

Abstract
The syntheses of a new class of barbiturate-based inhibitors for human and E. Coli Methionine Aminopeptidase -1 (MetAP-1) are described. Some of the synthesized inhibitors show selective inhibition of the human enzyme with high potency.
Keywords: Type-1 MetAP inhibitors, barbiturates
Cellular protein synthesis starts with an N-terminal methionine in the eukaryotic cells or with a formylmethionine in the prokaryotic cells. Removal of the methionine residue is essential for proper folding, post-translational modifications and translocation of the synthesized proteins.1 The metalloenzymes methionine aminopeptidases (MetAPs) catalyze the hydrolytic removal of the N-terminal methionine residue from the newly synthesized proteins.1 While the prokaryotic cells contain only the type-1 enzyme, eukaryotic cells contain both the type-1 and -2 enzymes in the cytosol.2 Deletion of the MetAP is shown to be lethal in various bacteria.2
Since the discovery that the irreversible MetAP-2 inhibitor fumagillin prevents angiogenesis, both reversible and irreversible inhibitors for this enzyme have been extensively studied as potential anti-angiogenesis and anti-cancer agents.3 Selective inhibition of MetAP-1 in bacteria (or in parasites) over the human enzymes has been shown to be a promising approach in designing new antibacterial4 and antimalarial5 agents. The role of MetAP-1 in human cell cycle progression has been elucidated recently.6 Selective inhibition of the human MetAP-1 (over MetAP-2) led to the arrest of cell division and induction of apoptosis in leukemia cells.6
In contrast to MetAP-2, there are only a few reports in the literature on inhibition of human MetAP-1. Peptide hydroxamic acids are reported to be competitive inhibitors for human MetAP-1 with moderate potency.7 Substituted pyridines were identified as another class of human MetAP-1 inhibitors using high-throughput screening of 175,000 small organic molecules.3b Ovalicin, the highly-potent, non-competitive, natural product inhibitor for MetAP-2, was found to bind human MetAP-1 weakly.8 We have recently initiated a research program to design and synthesize selective inhibitors for bacterial MetAP-1 as antibacterial agents and selective inhibitors for human MetAP-1 as anti-cancer agents.
Under physiological conditions, the metal ions in the active site of MetAPs are not firmly established. The active site binds to two divalent transition metal ions and the residues responsible for binding to the metal ions are conserved.4 One of these two bound metal ions undergoes a rapid exchange with free metal ions in solution.4 Since Co (II) ions activate all MetAPs, usually the inhibitors are screened using Co (II) form of the enzyme. Besides Co (II), Mn (II), Fe (II), Ni (II) and Zn (II) are proposed as possible metal ions under physiological conditions.4 To complicate inhibitor design, the potency of MetAP inhibitors depend on the identity of the metal ions in the active site of the enzyme.9 In addition, recent NMR spectroscopic studies have demonstrated that in the absence of any substrate or inhibitor, the enzyme in solution has several structures in dynamic equilibrium.4 For our studies, we added CoCl2 (100 μM) in the assay buffer; hence the reported compounds are Co (II) coordinating inhibitors.
All of the reported competitive inhibitors for MetAPs contain a Lewis base to coordinate to the transition metal ions in the active sites of the enzymes. In pursuit of designing new types of MetAP inhibitors, we noted that barbiturate-based potent inhibitors are known for the matrix metalloproteinases10 and for other metallo-enzymes.11 Herein, we report that potent inhibitors for MetAP-1 can be synthesized in one or two high-yielding steps starting from barbituric acid. To the best of our knowledge, this is the first report of MetAP inhibition by the barbituric acid derivatives.
We observed that barbituric acid, 2-thiobarbituric acid and 1,3-dimethylbarbituric acid were weak inhibitors for recombinant E. Coli and human MetAP-1 (Ki > 100μM). Preliminary docking studies suggested that barbituric acid is binding to one of the Co (II) ions in the active site through the oxygen atom on C-2. Our goal was to introduce substituents on the barbiturate moiety to produce more potent inhibitors. Knoevenegal condensation of aromatic aldehydes appeared to be an attractive method to achieve this goal as the products will have alkenes conjugated to the aromatic benzene ring. The general structures and the syntheses of the barbiturate inhibitors are shown in Scheme 1.
Scheme 1.
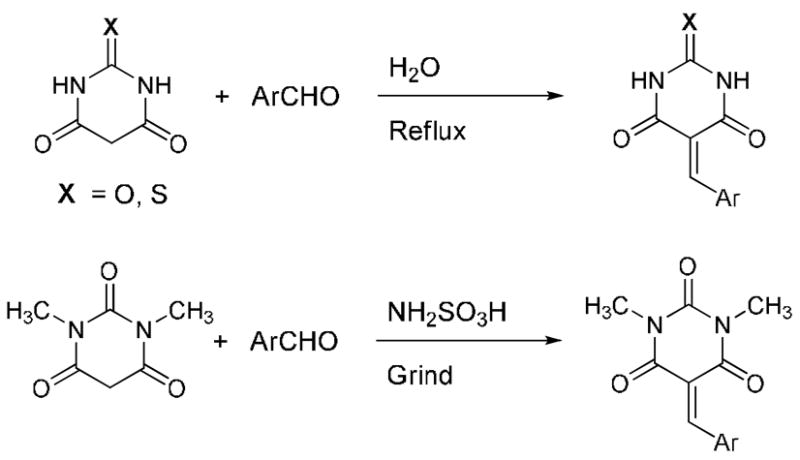
General structures and the syntheses of the barbiturate-based MetAP-1 inhibitors.
We performed the Knoevenegal condensation by heating to reflux the barbituric acid (or thiobarbituric acid) and the aromatic aldehydes in water.12 For 1,3-dimethylbarbituric acid, the condensation reaction was more conveniently performed by grinding the two solid reactants at room temperature in the presence of amidosulfonic acid.13 The residue was dissolved in dimethylsulfoxide and poured into water to precipitate the crude products. The solids obtained were recrystallized from dimethylformamide to give the pure products.
E. Coli and human MetAP-1 were expressed and purified as described previously, from expression systems kindly provided by Dr. Anthony Addlagatta14 and Dr. Brian Matthews.15 No attempt was made to remove the His-tags from either protein. Purity of the proteins was confirmed by SDS-PAGE. Protein concentration was determined using a BCA protein assay kit from Pierce, with BSA as the standard.
The inhibitory potencies of the synthesized compounds were determined by using the reported chromogenic substrate for MetAPs, Met-Pro-p-nitroanilide.16 Proline aminopeptidase was used as the coupling enzyme and we ascertained that the synthesized compounds are not inhibiting this enzyme.17 The yields obtained during the syntheses of the barbiturate derivatives and the measured inhibition constants with E. Coli and human MetAP-1 are shown in Table 1.
Table 1.
Structures, synthetic yields and the inhibition constants of the barbiturate derivatives.
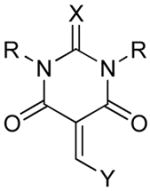 | ||||||
|---|---|---|---|---|---|---|
| Product | X | R | Y | Yield(%) | Ki (μM), MetAP-1 | |
| E. Coli | Human | |||||
| 1 | O | H | C6H5 | 85 | 517 ± 63 | 162 ± 13 |
| 2 | O | H | C6H4-pNO2 | 86 | 105 ± 18 | 8 ± 1 |
| 3 | O | H | C6H4-mNO2 | 61 | 29 ± 2 | 2 ± 0.3 |
| 4 | O | H | C6H4-pCO2H | 79 | 335 ± 47 | 5 ± 1 |
| 5 | O | H | C6H4-pOH | 86 | 4 ± 1 | 10 ± 2 |
| 6 | O | H | C6H4-pF | 89 | 89 ± 12 | 2 ± 0.2 |
| 7 | O | H | C6H4-pNMe2 | 96 | 13 ± 1 | 10 ± 2 |
| 8 | O | H | C6H4-pOMe | 94 | 25 ± 2 | 2 ± 0.4 |
| 9 | O | H | CH=CH2C6H5 | 88 | 12 ± 3 | 9 ± 2 |
| 10 | O | H | CH=CH2C6H4-pNMe2 | 83 | 0.05 ± 0.03 | 0.01 ± 0.002 |
| 11 | S | H | C6H5 | 67 | > 100 | > 100 |
| 12 | S | H | C6H4-pNO2 | 75 | > 100 | > 100 |
| 13 | S | H | C6H4-mNO2 | 56 | > 100 | > 100 |
| 14 | S | H | C6H4-pCO2H | 23 | > 100 | > 100 |
| 15 | S | H | C6H4-pOH | 94 | > 100 | > 100 |
| 16 | S | H | C6H4-pF | 78 | > 100 | 6 ± 0.6 |
| 17 | S | H | C6H4-pNMe2 | 96 | > 100 | > 100 |
| 18 | S | H | C6H4-pOMe | 50 | 87 ± 31 | > 100 |
| 19 | S | H | CH=CH2C6H5 | 89 | > 100 | 98 ± 4 |
| 20 | S | H | CH=CH2C6H4-pNMe2 | 63 | 17 ± 1 | 1 ± 0.1 |
| 21 | O | Me | C6H5 | 42 | > 100 | > 100 |
| 22 | O | Me | C6H4-pNO2 | 56 | > 100 | > 100 |
| 23 | O | Me | C6H4-mNO2 | 87 | > 100 | > 100 |
| 24 | O | Me | C6H4-pCO2H | 89 | > 100 | > 100 |
| 25 | O | Me | C6H4-pOH | 69 | > 100 | > 100 |
| 26 | O | Me | C6H4-pF | 87 | > 100 | > 100 |
| 27 | O | Me | C6H4-pNMe2 | 89 | > 100 | > 100 |
| 28 | O | Me | C6H4-pOMe | 54 | > 100 | > 100 |
| 29 | O | Me | CH=CH2C6H5 | 70 | > 100 | > 100 |
| 30 | O | Me | CH=CH2C6H4-pNMe2 | 45 | > 100 | > 100 |
We observed that the barbituric acid derivatives were more potent inhibitors of MetAP-1 compared to the derivatives of 2-thiobarbituric acid or 1,3-dimethylbarbituric acid. Under the enzyme assay conditions (25 mM HEPES buffer, pH = 7.5), the barbituric and thiobarbituric acid derivatives are expected to exist as the corresponding conjugate bases.18 It is likely that the conjugate bases from the compounds 1–20 are the active forms of these inhibitors. From our preliminary modeling studies, it is anticipated that the oxygen atom at C-2 of the barbituric acid moiety is binding to the active site metal ion of MetAP-1.
The barbituric acid derivatives 1–9 showed moderate to excellent potency in inhibiting the enzyme. Some of these inhibitors were found to be selective for the human MetAP-1 over the E. Coli enzyme. For example, compound 4 was 67 times more potent in inhibiting the human enzyme (Ki = 5 μM) compared to the E. Coli MetAP-1 (Ki = 335 μM). In order to determine the effect of an additional potential coordinating atom to the active site Co (II) atoms of MetAP, we synthesized the barbiturate derivative of 2-hydroxy-4-methoxybenzaldehyde. However, the resultant compound was found to be a weak inhibitor for both E. Coli and human MetAP-1 (Ki > 100 μM for both enzymes).
Inhibitors with substituents on the benzene ring were more effective compared to the molecule containing the unsubstituted benzene ring. In general, for E. Coli MetAP-1, compounds with electron releasing groups at the para-position of the benzene ring showed higher inhibitory potency compared to compounds with electron withdrawing groups on the aromatic ring. We did not observe any such trend for the inhibition of the human MetAP-1. Based on our calculations employing the semi-empirical PM3 force field (Spartan 06, Wavefunction Inc.), the charge densities at the oxygen atoms of the barbituric acid moiety are not perturbed by the nature of the substituents on the benzene ring. Currently, we are performing quantitative structure activity relationship studies with the synthesized inhibitors and MetAP-1 to determine the origin of this observed selectivity.
All of the compounds excepting 10 were competitive inhibitors for both E. Coli and human MetAP-1. Compound 10 was the most effective inhibitor synthesized (Ki = 50 nM and 10 nM for the E. Coli and human MetAP-1, respectively) and it demonstrated a mixed mode of inhibition for both of the enzymes. In fact, 10 is one of the most potent inhibitor reported for human MetAP-1 so far. In addition to any electronic effect, the hydrophobic alkene moieties also possibly contribute to the excellent inhibitory potency exhibited by compound 10. Structurally, it appears that the addition of the alkenyl spacer to compound 7 (i.e., compound 10) leads to substantial improvement in the inhibitory potency. A similar trend was observed for the inhibitors 1 and 9. Currently, we are evaluating the effect of this structural modification on the inhibitory potency for the compounds 2 – 6 and 8.
In contrast, most of the thiobarbiturate derivatives synthesized did not inhibit MetAP-1. The thiobarbiturate derivatives 18 and 20 showed weak inhibition of E. Coli MetAP-1 (Ki = 87 and 17 μM, respectively). Compounds 16 and 20 demonstrated moderate and selective inhibition of human MetAP-1 (Ki = 6 and 1 μM, respectively). The synthesized derivatives of 1,3-dimethylbarbituric acid (21–30) failed to inhibit the enzyme. We do not yet understand the molecular basis for this lack of inhibition exhibited by this series of compounds. The compounds 21–30 cannot deprotonate to generate the conjugate base under the enzyme assay conditions. In addition, the two methyl groups on the barbiturate moiety may sterically interfere with the binding process in the enzyme active site. Either of these two factors (or a combination) may lead to the lack of inhibition of MetAP-1 by this class of compounds.
In conclusion, we have synthesized several derivatives of barbituric acid and demonstrated their effectiveness in inhibiting MetAP-1. All of the reported inhibitors were synthesized in one step from commercially available starting materials. One of the synthesized compounds (e.g., 10) showed excellent inhibitory potency for E. Coli and human MetAP-1 (Ki < 100 nM). Another moderate inhibitor (e.g., 4) showed very good selectivity (> 60) for human MetAP-1 compared to the E. Coli enzyme. Detailed molecular modeling and cellular assay studies are currently in progress and these results will be reported later.
Supplementary Material
Supplementary data: Experimental details and characterization data for the synthesis of the compounds reported in Table 1 are available as Supplementary data. Supplementary data associated with this article can be found, in online version, at doi:
Acknowledgments
This research was supported by the NIH grant 1R01 CA113746 and NSF DMR-0705767 to SM and DKS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Frottin F, Martinez A, Peynot P, Mitra S, Holz RC, Giglione C, Meinnel T. Mol Cell Proteomics. 2006;5:2336. doi: 10.1074/mcp.M600225-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Swierczek K, Copik AC, Swierczek SI, Holz RC. Biochemistry. 2005;44:12049. doi: 10.1021/bi047752g. [DOI] [PubMed] [Google Scholar]
- 3.(a) Cooper AC, Karp RM, Clark EJ, Taghizadeh NR, Hoyt JG, Labenski MT, Murray MJ, Hannig G, Westin WF, Thompson CD. Clin Cancer Res. 2006;12:2583. doi: 10.1158/1078-0432.CCR-05-0871. [DOI] [PubMed] [Google Scholar]; (b) Hu X, Addlagatta A, Matthews BM, Liu JO. Angew Chem Int Ed. 2006;45:3772. doi: 10.1002/anie.200600757. [DOI] [PubMed] [Google Scholar]; (c) Sheppard GS, Wang J, Kawai M, Fidanze SD, BaMaung NY, Erickson SA, Barnes DM, Tedrow JS, Kolaczkowski L, Vasudevan A, Park DC, Wang GT, Sanders WJ, Mantei RA, Palazzo F, Tucker-Gracia L, Lou P, Zhang Q, Park CH, Kim KH, Petros A, Olejniczak E, Nettesheim D, Hajduk P, Henkin J, Lesniewski R, Davidsen SK, Bell RL. J Med Chem. 2006;49:3832. doi: 10.1021/jm0601001. [DOI] [PubMed] [Google Scholar]
- 4.(a) Evdokimov AG, Pokross M, Walter RL, Mekel M, Barnett BL, Amburgey J, Seibel WL, Soper SJ, Djung JF, Fairweather N, Diven C, Rastogi V, Grinius L, Klanke C, Siehnel R, Twinem T, Andrews R, Curnow A. Proteins. 2007;66:538. doi: 10.1002/prot.21207. [DOI] [PubMed] [Google Scholar]; (b) Schiffman R, Neugebauer A, Klein CD. J Med Chem. 2006;49:511. doi: 10.1021/jm050476z. [DOI] [PubMed] [Google Scholar]; (c) Cui YM, Huang QQ, Xu J, Chen LL, Li JY, Ye QZ, Li J, Nan FJ. Bioorg Med Chem Lett. 2005;15:4130. doi: 10.1016/j.bmcl.2005.06.005. [DOI] [PubMed] [Google Scholar]; (c) Luo QL, Li JY, Liu ZY, Chen LL, Li J, Ye QZ, Nan FJ. Bioorg Med Chem Lett. 2005;15:635. doi: 10.1016/j.bmcl.2004.11.034. [DOI] [PubMed] [Google Scholar]
- 5.Chen X, Chong CR, Shi L, Yoshimoto T, Sullivan DJ, Jr, Liu JO. Proc Natl Acad Sci USA. 2006;103:14548–14553. doi: 10.1073/pnas.0604101103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu X, Addlagatta A, Lu J, Matthews BW, Liu JO. Proc Natl Acad Sci USA. 2006;103:18148. doi: 10.1073/pnas.0608389103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu X, Zhu J, Srivathan S, Pei D. Bioorg Med Chem Lett. 2004;14:77. doi: 10.1016/j.bmcl.2003.10.031. [DOI] [PubMed] [Google Scholar]
- 8.Addlagatta A, Matthews BW. Protein Science. 2006;15:1842. doi: 10.1110/ps.062278006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang M, Xie SX, Ma ZQ, Hanzlik RP, Ye QZ. Biochem Biophys Res Commun. 2006;339:506. doi: 10.1016/j.bbrc.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 10.(a) Hutchings S, Liu W, Radinov R. Heterocycles. 2006;67:763. [Google Scholar]; (b) Kim SH, Pudzianowski AT, Leavitt KJ, Barbosa J, McDonnell PA, Metzler WJ, Rankin BM, Liu R, Vaccaro W, Pitts W. Bioorg Med Chem Lett. 2005;5:1101. doi: 10.1016/j.bmcl.2004.12.016. [DOI] [PubMed] [Google Scholar]; (c) Daniewski AR, Liu W, Okabe M. Org Proc Res Dev. 2004;8:411. [Google Scholar]
- 11.(a) Suzuki H, Kneller MB, Rock DA, Jones JP, Trager WF, Rettie AE. Arch Biochem Biophys. 2004;429:1. doi: 10.1016/j.abb.2004.05.015. [DOI] [PubMed] [Google Scholar]; (b) Soong CL, Ogawa J, Sakuradani E, Shimizu S. J Biol Chem. 2002;277:7051. doi: 10.1074/jbc.M110784200. [DOI] [PubMed] [Google Scholar]
- 12.Deb ML, Bhuyan PL. Tetrahedron Lett. 2005;46:6453. A representative procedure is provided here. To a solution of barbituric acid or thiobarbituric acid (3 mmol) in hot water (20 mL), the aromatic aldehyde (3 mmol) was added. After stirring at room temperature for 30 minutes the solution was heated to reflux for 1 h. The precipitated solid was filtered, washed with methanol and dried under vacuum. The crude product was recrystallized from N, N-dimethylformamide and was characterized by 1H, 13C NMR and mass spectroscopy
- 13.Li JT, Dai HG, Liu D, Li TS. Synth Commun. 2006;36:789. A representative procedure is provided here. Solid 1,3-dimethylbarbituric acid (3 mmol), aromatic aldehyde (3 mmol), and amidosulfonic acid (3 mmol) were ground in a mortar for 10 minutes and then placed in a desiccator for 2 h. The paste or the solid (depending on the aromatic aldehyde) was dissolved in 15 mL dimethylsulfoxide and was poured into 50 mL water. The precipitated solid was filtered, washed with methanol and then dried under vacuum. The crude product was recrystallized from N, N-dimethylformamide and was characterized by 1H, 13C NMR and mass spectroscopy
- 14.Addlagatta A, Hu X, Liu JO, Matthews BW. Biochemistry. 2005;44:14741. doi: 10.1021/bi051691k. [DOI] [PubMed] [Google Scholar]
- 15.Lowther WT, McMillen DA, Orville AM, Matthews BW. Proc Natl Acad Sci USA. 1998;95:12153. doi: 10.1073/pnas.95.21.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou Y, Guo XC, Yi T, Yoshimoto T, Pei D. Anal Biochem. 2000;280:159. doi: 10.1006/abio.2000.4513. [DOI] [PubMed] [Google Scholar]
- 17.Enzyme assay procedure: Coupled assays were carried out at room temperature using chromogenic substrate Met-Pro-pNA and proline aminopeptidase as the coupling enzyme in 96 well microplates. A 100 μL assay mixture contained 50 mM HEPES buffer (pH = 7.5), 100 μM CoCl2, 400 μM Met-Pro-pNA, 0.25–4.0 μM MetAP-1, 10–25 μM proline aminopeptidase and varying concentrations of inhibitor. Release of pNA was monitored at 405 nm on a Spectramax microplate reader. The Ki values were determined by non-linear regression analysis software Grafit 4.0, using the competitive steady-state model as described in: Banerjee AL, Swanson M, Roy BC, Jia X, Haldar MK, Mallik S, Srivastava DK. J Am Chem Soc. 2004;126:10875. doi: 10.1021/ja047557p.
- 18.Krasnov KA, Kartsev VG, Yurova MN. Chem Natural Compounds. 2001;6:543. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data: Experimental details and characterization data for the synthesis of the compounds reported in Table 1 are available as Supplementary data. Supplementary data associated with this article can be found, in online version, at doi: