- 14.Makabe K, McElheny D, Tereshko V, Hilyard A, Gawlak G, Yan S, Koide A, Koide S. Atomic structures of peptide self-assembly mimics. Proc Natl Acad Sci U S A. 2006;103:17753–17758. doi: 10.1073/pnas.0606690103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan S, Gawlak G, Makabe K, Tereshko V, Koide A, Koide S. Hydrophobic Surface Burial Is the Major Stability Determinant of a Flat, Single-layer beta-Sheet. J Mol Biol. 2007;368:230–43. doi: 10.1016/j.jmb.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohnishi S, Koide A, Koide S. Solution Conformation and Amyloid-like Fibril Formation of a Polar Peptide Derived from a β-Hairpin in the OspA Single-Layer β-Sheet. J Mol Biol. 2000;301:477–489. doi: 10.1006/jmbi.2000.3980. [DOI] [PubMed] [Google Scholar]
- 17.Koide S, Huang X, Link K, Koide A, Bu Z, Engelman DM. Design of single-layer beta-sheets without a hydrophobic core. Nature. 2000;403:456–460. doi: 10.1038/35000255. [DOI] [PubMed] [Google Scholar]
- 18.Richardson JS, Richardson DC. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci U S A. 2002;99:2754–9. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon DJ, Balbach JJ, Tycko R, Meredith SC. Increasing the amphiphilicity of an amyloidogenic peptide changes the beta-sheet structure in the fibrils from antiparallel to parallel. Biophys J. 2004;86:428–34. doi: 10.1016/S0006-3495(04)74119-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fandrich M, Dobson CM. The behaviour of polyamino acids reveals an inverse side chain effect in amyloid structure formation. Embo J. 2002;21:5682–90. doi: 10.1093/emboj/cdf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J Mol Biol. 2005;350:379–92. doi: 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 22.Rousseau F, Schymkowitz J, Serrano L. Protein aggregation and amyloidosis: confusion of the kinds? Curr Opin Struct Biol. 2006;16:118–26. doi: 10.1016/j.sbi.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 23.Ganesh S, Jayakumar R. Structural transitions involved in a novel amyloid-like beta-sheet assemblage of tripeptide derivatives. Biopolymers. 2003;70:336–45. doi: 10.1002/bip.10474. [DOI] [PubMed] [Google Scholar]
- 24.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CC. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 25.Ivanova MI, Thompson MJ, Eisenberg D. A systematic screen of beta(2)-microglobulin and insulin for amyloid-like segments. Proc Natl Acad Sci U S A. 2006;103:4079–82. doi: 10.1073/pnas.0511298103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan S, Gawlak G, Smith J, Silver L, Koide A, Koide S. Conformational heterogeneity of an equilibrium folding intermediate quantified and mapped by scanning mutagenesis. J Mol Biol. 2004;338:811–25. doi: 10.1016/j.jmb.2004.02.063. [DOI] [PubMed] [Google Scholar]
- 27.Makabe K, Tereshko V, Gawlak G, Yan S, Koide S. Atomic-resolution crystal structure of Borrelia burgdorferi outer surface protein A via surface engineering. Protein Sci. 2006;15:1907–1914. doi: 10.1110/ps.062246706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wouters MA, Curmi PM. An analysis of side chain interactions and pair correlations within antiparallel beta-sheets: the differences between backbone hydrogen-bonded and non-hydrogen-bonded residue pairs. Proteins. 1995;22:119–131. doi: 10.1002/prot.340220205. [DOI] [PubMed] [Google Scholar]
- 29.Finkelstein AV, Nakamura H. Weak points of antiparallel beta-sheets. How are they filled up in globular proteins? Protein Eng. 1993;6:367–72. doi: 10.1093/protein/6.4.367. [DOI] [PubMed] [Google Scholar]
- 30.Esteras-Chopo A, Serrano L, Lopez de la Paz M. The amyloid stretch hypothesis: recruiting proteins toward the dark side. Proc Natl Acad Sci U S A. 2005;102:16672–7. doi: 10.1073/pnas.0505905102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim W, Hecht MH. Generic hydrophobic residues are sufficient to promote aggregation of the Alzheimer's Abeta42 peptide. Proc Natl Acad Sci U S A. 2006;103:15824–9. doi: 10.1073/pnas.0605629103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Collaborative_Computational_Project. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–55. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]