

Abstract
Hydrogels formed from the photoinitiated, solution polymerization of macromolecular monomers present distinct advantages as cell delivery materials and are enabling researchers to three-dimensionally encapsulate cells within diverse materials that mimic the extracellular matrix and support cellular viability. Approaches to synthesize gels with biophysically and biochemically controlled microenvironments are becoming increasingly important, and require strategies to control gel properties (e.g., degradation rate and mechanism) on multiple time and size scales. Furthermore, biological responses of gel-encapsulated cells can be promoted by hydrogel degradation products, as well as by the release of tethered biologically relevant molecules.
Keywords: Photopolymerization, multifunctional monomers, hydrogels, tissue engineering
1. Introduction
Hydrogels synthesized from the chain polymerization of multifunctional macromolecular monomers represent an important class of biomaterials that are increasingly used as in situ forming materials for cell delivery and tissue regeneration 1–5. When combined with photoinitiated polymerization, gels can be fabricated under physiological conditions, enabling gelation in the presence of tissues, cells, and DNA 6–8. Highly swollen gels provide a tissue-like microenvironment for cells; the high water content promotes facile transport and can be tuned to permit long-term cell survival. Oftentimes, the gel chemistry is systematically varied to introduce biological cues, creating tailored hydrogel niches that promote or suppress targeted cell interactions or functions. In essence, hydrogels provide a tailorable microenvironment or niche that can support cell survival and function. These hydrogel niches represent an important departure from classic two-dimensional cell culture to three-dimensional cell culture, and are enabling researchers to answer questions of both fundamental biological and clinical importance. Specifically, biomaterials research spans the spectrum of fundamental studies to better understand the role of the gel environment on cellular functions and the biology of tissue formation to targeted clinical applications in the design of in situ forming cell carriers that promote healing. Critical to these approaches is the ability to control the gel properties on many times scales, from seconds to months, and on many size scales, from the molecular to macroscopic.
When designing hydrogels from macromolecular monomers, several factors must be taken into account to create a suitable niche for 3D cell culture, and these requirements are often quite stringent when forming the gel in the presence of cells. Traditionally, covalently crosslinked gels have been formed through the step polymerization of high molecular weight polymers with highly reactive low molecular weight, and often toxic, crosslinkers. These techniques include the use of aldehydes (e.g., gluteraldehyde), high energy irradiation, and the application of addition and condensation reactions 9. Most of these methods are simply incompatible with the encapsulation of cells. Thus, gels formed from the solution polymerization of water soluble precursors are becoming more widely explored for cell encapsulation, and in these systems, cytocompatibility of the monomer is a major concern. A general guideline is to synthesize macromolecular monomers with molecular weights between 2–40 kDa, as lower molecular weights are more readily internalized by cells and can lead to problems with cytotoxicity and higher molecular weights are more difficult for the body to absorb and excrete, which can lead to accumulation of unreacted or degraded products 10, 11. These water soluble macromolecules are often functionalized with vinyl end groups, and the presence of two or more polymerizable moieties leads to the formation of a crosslinked network. In biomaterial applications, methacrylate and acrylate functionalities are the most widely studied; methacrylates have the advantages of lower toxicity 12, 13, but acrylates have higher reactivities 14.
Formation of gels from these macromolecular monomers often involves the chain polymerization of multifunctional monomers in solution (e.g., 10–20 wt% solutions are common conditions). Photoinitiated polymerizations are becoming increasingly popular as an approach to form these types of hydrogel biomaterials because of the ability to form gels under physiological conditions with spatial and temporal control of the initiation process. Regardless of the initiation mechanism employed, however, care must be taken when conducting polymerizations in the presence of tissues, cells, proteins, and other biologics. Oftentimes the active specie used to initiate polymerization is highly reactive, and side reactions to polymerization can include chain transfer to proteins and/or molecules on the cell membrane. This in turn can lead to deleterious effects, eventually compromising compatibility of the material for the intended application. With this in mind, our group and others have demonstrated that under mild initiation conditions 6, 8, the polymerization rate can be tuned to allow gel formation on a clinically acceptable timescale (< 10 min) with undetectable or minimal damage to tissues, cells, proteins, or DNA 1, 2, 7, 15–19.
Another important consequence when designing crosslinked biomaterials is that, upon network formation, the network structure and materials properties are fixed. For hydrogel systems, this means that the liquid precursors are converted directly into a solid biomaterial that must be in its useful form. Manipulating the properties of the gel requires careful design and control of the initial macromolecular monomer structure, as well as the influence of the polymerization conditions on the evolution of the final network structure. Thus, versatile synthetic approaches that allow precise control of the monomer molecular weight and its distribution, functionality and architecture (e.g., linear, hyperbranched), and the distribution of chemical functionality are critical, along with understanding how reaction conditions can be used to influence the connectivity of the monomers in the final network structure. For example, recent interest has grown in the area of biomaterial formation using mixed-mode polymerizations 20–25 or controlled radical polymerizations 26–30 that lead to dramatically different network structures than classical chain polymerizations. Such approaches afford users greater versatility in tailoring the biomaterial properties from a given macromolecular monomer chemistry. Finally, numerous applications require the eventual degradation and erosion of the hydrogel system. Degradation is often introduced into crosslinked hydrogel systems by incorporation of hydrolytically or enzymatically degradable functionalities that reside within the backbone of the macromolecular monomer, leading to temporal variations in the network crosslinking density as degradation occurs. A particularly challenging aspect of biomaterial design is tuning this degradation rate, and ultimate mass loss, to match healing or the regeneration of a tissue 2, 31, 32.
In this article, we aim to demonstrate some of the needs and current directions in developing hydrogel biomaterials from the photoinitiated polymerization of multifunctional macromolecular monomer precursors. These directions relate primarily to the requirements for facile approaches to modulate network structure and incorporate diverse biological functionalities. First, by fine tuning the chemistry of macromolecular triblock monomers in combination with an externally-triggerable degradation factor, namely lipase, the degradation of poly(ethylene glycol) (PEG)-based hydrogels can be easily controlled to accommodate extracellular matrix (ECM) elaboration by PEG-encapsulated chondrocytes. Second, the network structure of PEG hydrogels formed through a photoinitiated polymerization can be dramatically affected by the incorporation of a multi-functional thiol through a mixed-mode polymerization mechanism. In addition to several advantages afforded by this polymerization mechanism, the degradation behavior of these networks is far different than that of traditional homopolymerizations. Third, photopolymerized networks can be rationally designed such that degradation products of the network itself lead to important cellular responses of gel-encapsulated cells. A striking example is discussed, in which degradation products of a PEG-hyaluronic acid (HA) copolymer gel can lead to differential responses of valvular interstitial cells (VICs), depending upon the molecular weight of the released HA fragments. This response can be altered by varying the network structure and, consequently, the size of the degradation products. Finally, polymerizable drug conjugates can be incorporated into the network during photopolymerization, and subsequent release of the tethered drug molecule can lead to a cellular response of encapsulated cells. For example, a methacrylated dexamethasone conjugate is presented; this molecule is copolymerized into a PEG gel to form dexamethasone-containing tethers, and hydrolytic degradation of ester bonds in the tethers results in the release of dexamethasone from the network. The liberated dexamethasone induces osteogenic differentiation of encapsulated human mesenchymal stem cells (hMSCs). In sum, these examples were chosen to illustrate collectively how alterations in the chemistry of the macromolecular monomers used in the preparation of photoinitiated hydrogels are crucial for developing biochemically and biophysically defined gel niches that support and promote specific cell functions important to tissue engineers.
2. Design of Multifunctional Degradable Macromers: Synthesis of Gels with Controlled Degradation Profiles
Numerous groups are interested in exploiting the photoinitiated polymerization of poly(ethylene glycol) (PEG)-based macromolecular monomers to create hydrogels for applications ranging from cell delivery vehicles [2, 15, 16, 18, 33–48] for tissue regeneration to in situ forming tissue adhesives 33–35. In many of these biomaterial applications, degradation of the final gel is desired, and the mechanism of degradation and subsequent mass loss profile are critical parameters that dictate the usefulness of the material for its intended application. Since the early work of Sawhney et al. 36, researchers have exploited the usefulness of triblock copolymer structures in controlling the final gel’s degradation-dependent properties. Specifically, lactide, glycolide, and caprolactone blocks have been grafted from PEG cores of varying molecular weight. These triblock macromolecules were then modified with polymerizable end groups to form covalently crosslinked gels with hydrolyzable crosslinks. The chemistry and structure of the degradable blocks, along with the connectivity of the final gel structure (e.g., kinetic chain lengths and cyclization), influence temporal changes in the crosslinking density and subsequent erosion of mass from the gel. Metters et al. 37–39 developed a statistical-kinetic model to describe the degradation of these types of gels, and Bryant et al. 6, 37 further demonstrated how the gel’s degradation profile dramatically influences the distribution of cell-secreted extracellular matrix molecules in a cartilage tissue engineering application. Because of the predominantly PEG-like character and high water content of these gels, degradation proceeds via a pseudo-first order hydrolysis mechanism, and mass loss results from release of degraded PEG crosslinks at early stages and release of unattached kinetic chains at later stages. While the shape and time scale of the mass loss profile can be tuned through changes in the block copolymer structure and chemistry, this approach relies on pre-selecting the appropriate degradation profile, which oftentimes can be quite difficult to know without extensive experimentation. Thus, recent research has focused on alternative approaches to pre-engineering the gel degradation, and these include the design of macromers with blocks that can be degraded by cell-secreted proteases (e.g., peptide sequences that are cleaved by matrix metalloproteases) or incorporating chemistries that degrade in response to an exogenously delivered signal 18, 40, 41. Both of these approaches can be particularly beneficial for tissue engineering strategies. In the former system, cells decide when to degrade the biomaterial, and in the latter system, degradation can be externally triggered in response to a monitored change in the biomaterial environment (e.g., temperature or pH). The following paragraph illustrates one example of the usefulness of such a system in monitoring and controlling the regeneration of cartilaginous tissue in a PEG-based gel.
Recent publications have demonstrated that lipase, a general class of enzymes that catalyze the degradation of ester bonds, will cleave caprolactone ester bonds 42, 43. Since lipase is not normally present in the cartilage microenvironment and is not secreted by cartilage forming cells, chondrocytes, this approach provides a relatively simple strategy to design gels where the degradation and mass loss can be controlled by external delivery of lipase. To capitalize on the lipase-catalyzed hydrolysis of caprolactone ester bonds, a dimethacrylated tri-block copolymer, polycaprolactone-b-poly(ethylene glycol)-b-polycaprolactone (PEG-CAP-DM) (Figure 1A) was synthesized. After polymerization of PEG-CAP-DM and the formation of a network, degradation of the polycaprolactone regions can be achieved through exogenous delivery of lipase 44 (Figure 1B). Thus, there is a high-degree of external control over the degradation of the material; the rate of degradation can be increased or decreased simply by controlling the amount of exogenously-delivered lipase, as well as by the number of lipase-degradable caprolactone ester bonds per crosslink. In this way, the temporal delivery of lipase can be used to carefully control the rate of network degradation to match the rate of extracellular matrix production by gel-encapsulated chondrocytes. As shown in Figure 1C, control over the mass loss of PEG-CAP-DM hydrogels can be achieved by controlling the concentration of lipase in the surrounding solution. The enzyme becomes inhibited periodically, so fresh lipase enzyme must be added, and this accounts for the non-continuous mass loss profiles shown in Figure 1.
Fig. 1.
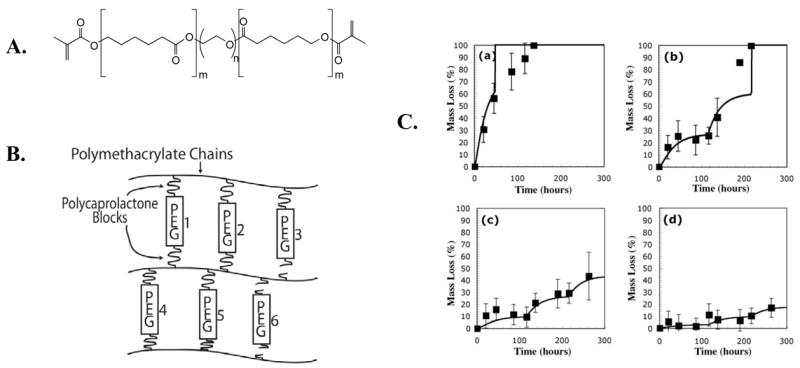
PEG-CAP-DM macromonomers (A) were photopolymerized to create a cross-linked hydrogel network (B). Shown is an ideal network in which crosslinks 3 and 6 have been enzymatically cleaved by exogenously added lipase. (C) Experimental mass loss data and calculated mass loss profiles (solid lines) for hydrogels synthesized from PEG-CAP-DM macromonomers and incubated in solutions with (a) 1.0 mg/ml, (b) 0.4 mg/ml, (c) 0.2 mg/ml, and (d) 0.1 mg/ml Lipase PS 44.
This externally-triggerable gel mechanism provides numerous advantages for both fundamental and applied studies in cartilage tissue engineering research. Degradation is critical to the evolution of biochemically and biomechanically robust tissue, which requires that the gel’s crosslinking density and mass loss be tuned to match the rate at which chondrocytes secrete extracellular matrix molecules while simultaneously allowing for their assembly in 3D. Gels formed from these PEG-CAP-DM macromolecules afford new opportunities to systematically study the influence of the gel degradation profile on cartilaginous tissue evolution. Further, one might envision the application of such a system in strategies to regenerate cartilage in advanced bioreactors. Cell-gel constructs could be mechanically stimulated, and the delivery of the enzyme correlated to the evolution of a particular property, such as mechanics, that would be monitored in real time. In sum, strategies to develop macromolecular monomers that afford greater control over the degradation-dependent properties of the resulting gel will provide significant benefits to numerous tissue engineering endeavors.
3. Using Polymerization Mechanism to Control Biomaterial Properties: Thiol-Acrylate Mixed-Mode Polymerizations
Traditional interest in biomaterials formed from multifunctional macromolecular monomers has focused on networks synthesized through photoinitiated chain polymerizations. Such strategies result in the formation of a relatively highly crosslinked network where degradation is typically introduced through functional groups incorporated into the network crosslinks. A hydrogel synthesized from di(meth)acrylated PEG macromolecular monomers provides one example (Figure 1B). The final degradation products of this type of system include a distribution of relatively high molecular weight kinetic chains in addition to the lower molecular weight fragments that are released from the crosslinks. The molecular weight of the degradation products is intimately linked to the biocompatibility of the material. Equally important is the resulting material’s degradation behavior and mass loss profile. Thus, strategies to control the structure of the network, where degradation occurs, as well as the size and composition of the ultimate degradation products represent important new directions in the development of biomaterials from macromolecular monomers.
As an alternative to the classic chain polymerization of multifunctional (meth)acrylate monomers, (meth)acrylate monomers can be copolymerized with multifunctional thiols to form networks via a radically-mediated mixed chain-growth and step-growth mechanism 20, 23–25. Others have used step-growth polymerization of thiol and vinyl groups to synthesize degradable networks (e.g., Michael-addition reactions) 45–47. In complementary work, specific advantages of the photoinitiated mixed-mode polymerization of thiol-acrylates have been recently demonstrated. For example, the photopolymerization of poly(ethylene glycol)-b-poly(lactic acid)-diacrylate (PEG-PLA-DA) with a tetrathiol monomer leads to a crosslinked network with a substantially different structure than the homopolymerization of PEG-PLA-DA. There are no high molecular weight kinetic chains that evolve during a chain polymerization, because propagation and chain transfer occur simultaneously, leading to network structures that are highly dependent on the thiol:acrylate functional group ratio and relative concentrations. Further, the network structure and bulk network chemistry are easily modified through changes in the thiol monomer functionality, chemistry, and functional group concentration. For example, thiol-containing peptide sequences and/or crosslinkers that are included in the initial monomer formulation are covalently tethered into the network as it polymerizes [65]. Thus, mixed-mode polymerizations offer a higher degree of control of the network structure, and this control is significant since network structure is closely coupled to the ultimate degradation behavior and mechanical properties of the material.
Beyond advantages in controlling the evolution of the network structure, thiol-acrylate photopolymerizations afford specific processing advantages. In a typical photoinitiated chain polymerization, an initiator is included in the formulation that absorbs light of the appropriate wavelength and is converted to an active specie that initiates polymerization through the (meth)acrylate moieties. While the polymerization occurs under physiological conditions with temporal and spatial control, the use of photoinitiators presents two additional challenges. First, thick samples are difficult to fabricate due to light attenuation through the sample as initiator molecules absorb light; and secondly, initiators and their activated species can be toxic to cells and damaging to proteins, depending upon their concentration 6, 8. Thiol-acrylate photopolymerizations address both of these limitations. When a thiol-containing monomer is combined with a di-acrylated PEG monomer, photopolymerization rapidly occurs in the absence of any added photoinitiator, eliminating any initiator toxicity issues and allowing the fabrication of thick samples (in excess of 10 cm) 24.
Figure 2 demonstrates the unique polymerization and degradation behavior exhibited by these mixed-mode thiol-acrylate networks in relation to pure acrylate chain-growth polymers. In the absence of photoinitiator, the polymerization of poly(ethylene glycol)-b-poly(lactic acid)-diacrylate (PEG-LA-DA, structure shown in Figure 2A) is characterized by very low conversion of acrylate groups over time [curve (a) in Figure 2B]. However, when a tetrathiol [pentaerythritol tetrakis(3-mercaptopropionate), structure shown in Figure 2A] is added at 15 mol% (thiol functional groups relative to total acrylate and thiol functional groups) to the same PEG-LA-DA composition [curve (b)], the reaction reaches high conversions after 12 minutes of light exposure (5 mW/cm2); the rate of reaction is enhanced additionally by increasing the light intensity to 50 mW/cm2 [curve (c)]. Due to the dependence of mass loss behavior on network structure for these networks, the thiol-acrylate mass loss profile is altered substantially by increasing the composition of tetrathiol in the initial monomer solution; increasing tetrathiol content leads to a more rapid degradation of the network as shown in Figure 2C. Figure 2D depicts a pure acrylate chain-growth systems (a) and a thiol-acrylate mixed-mode system (b) as monomer solutions, crosslinked polymer networks, and final degradation products. Network structure can be easily modified by altering the initial ratio of thiol and acrylate monomers in the monomer precursor solution. Finally, if the thiol monomer is a cysteine-containing peptide, hydrogel networks can be synthesized with a diverse array of peptide functionalities. Figure 2E shows human mesenchymal stem cells (hMSCs) encapsulated in a gel formed from the photopolymerization of a diacrylated PEG with a cysteine-containing peptide sequence, CRGDSG. A viability stain illustrates the ability of the incorporated RGDS functionality to promote cell survival.
Fig. 2.

(A) Materials were created from the reaction of poly(ethylene glycol)-b-poly(lactic acid)-diacrylate (PEG-PLA-DA) and pentaerythritol tetrakis(3-mercaptopropionate) (tetrathiol); (B) Conversion profiles of initiatorless photopolymerizations: (a) 100% PEG-PLA-diacrylate, light intensity = 5 mW/cm2, (b) 85 mol% PEG-PLA-diacrylate, 15 mol% tetrathiol, light intensity = 5 mW/cm2, (c) 85 mol% PEG-PLA-diacrylate, 15 mol% tetrathiol, light intensity = 50 mW/cm2; (C) Experimental mass loss profiles for degradable thiol-acrylate networks made from a PEG-PLA-DA and 10 (□), 30 (○), and 50 (△ ) mol% tetrathiol. (D) Pictorial representation of the initial monomer molecules, crosslinked polymer networks, and degradation products for materials formed from (a) chain-growth polymerization mechanism and (b) mixed-mode mechanism 25. (E) Encapsultion of human mesenchymal stem cells in PEG-peptide gels synthesized through the thiol-acrylate chemistry. Micrographs are from day 7 of culture of hMSCs encapsulated at a density of 5×106 cells/ml in gels formed from (a) PEG-DA and (b) PEG-DA with 5mM CRGDSG. The images are stained with LIVE/DEAD to differentiate living cells (gray) from dead (black), and illustrate the ability of the CRGDSG functionalized gels to promote hMSC survival. Scale bar=50μm.
Control of the polymerization mechanism in the development of hydrogels provides specific advantages in manipulating properties through the control of polymer structure. Through the copolymerization of a small set of macromolecular monomers, a diverse array of biomaterial properties can be realized. When thiol-acrylate systems are employed, further advantages include the facile ability to integrate synthetic functionalitites with biological functionalities (e.g., oligopeptides), which impart desirable characteristics for 3D cell culture platforms, tissue engineering applications, and guided wound healing.
4. Synthesizing Macromolecular Monomers of Varying Functionality: Controlling the Molecular Weight of Degradation Products to Influence Cell Function
In addition to macromolecular monomers based on synthetic polymers, many biologically based polymers provide an alternative direction to fabricate gels. For example, tissues are comprised of numerous polysaccharides, including hyaluronan, chondroitin sulfate, and keratin sulfate, as well as a water swollen network of cell-secreted extracellular matrix (ECM) proteins including collagens and other associated ECM components. These molecules can serve as the basis for the synthesis of chain crosslinkable macromers. Typically, (meth)acrylate-functionalized biological monomers will react to form gels that cells can degrade through secretion of enzymes, and often the released products have a potent effect on the activity and function of the cells. For example, in addition to hyaluronan’s role as a structural element, high molecular weight hyaluronan is often degraded by migrating cells during wound healing events, and the lower molecular weight fragments can be bound or intracellularized to influence cell proliferation, matrix secretion, and gene expression 48–50.
Given the structure and high level of functionality of most natural polymers, approaches to modify the polymer pendant groups with polymerizable moieties are robust and diverse. Thus, one can readily envision multiple strategies to create multivinyl macromers and control the ultimate connectivity of the reacted macromolecule in the final network structure. The importance of this is two-fold. First, structure-property relationships can be further tuned for a particular application. Secondly, the molecular weight distribution of the degradation products is influenced by the chemical modification and crosslinking density. Wang et al. [69] recently demonstrated the benefits of multifunctional chondroitin sulphate monomers to create bioadhesive gels that promoted integration of regenerated cartilage tissue. The chondroitin sulphate was functionalized with chain polymerizable methacrylate groups to allow in situ gelation and aldehyde groups to increase adhesion through conjugation with amines present on the tissue surface. Prestwich and collaborators have a long-standing interest in the functionalization of hyaluronic acid macromolecules to create covalent crosslinked gels with unique biological activity [70], as well as approaches to create multicomponent gels through reactions between functionalized hyaluronan, chondroitin sulfate, and gelatin [71]. Here, we illustrate a striking example, in which a multivinyl hyaluronan (HA) macromer is used to encapsulate valvular interstitial cells found in the heart valve, to demonstrate the effect of gel composition and connectivity on cell function.
Specifically, the primary alcohol on hyaluronan was modified with methacrylate groups (e.g., 1.5% to 100% substitution) to synthesize multimethacrylated hyaluronan (HA-MA, Figure 3A). This macromolecular monomer was used to encapsulate valvular interstitial cells (VICs) through a photoinitiated polymerization. HA based gels provide an advantageous system for the 3D culture of VICs and heart valve regeneration because of HA’s importance in heart valve development. For example, embryonic heart valves develop from a HA-rich “cardiac jelly” 51, and heart valves fail to develop in embryos that are genetically deficient in hyaluronic acid synthase 2 52. VIC viability within these HA gels remained high (>90% after 1 week in culture), and significant matrix production was observed after 6 weeks 53. Concurrently, results demonstrated that cell proliferation and extracellular matrix (ECM) production by VICs were both significantly affected by HA fragments that were exogeneously added to VIC cultures 53. For example, total ECM production, as measured by 1H-glycine incorporation, was greatest when VICs were cultured for 20 days in the presence of low molecular weight HA fragments (Mn¯~6700 g/mol, as shown in Figure 3B).
Fig. 3.

(A) Structure of methacrylated hyaluronic acid (HA). (B) Soluble HA fragments of low and high molecular weight (Mn¯) have profound effects on total ECM production, as measured by 1H-glycine incorporation at 3 days (striped bars) and 20 days (white bars). (C) Schematic showing structure and degradation products of HA-MA networks containing a high crosslinking density (a) or low crosslinking density (b) 53.
From a tissue-engineering standpoint, the ability to control ECM production over time provides distinct advantages. As such, future work that tunes the functionality and connectivity of the HA to the gel will be important, so that one can engineer the release of HA fragments of different sizes on different time scales. Cells remodel the HA-MA gels through enzymatic degradation. Depending upon crosslinking density of the hydrogels, there is a complex distribution of degradation products that can result (Figure 3C). Degradation of gels containing a high crosslinking density (a) will lead to a greater distribution of lower molecular weight HA fragments than gels containing a low crosslinking density (b). As such, by controlling the structure of HA-MA gels, one can potentially control the outcome of enzymatic degradation such that degradation products will have a profound cellular effect on ECM production by gel-encapsulated VICs. For example, initially it might be important for ECM production to be high; thus, the release of low molecular weight HA fragments during degradation of a HA-MA gel would stimulate ECM production. However, once sufficient ECM has been formed, ECM production should cease (to prevent fibrosis of the tissue) in the latter stages of tissue evolution and remodeling. In general, the ability to manipulate the structure of gels containing natural matrix components affords an excellent opportunity to promote or suppress selected functions of cells residing in these niches. Models that predict the release and molecular weight distribution of networks formed from these types of macromolecular monomers will provide useful tools for the design of experiments and hypothesis testing.
5. Asymmetric Macromolecular Monomers: Synthesis of Gels with Drug-Releasing Tethers
Functionalizing gels with biological signals is critical for many applications ranging from cell carriers to drug delivery devices. While most synthetic monomers allow one to create and engineer gels with carefully tuned structures and desired matrix properties, synthetic polymers lack any biologically recognized functionality, and at best, facilitate non-specific interactions (e.g., protein adsorption) that can be exploited with limited control. Thus, there is a great deal of interest and opportunity in the development of strategies to tether biological signals or therapeutic molecules to hydrogel systems. One obvious approach is the synthesis of multifunctional monomers that are asymmetric in their functionality, containing at least one polymerizable group and at least one conjugated drug, peptide, or protein. The bioactive molecule of interest can be either permanently linked or released over time through degradable linkers, depending on the requirements for the application. Examples of this type of approach include the tethering of transforming growth factor beta (TGFβ) to gels through polymerization of monoacrylated PEG conjugates; the conjugated TGFβ was found to be active and increased ECM production by smooth muscle cells [75]. Numerous others use this type of approach to create asymmetric monomers with acrylate functionalities conjugated to peptide sequences of interest, especially those that promote cell adhesion (e.g., RGD, YIGSR, REDV, and IKVAV) [reviewed in 76]
Design of synthetic-biologic monomer conjugates allows one to manipulate the concentration of the functional group (i.e., by proper selection of the comonomer ratio); however, high loadings are difficult to achieve, so such strategies are best suited for potent molecules that are therapeutically effective at low doses. One such example is the development of functionalized gels that direct the differentiation of adult stem cells. Recent interest has emerged in the use of bone-marrow derived stem cells for tissue engineering applications. Human mesenchymal stem cells (hMSCs), also commonly referred to as bone marrow stromal cells, are a unique class of multipotent cells that are non-committed and remain in an undifferentiated state. When treated with the right chemicals, hormones, and growth factors, hMSCs can be transformed into the cells of bone, cartilage, fat, tendon, muscle, and other tissues 54–57. For example, the synthetic glucocorticosteroid, dexamethasone, is a potent inducer of osteogenic differentiation of hMSCs when it is added to two-dimensional cultures. However, interesting questions remain for investigators developing three-dimensional culture niches for hMSCs and bone regeneration, especially how one might use the scaffold chemistry to integrate osteogenic signals in a manner that delivers these factors in the right context at the right time, dose, and location.
As one example, dexamethasone has been covalently linked into PEG hydrogels by copolymerization of a dexamethasone-PEG conjugate [PEG526MMA-nLac-Suc-Dex, (a) in Figure 4A] with di-acrylated PEG (PEG3400DA) in the presence of a photoinitiating molecule. A small concentration (2.8 mM) of a mono-acrylated PEG-cell adhesive peptide sequence (Acryl-PEG-RGD) was included to promote cell adhesion and viability 15. This copolymerization results in the tethering of dexamethasone to the network through a degradable bond [(b) in Figure 4A]. Over time, these degradable lactide bonds hydrolyze and release soluble dexamethasone [(c) in 4A]. The rate of dexamethasone release is controlled by varying the chemistry of the linking spacer. Only one of the hydrolyzable bonds must be degraded for dexamethasone to be liberated; in general, increasing the number of hydrolyzable lactic acid repeat units improves the chances that just one of those bonds will cleave to release dexamethasone. Importantly, a variety of dexamethasone conjugates can be released depending upon which ester bond hydrolyzes first. This does not necessarily lead to the release of pure dexamethasone per se; rather, the released dexamethasone conjugates may or may not have biological activity. However, there is evidence that carboxylic acid-containing dexamethasone conjugates are capable of autocatalysis of the remaining ester bonds, leading to rapid degradation of dexamethasone conjugates to pure dexamethasone upon hydrolysis of a single ester bond 58, 59. Further control over dexamethasone release can be achieved by manipulating the chemistry of the ester bond. For example, a caprolactone ester bond degrades much more slowly than a lactide ester bond; thus, incorporating caprolactone bonds results in slower dexamethasone release. Similar control can potentially be achieved using other types of degradable bonds.
Fig. 4.

(A) Dexamethasone was covalently linked to a photoreactive poly(ethylene glycol) molecule through a degradable poly(lactide) bond [PEG526MMA-nLac-Suc-Dex, (a)] and incorporated into a hydrogel network through photopolymerization (b). Over time, dexamethasone is released from its insoluble, hydrogel-bound form (b) to a soluble form (c) where it is able to interact with cells and cause osteogenic differentiation of gel-encapsulated hMSCs. (B) Human MSCs were seeded on tissue culture plastic and cultured in the presence of control media (CON, top left), 100 nM dexamethasone (top right), and supernatant from dexamethasone that had been released from PEG-Dex hydrogels (bottom). (C) Human MSCs were photoencapsulated in PEG3400DA hydrogels (solid bars) containing 2.8 mM of a mono-acrylated cell adhesive sequence (Acryl-PEG-RGD), which promotes cell viability, and cultured in CON media or Dex media. Cells were also encapsulated in PEG3400DA hydrogels in the presence of PEG526MMA-4Lac-Suc-Dex and cultured in CON media (solid bar, “Released Dex”). After two weeks in culture, total mRNA was isolated and gene expression of core binding factor alpha (Cbfa1) was assessed using real-time RT-PCR. An asterisk denotes that results are significant when compared to CON results (p<0.05) 58.
Released dexamethasone causes monolayer hMSCs to attain the osteogenic phenotype, similar to when the cells are cultured in dexamethasone-containing media (Figure 4B). Osteoblastic cells are characterized by a more stellate and flattened morphology as opposed to the elongated, spindle-shaped hMSCs. Furthermore, cells exhibit increased gene expression of core binding factor alpha 1 (Cbfa1), an important transcription factor implicated in osteogenic differentiation, in gel-encapsulated (3D) hMSCs (Figure 4C). This example shows a compelling case for the importance of introducing functionality into gels in a tissue engineering application, and a plethora of future work remains in order to develop more sophisticated gel platforms that integrate multiple functionalities that can be displayed in a manner that is controlled in both time and space. This will become increasingly important as new classes of therapeutic molecules emerge (e.g., miRNAs), and more sophisticated delivery platforms evolve where the gel itself can amplify or offset the cells response to the delivered signal.
6. Conclusions
In situ forming hydrogel materials are promising candidates for cell delivery applications. While many hydrogels can not be formed in the presence of cells, photoinitiated polymerizations of aqueous macromolecules, such as PEG-based macromolecular monomers, are enabling researchers to three-dimensionally encapsulate cells in hydrogel materials that mimic the extracellular matrix and support cellular viability. As exhibited by the examples in this article, chemical control over macromolecular monomers used in photoinitiated polymerizations for hydrogel cell delivery materials affords substantial control over diverse gel properties, including degradation rate and mechanism of gel degradation. While traditional polymeric scaffolds have generally been thought of as passive materials that provide little more than a structural element to support three dimensional cell growth and survival, more recent hydrogel scaffolds are being synthesized through photoinitiated polymerizations of aqueous macromolecules to actively promote targeted cell responses to degradation products, as well as providing control over the release of biologically relevant small molecules. Future biomaterial platforms will benefit from advances in polymer chemistry to synthesize hydrogel niches with highly defined biochemical and biophysical microenvironments.
Acknowledgments
The authors wish to acknowledge financial support for this broad research effort from the National Institutes of Health (Grants DE12998, ARS3126, and DE16523), the National Science Foundation (Grant EEC444771), and the American Heart Association (Grant 355488Z).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cruise GM, Hegre OD, Lamberti FV, Hager SR, Hill R, Scharp DS, Hubbell JA. In vitro and in vivo performance of porcine islets encapsulated in interfacially photopolymerized poly(ethylene glycol) diacrylate membranes. Cell Transplant. 1999;8(3):293–306. doi: 10.1177/096368979900800310. [DOI] [PubMed] [Google Scholar]
- 2.Elisseeff J, McIntosh W, Anseth K, Riley S, Ragan P, Langer R. Photoencapsulation of chondrocytes in poly(ethylene oxide)-based semi-interpenetrating networks. J Biomed Mater Res. 2000;51(2):164–71. doi: 10.1002/(sici)1097-4636(200008)51:2<164::aid-jbm4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 3.Fisher JP, Dean D, Engel PS, Mikos AG. Photoinitiated polymerization of biomaterials. Annual Review of Materials Research. 2001;31:171–181. [Google Scholar]
- 4.Randolph MA, Anseth K, Yaremchuk MJ. Tissue engineering of cartilage. Clin Plast Surg. 2003;30(4):519–37. doi: 10.1016/s0094-1298(03)00070-1. [DOI] [PubMed] [Google Scholar]
- 5.Sittinger M, Hutmacher DW, Risbud MV. Current strategies for cell delivery in cartilage and bone regeneration. Curr Opin Biotechnol. 2004;15(5):411–8. doi: 10.1016/j.copbio.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 6.Bryant SJ, Nuttelman CR, Anseth KS. Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH/3T3 fibroblasts in vitro. J Biomater Sci Polym Ed. 2000;11(5):439–57. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 7.Quick DJ, Anseth KS. DNA delivery from photocrosslinked PEG hydrogels: encapsulation efficiency, release profiles, and DNA quality. J Control Release. 2004;96(2):341–51. doi: 10.1016/j.jconrel.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 8.Williams CG, Malik AN, Kim TK, Manson PN, Elisseeff JH. Variable cytocompatibility of six cell lines with photoinitiators used for polymerizing hydrogels and cell encapsulation. Biomaterials. 2005;26(11):1211–8. doi: 10.1016/j.biomaterials.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 9.Hennink WE, van Nostrum CF. Novel crosslinking methods to design hydrogels. Adv Drug Deliv Rev. 2002;54(1):13–36. doi: 10.1016/s0169-409x(01)00240-x. [DOI] [PubMed] [Google Scholar]
- 10.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2(5):347–60. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 11.Rihova B. Biocompatibility of biomaterials: Hemocompatibility, immunocompatibility and biocompatibility of solid polymeric materials and soluble targetable polymeric carriers. Adv Drug Deliv Rev. 1996;21(2):157–176. [Google Scholar]
- 12.Yoshii E. Cytotoxic effects of acrylates and methacrylates: relationships of monomer structures and cytotoxicity. J Biomed Mater Res. 1997;37(4):517–24. doi: 10.1002/(sici)1097-4636(19971215)37:4<517::aid-jbm10>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 13.Geurtsen W, Leyhausen G. Chemical-Biological Interactions of the resin monomer triethyleneglycol-dimethacrylate (TEGDMA) J Dent Res. 2001;80(12):2046–50. doi: 10.1177/00220345010800120401. [DOI] [PubMed] [Google Scholar]
- 14.Odian G. Principles of Polymerization. 3. John Wiley & Sons; New York: 1991. Vol. [Google Scholar]
- 15.Nuttelman CR, Tripodi MC, Anseth KS. Synthetic hydrogel niches that promote hMSC viability. Matrix Biol. 2005;24(3):208–18. doi: 10.1016/j.matbio.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Burdick JA, Anseth KS. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23(22):4315–23. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 17.Li Q, Williams CG, Sun DD, Wang J, Leong K, Elisseeff JH. Photocrosslinkable polysaccharides based on chondroitin sulfate. J Biomed Mater Res A. 2004;68(1):28–33. doi: 10.1002/jbm.a.20007. [DOI] [PubMed] [Google Scholar]
- 18.Williams CG, Kim TK, Taboas A, Malik A, Manson P, Elisseeff J. In vitro chondrogenesis of bone marrow-derived mesenchymal stem cells in a photopolymerizing hydrogel. Tissue Eng. 2003;9(4):679–88. doi: 10.1089/107632703768247377. [DOI] [PubMed] [Google Scholar]
- 19.Hubbell JA. Hydrogel systems for barriers and local drug delivery in the control of wound healing. Journal of Controlled Release. 1996;39(2–3):305–313. [Google Scholar]
- 20.Posner T. Unsaturated compounds. II. Addition of mercaptans to unsaturated hydrocarbons. Ber Deut Chem Ges. 1905;38:646–57. [Google Scholar]
- 21.Woods JG, editor. Plenum; New York: 1992. pp. 333–398. [Google Scholar]
- 22.Jacobine AF, editor. Elsevier Applied Science; London: 1993. p. 219. [Google Scholar]
- 23.Cramer NB, Bowman CN. Kinetics of thiol-ene and thiol-acrylate photopolymerizations with real-time Fourier transform infrared. 2001 [Google Scholar]
- 24.Cramer NB, Scott JP, Bowman CN. Photopolymerizations of thiol-ene polymers without photoinitiators. Macromolecules. 2002;35(14):5361–5365. [Google Scholar]
- 25.Rydholm AE, Bowman CN, Anseth KS. Degradable thiol-acrylate photopolymers: polymerization and degradation behavior of an in situ forming biomaterial. Biomaterials. 2005;26(22):4495–506. doi: 10.1016/j.biomaterials.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 26.Otsu T. Iniferter concept and living radical polymerization. Journal of Polymer Science Part A-Polymer Chemistry. 2000;38(12):2121–2136. [Google Scholar]
- 27.Anseth KS, Newman SM, Bowman CN. Polymeric dental composites: Properties and reaction behavior of multimethacrylate dental restorations. Biopolymers II Advances in Polymer. Science. 1995;122:177–217. [Google Scholar]
- 28.Higashi J, Nakayama Y, Marchant RE, Matsuda T. High-spatioresolved microarchitectural surface prepared by photograft copolymerization using dithiocarbamate: Surface preparation and cellular responses. Langmuir. 1999;15(6):2080–2088. [Google Scholar]
- 29.Nakayama Y, Matsuda T, Irie M. A novel surface photo-graft polymerization method for fabricated devices. Asaio J. 1993;39(3):M542–4. [PubMed] [Google Scholar]
- 30.Nakayama Y, Miyamura M, Hirano Y, Goto K, Matsuda T. Preparation of poly(ethylene glycol)-polystyrene block copolymers using photochemistry of dithiocarbamate as a reduced cell-adhesive coating material. Biomaterials. 1999;20(10):963–70. doi: 10.1016/s0142-9612(98)00252-x. [DOI] [PubMed] [Google Scholar]
- 31.Griffith LG. Polymeric biomaterials. Acta Biomater. 2000;48(1):263–277. [Google Scholar]
- 32.Hubbell J. Synthetic biodegradable polymers for tissue engineering and drug delivery. Current Opinion in Solid State & Materials Science. 1998;3(3):246–251. [Google Scholar]
- 33.Elbert DL, Hubbell JA. Reduction of fibrous adhesion formation by a copolymer possessing an affinity for anionic surfaces. J Biomed Mater Res. 1998;42(1):55–65. doi: 10.1002/(sici)1097-4636(199810)42:1<55::aid-jbm8>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 34.Halstenberg S, Panitch A, Rizzi S, Hall H, Hubbell JA. Biologically engineered protein-graft-poly(ethylene glycol) hydrogels: a cell adhesive and plasmin-degradable biosynthetic material for tissue repair. Biomacromolecules. 2002;3(4):710–23. doi: 10.1021/bm015629o. [DOI] [PubMed] [Google Scholar]
- 35.Seliktar D, Zisch AH, Lutolf MP, Wrana JL, Hubbell JA. MMP-2 ensitive, VEGF-bearing bioactive hydrogels for promotion of vascular healing. J Biomed Mater Res A. 2004;68(4):704–16. doi: 10.1002/jbm.a.20091. [DOI] [PubMed] [Google Scholar]
- 36.Sawhney AS, Hubbell JA. Poly(ethylene oxide)-graft-poly(L-lysine) copolymers to enhance the biocompatibility of poly(L-lysine)-alginate microcapsule membranes. Biomaterials. 1992;13(12):863–70. doi: 10.1016/0142-9612(92)90180-v. [DOI] [PubMed] [Google Scholar]
- 37.Martens PJ, Bryant SJ, Anseth KS. Tailoring the degradation of hydrogels formed from multivinyl poly(ethylene glycol) and poly(vinyl alcohol) macromers for cartilage tissue engineering. Biomacromolecules. 2003;4(2):283–92. doi: 10.1021/bm025666v. [DOI] [PubMed] [Google Scholar]
- 38.Metters AT, Anseth KS, Bowman CN. A statistical kinetic model for the bulk degradation of PLA-b-PEG-b-PLA hydrogel networks: Incorporating network non-idealities. J Phys Chem B. 2001;105(34):8069–8076. [Google Scholar]
- 39.Metters AT, Bowman CN, Anseth KS. A statistical kinetic model for the bulk degradation of PLA-b-PEG-b-PLA hydrogel networks. J Phys Chem B. 2000;104(30):7043–7049. [Google Scholar]
- 40.Mahkam M. Using pH-sensitive hydrogels containing cubane as a crosslinking agent for oral delivery of insulin. J Biomed Mater Res B Appl Biomater. 2005;75(1):108–12. doi: 10.1002/jbm.b.30279. [DOI] [PubMed] [Google Scholar]
- 41.Cao LQ, Xu SM, Feng S, Wang JD. Swelling and thermal behaviors of starch-based superadsorbent hydrogel with quaternary ammonium and carboxyl groups. J Applied Poly Science. 2005;96(6):2392–2398. [Google Scholar]
- 42.Zhao Y, Liang HJ, Wang SG, Wu C. Self-assembly of poly(caprolactone-b-ethylene oxide-b-caprolactone) via a microphase inversion in water. J Phys Chem B. 2001;105:848–851. [Google Scholar]
- 43.Nie T, Zhao Y, Xie ZW, Wu C. Micellar formation of poly(caprolactone-block-ethylene oxide-block-caprolactone) and its enzymatic biodegradation in aqueous dispersion. Macromolecules. 2003;36:8825–8829. [Google Scholar]
- 44.Rice MA, Sanchez-Adams J, Anseth KS. Exogenously triggered, enzymatic degradation of photopolymerized hydrogels with polycaprolactone subunits: experimental observation and modeling of mass loss behavior. Biomacromolecules. 2006;7(6):1968–75. doi: 10.1021/bm060086+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nature Biotechnology. 2005;23(1):47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 46.DuBose JW, Cutshall C, Metters AT. Controlled release of tethered molecules via engineered hydrogel degradation: model development and validation. J Biomed Mater Res A. 2005;74(1):104–16. doi: 10.1002/jbm.a.30307. [DOI] [PubMed] [Google Scholar]
- 47.Metters A, Hubbell J. Network formation and degradation behavior of hydrogels formed by Michael-type addition reactions. Biomacromolecules. 2005;6(1):290–301. doi: 10.1021/bm049607o. [DOI] [PubMed] [Google Scholar]
- 48.West DC, Hampson IN, Arnold F, Kumar S. Angiogenesis induced by degradation products of hyaluronic acid. Science. 1985;228(4705):1324–6. doi: 10.1126/science.2408340. [DOI] [PubMed] [Google Scholar]
- 49.Chen WY, Abatangelo G. Functions of hyaluronan in wound repair. Wound Repair Regen. 1999;7(2):79–89. doi: 10.1046/j.1524-475x.1999.00079.x. [DOI] [PubMed] [Google Scholar]
- 50.Toole BP. Hyaluronan in morphogenesis. Cell Dev Biol. 2001;12:79–87. doi: 10.1006/scdb.2000.0244. [DOI] [PubMed] [Google Scholar]
- 51.Eisenberg LM, Markwald RR. Molecular regulation of atrioventricular valvuloseptal morphogenesis. Circ Res. 1995;77(1):1–6. doi: 10.1161/01.res.77.1.1. [DOI] [PubMed] [Google Scholar]
- 52.Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest. 2000;106(3):349–60. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masters KS, Shah DN, Leinwand LA, Anseth KS. Crosslinked hyaluronan scaffolds as a biologically active carrier for valvular interstitial cells. Biomaterials. 2005;26(15):2517–25. doi: 10.1016/j.biomaterials.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 54.Caplan A, Bruder S, editors. R.G. Landes; New York: 1997. [Google Scholar]
- 55.Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991;9(5):641–50. doi: 10.1002/jor.1100090504. [DOI] [PubMed] [Google Scholar]
- 56.Haynesworth SE, Baber MA, Caplan AI. Cell surface antigens on human marrow-derived mesenchymal cells are detected by monoclonal antibodies. Bone. 1992;13(1):69–80. doi: 10.1016/8756-3282(92)90363-2. [DOI] [PubMed] [Google Scholar]
- 57.Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–7. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 58.Nuttelman CR, Tripodi MC, Anseth KS. Dexamethasone-functionalized gels induce osteogenic differentiation of encapsulated hMSCs. Journal of Biomedical Materials Research Part A. 2006;76A(1):183–195. doi: 10.1002/jbm.a.30537. [DOI] [PubMed] [Google Scholar]
- 59.Siparsky GL, Voorhees KJ, Miao F. Hydrolysis of polylactic acid (PLA) and polycaprolactone (PCL) in aqueous acetonitrile solutions: autocatalysis. Journal of Environmental Polymer Degradation. 1998;6:31–41. [Google Scholar]