

Abstract
The distinctive structure of the [(his)7Cu4(μ-S)]n+ cluster in the “CuZ” active site of nitrous oxide reductase and the intriguing mechanistic hypotheses for its catalytic reactivity provide inspiration for synthetic model studies aimed at characterizing relevant copper-sulfur compounds and obtaining fundamental insights into structure and bonding. In this brief review, we summarize such studies that have focused on the synthesis and characterization of a range of copper-sulfur complexes supported by N-donor ligands. Compounds with variable nuclearities and sulfur redox levels have been isolated, with the nature of the species obtained being dependent on the supporting ligand, sulfur source, and the reaction conditions. Spectroscopic data and theoretical calculations, often performed with a view toward drawing comparisons to oxygen analogs, have provided insight into the nature of the copper-sulfur bonding interactions in the complexes.
Keywords: copper, sulfur, nitrous oxide, disulfide
1. Introduction
Nitrous oxide (N2O) is of particular interest as an energy-storing small molecule because of its potential as an environmentally clean oxidant and its role as a harmful greenhouse gas [1]. Its propensity for reduction and O-atom transfer to release the inert product N2 is highly favorable (E° = 1.76 V), but high kinetic barriers limit its utility as an oxidant, notwithstanding successful heterogeneous catalytic reactions that occur at temperatures much higher than ambient [2]. In general, binding and activation of N2O at a transition metal center is required in order to obtain reasonably rapid rates of reaction under mild conditions [3]. However, knowledge of the fundamental underlying coordination chemistry with which to develop useful catalytic systems has lagged behind similar efforts to understand activation of other small molecules like dioxygen, for example [4]. This problem stems in large part from the weak σ-donor and π-acceptor capabilities of N2O that make it a poor ligand. Indeed, only one metal-N2O complex ([Ru(NH3)5(N2O)]2+) has been isolated and characterized [5].
In biology, N2O reduction is performed enzymatically under mild conditions during denitrification, a microbial respiratory process that is a key component of the biogeochemical nitrogen cycle [6]. The enzyme responsible for the reaction in eq. 1, nitrous oxide reductase (N2OR), has been shown by X-ray crystallography [7] and spectroscopy [8,9] to contain two multicopper sites. One is a bis(cysteinato)dicopper moiety that is delocalized mixed-valent (Cu+1.5Cu+1.5) in its oxidized state and operates as an electron transfer center (“CuA”; not shown) [10]. The other site (“CuZ”; Figure 1, top) contains a [Cu4(μ-S)]n+ cluster ligated by 7 histidine imidazoles that is unprecedented; no other examples of such a tetracopper arrangement or a copper site with inorganic sulfur have been observed in biology [11]. This site is believed to be where N2O binds and is reduced. Spectroscopic studies have demonstrated that the active form of the enzyme is in a fully-reduced state (4Cu+1), and calculations suggest that the N2O molecule
| eq. 1 |
binds through the terminal N-atom to CuI [9,12]. These theoretical studies also suggest that the lowest energy pathway for N2O reduction involves binding of the O-atom to CuIV to yield a bent μ-1,3-bridged fragment (Figure 1, bottom). This binding mode is suggested to maximize CuZ → N2O π backbonding and activate the molecule for protonation. The unusual bridging sulfide is postulated to facilitate electronic delocalization throughout the tetracopper cluster, not only to increase the ability of the cluster to backbond into the N2O molecule, but also to lower the reorganization energy associated with the CuI/CuII redox change(s) during catalysis.
Figure 1.

(top) Representation of the X-ray structure of the CuZ site of N2OR. (bottom) Proposed mechanism for binding of activation of N2O by CuZ.
While theoretical calculations provide enticing hypotheses for the mechanism of N2O reduction by CuZ, experimental information is lacking about the nature of the bonding interactions between copper, inorganic sulfur, and/or N2O. A useful approach to obtain such knowledge involves the study of synthetic complexes designed to mimic the structural and/or spectroscopic properties of metalloenzyme active sites and proposed intermediates [13]. Such studies of copper-sulfur complexes are motivated by the inherent challenges involved in activating N2O, the novel structural topology of the CuZ site, and the provocative mechanistic ideas for N2O reduction by CuZ proposed on the basis of theory. In this brief review, we survey recent work aimed at synthesizing and characterizing copper-sulfur complexes relevant to the CuZ site. In progressing toward the difficult, and as of yet unattained goal of preparing an accurate model of the [(his)7Cu4(μ-S)]n+ core, a wide range of compounds with variable copper nuclearities and sulfur oxidation states have been prepared, opening new vistas into the inorganic chemistry of copper and sulfur.
2. Copper-Sulfur Complexes Lacking N-Donor Supporting Ligands
A survey of crystallographically characterized molecules having copper-sulfide bonds in the Cambridge Structure Database [14] reveals a large number of structures having Cu3(μ3-S) and Cu4(μ4-S) bonding motifs. Most of these contain copper ions in large clusters with other metals such as Mo [15] and W [16], however, and have little relevance to N2OR. Nonetheless, the isolation of these molecules illustrates that a wide variety of sulfur sources, including S8, Li2S, and Na2S2, are synthetically useful.
Few examples of discrete molecules with Cu3(μ3-S) and Cu4(μ4-S) cores have been reported and these can be classified as members of a large class of [CuIxS2−y] clusters supported by abiological phosphine ligands that have garnered interest due to their potential applications in material science [17]. One example is notable in the current context because it contains multicopper centers bridged by inorganic sulfide ions in a Cu4(μ4-S) geometry that is related to that observed in CuZ (Figure 2) [18]. The cluster [L4Cu4(μ4-S)]2+ (1, L = the bidentate ligand 1,3-diphenylphosphinomethane) was synthesized either through the addition of CS2 to LCuII(NO3)2 in MeOH [18a] or the addition of Na2S to the dicopper(I) complex [Cu2(μ-dppm)2(MeCN)2](PF6)2 [18b]. In the former route reduction of the starting CuII centers occurs, consistent with the known propensity of phosphines to stabilize the copper(I) oxidation state [19]. Indeed, this stabilization of the copper(I) state is reflected in the chemistry of these compounds, which are resistant to oxidation and are air stable (E1/2 = 0.188 V vs. SCE). Thus, while phosphine ligands allow for the synthesis and isolation of [CuIxS2−y] clusters by preventing redox chemistry that can lead to decomposition, this same redox inertness inhibits biomimetic reactivity, thus decreasing the relevance of these molecules as N2OR CuZ models. From another perspective, these results underscore the importance of using N-donors to model the His supporting ligands in CuZ and to enable access to higher copper oxidation states within the cluster that occur during the N2O reduction process.
Figure 2.
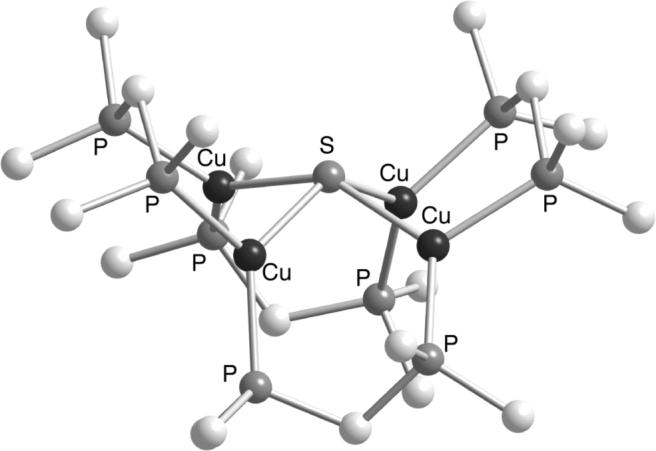
Representation of the X-structure of [(dppm)4Cu4(μ4-S)]2+ (1), with only the ipso C atoms of the phenyl rings of the dppm ligands shown [18].
3. Copper-Sulfur Complexes With N-Donor Supporting Ligands
3.1 Dinuclear Complexes
The first reported synthetic example of a copper-sulfide complex with N-donor ligation is (TpiPr2Cu)2(S2) (2, Figure 3), which was initially isolated from the thermal decomposition of a copper(II)-thiolate complex [20] and was later obtained from the reaction of TpiPr2CuI(MeCN) and S8 [21]. This reddish-brown dinuclear complex was shown by X-ray crystallography to contain a side-on bound μ−η2:η2−disulfido (S22−) moiety with an S-S bond length of 2.073(4) Å. An alternative end-on bound μ−η1:η1 binding mode for a disulfido ligand in a dicopper complex was revealed in {[(TMPA)Cu]2(S2)}(X)2 (3, Figure 3), which was isolated from the reaction of S8 with the copper(I) complex [(TMPA)Cu(MeCN)]X in acetonitrile (TMPA = tris(2-pyridylmethyl)amine, X = ClO4− or PF6−) [22]. X-ray crystallographic analysis of {[(TMPA)Cu]2(S2)}(ClO4)2 revealed an S-S bond length of 2.044(4) Å akin to that reported for 2 despite their differing disulfide binding modes.
Figure 3.
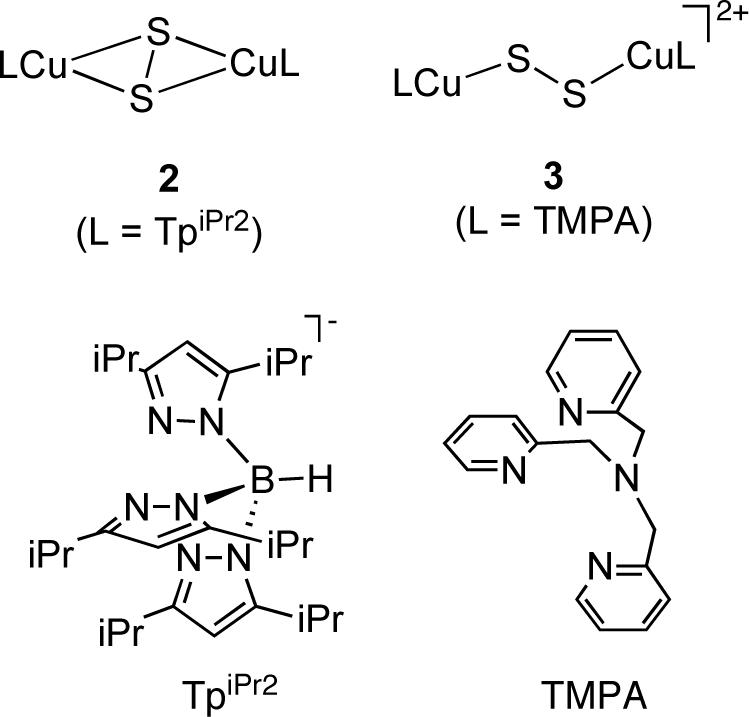
Disulfido-dicopper(II) complexes supported by TpiPr and TMPA.
Complexes 2 and 3 are structurally similar to previously characterized oxygen analogs [23, 24], and in order to gain an understanding of the differences in the bonding interactions for the Cu2E2 systems (E = O or S), as well as between side-on and end-on coordination of the bridging moieties, detailed spectroscopic and theoretical comparisons of these complexes were conducted [21]. Key spectroscopic features are summarized in Figure 4. Each complex displays a distinctive UV-vis absorption pattern dominated by intense E22− → CuII ligand-to-metal charge transfer (LMCT) features, irradiation into which results in the enhancement of E-isotope-sensitive ν(E-E) features in resonance Raman spectra. The complexes exhibit d → d bands indicative of copper in the +2 oxidation state, but they are EPR silent, consistent with strong antiferromagnetic coupling of the CuII ions.
Figure 4.
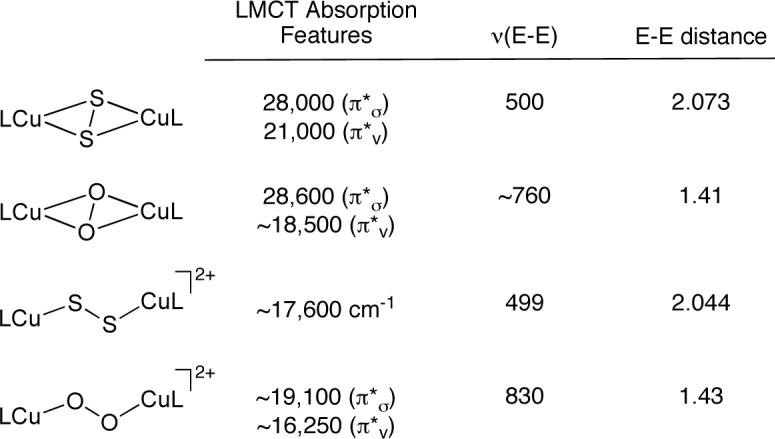
Summary of selected spectroscopic features of the μ−η2:η2 − and μ−η1:η1−(disulfido/peroxo)dicopper complexes with L = TpiPr2 or TMPA, respectively (E = S or O). The absorption features and ν(E-E) values are reported in cm−1, and the E-E distance is reported in Å. The data were obtained from refs. 21, 23, and 24.
Bonding pictures were developed in order to understand and compare the properties of the side-on and end-on Cu2E2 complexes, with that for the side-on species shown in Figure 5. In both geometries, the main bonding interaction involves strong σ-donation from the E22− π*σ orbital into the dxy orbitals of the two copper centers. A key finding was that the Cu-S bonding is stronger and more covalent than the Cu-O bonding within each pair of analogous structures, which may be traced to better overlap of the more diffuse and higher-energy S orbitals with the copper d-orbitals. Another important observation was that the E-E bonds in the side-on (μ−η2:η2) structures are weak, as illustrated by low ν(E-E) values that indicate a high degree of E-E bond activation. This bond weakening is ascribed to back-donation from the Cu dxy orbitals into the empty E22− σ* orbital.
Figure 5.
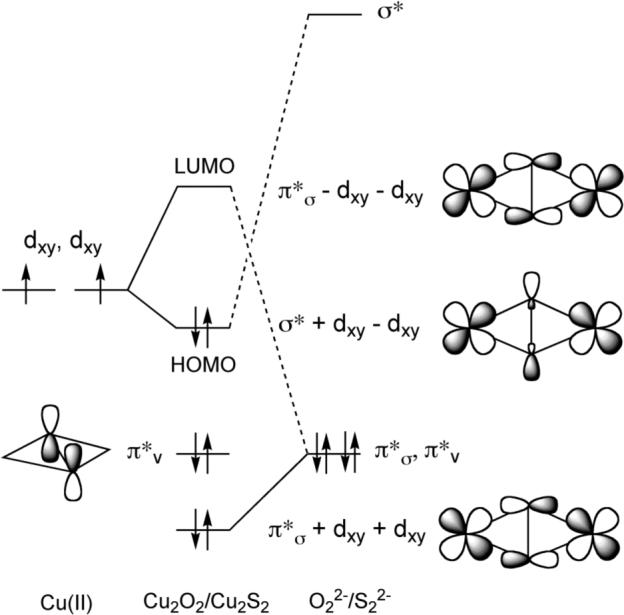
Orbital energy level diagram for (μ−η2:η2-peroxo/disulfido)dicopper complexes. Reproduced with permission from ref. 27.
Another example of a side-on (μ−η2:η2-S22−)dicopper(II) complex (4) was prepared using a tridentate N-donor suppporting ligand (Me2NPY2 = N,N-bis{2-[2-(N’N’-4-dimethylamino)pyridyl]ethyl}methylamine) [25]. X-ray crystallography showed the copper centers to be roughly square pyramidal, with an S-S bond length of 2.117(2). Particular emphasis in this study was placed on the reactivity of the complex. It was shown to quantitatively transfer a sulfide ion to both PPh3 and 2,6-dimethylphenylisocyanide to form S=PPh3 and the isothiocyanate, yet it did not react with benzyl bromide, suggesting that the disulfide bridge is electrophilic. This disulfide bridge is labile, as shown by the reaction of the complex with CO or O2 that resulted in the rapid formation of the LCuI-CO adduct or [(LCu)2(O22−)]2+, respectively.
A large class of side-on (μ−η2:η2-S22−)dicopper(II) complexes (5a-j) supported by bidentate, monoanionic N-donors was prepared using β-diketiminate and related anilido-imine ligands with variable substituents that conferred differing steric profiles (Figure 6) [26, 27]. The initial synthesis of these complexes was by addition of the sulfide reagent S(SiMe3)2 to LCuIICl in acetonitrile (L = β-diketiminate and related anilido-imine ligands). Yields of the complexes [(LCu)2(μ-S2)] synthesized by this metathesis approach were low (17−29%), with the concomitant formation of LCuI(NCCH3) being confirmed by NMR, consistent with a redox process between S22− ligands and the copper(II) centers. The syntheses were also complicated by the formation of copper(I)-thiolate byproducts resulting from attack of sulfur at the β-diketiminate methine position, exemplified by 6 (Figure 7). In an alternative synthetic route, 5a-j were synthesized in higher yields (40−91%) by addition of one equivalent of elemental sulfur to acetonitrile solutions of LCuI(NCCH3).
Figure 6.
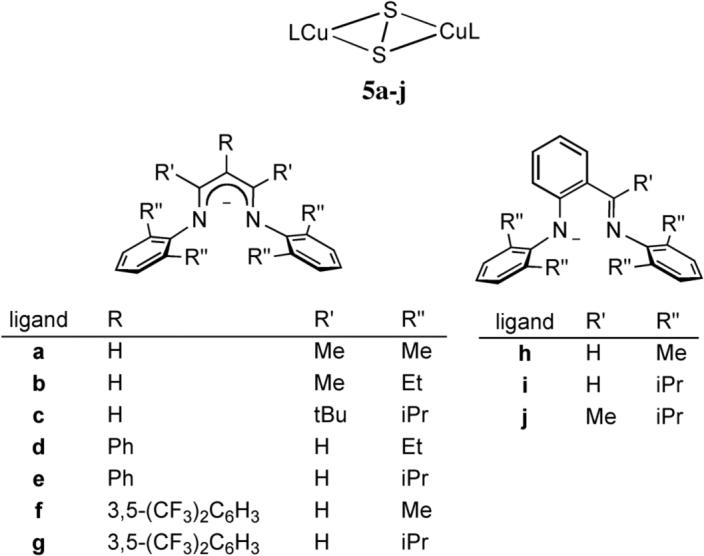
Complexes 5a-j.
Figure 7.
Complex 6 (Ar = aryl).
X-ray structure determinations for 5a-j revealed S-S bond distances ranging from 2.165(3) to 2.214(10) Å, which are significantly longer than those in 2 (2.073 Å) or 3 (2.044 Å). Within the series of (μ−η2:η2-S22−)dicopper(II) complexes 2, 4, and 5a-j, lengthening of the S-S bond correlates with shortening of the Cu-Cu and Cu-S distances, as plotted in Figure 8 [27]. It is notable that the compounds with β-diketiminate and anilido-imine supporting ligands are at the extreme of long S-S and short Cu-Cu and Cu-S distances, and only when sufficient steric hindrance is introduced (5c,i, and j) do these values approach those of 2 and 4. Consistent with these structural findings, the resonance Raman spectra of 5i-j (λex = 457.9 nm) contain a single strong, 34S-sensitive ν(S-S) at 428−454 cm−1 (Δ34S = 8−13 cm−1) that is ∼50 cm−1 lower than observed for 2 and 3 (∼ 500 cm−1), suggesting greater backbonding from the copper(II) centers into the σ*-orbitals of the S-S moiety with an accompanying weakening of this bond. Taken together, the structural and spectroscopic data support stronger and more covalent Cu-S bonding and greater backdonation into the S22− σ* orbital in 5i-j that results in a greater degree of S-S bond activation relative to 2−4. These effects are mitigated only when the powerfully electron donating β-diketiminate and anilido-imine supporting ligands are particularly sterically encumbered. Indeed, a more quantitative correlation of the S-S distance and ν(S-S) using Badger's rule [28] shows that the degree of S-S bond activation in 5i-j supercedes that of a large set of metal-disulfide complexes (Figure 8) [27]. Yet despite the electron donating capabilities of the supporting ligands in 5i-j, complete scission of the S-S bond to yield bis(sulfido)dicopper(III) species via a process akin to that identified previously in [Cu2(μ−η2:η2-O2)]2+/[Cu2(μ-O)2]2+ systems [4b,c, 29] has not been observed. This implicated instability of the [Cu2(μ-S)2]2+ core has been verified by theory [26].
Figure 8.

(Left) Correlation of Cu-S, S-S, and Cu-Cu distances in (μ−η2:η2-S22−)dicopper(II) complexes 2, 4, and 5a-j. (Right) Plot of S-S distances and 1/ν2/3 (ν = ν(S-S)) for species with S-S bonds, with a fit to Badger's rule shown as a dashed line. Adapted from ref. 27.
A dicopper(II) complex with two disulfido ligands at a different redox level than S22− was prepared upon treatment of [(Me4eda)Cu(CH3CN)]O3SCF3 with S8 (7a, Figure 9) [30]. A structurally similar compound with Cl− instead of CF3SO3− groups was recently obtained from a mixture of N,N,N’,N’-tetramethyl-trans-1R,2R-diaminocyclohexane (Me4chd), CuCl2, and Na2S2 (7b) [31]. The S-S distances of ∼1.95 Å in these compounds are shorter than in typical complexes of S22− (see above), yet are longer than that of doubly bonded S2 (1.892 Å [32]). In addition, resonance Raman spectra for 7 contain ν(S-S) at 613 cm−1 (Δ34S = 19 cm−1), which lies between those seen for S22− compounds and that of S2 (718 cm−1 [33]); note the position of the data point for 7a in the Badger's rule plot of Figure 8 [27]. The EPR silent complexes are thus best formulated as having a [CuII2(μ-S2•−)2]2+ core (i.e., with disulfido(•1−) ligands). This is a novel motif in copper chemistry, albeit one that has been identified in complexes of iron and ruthenium [34].
Figure 9.
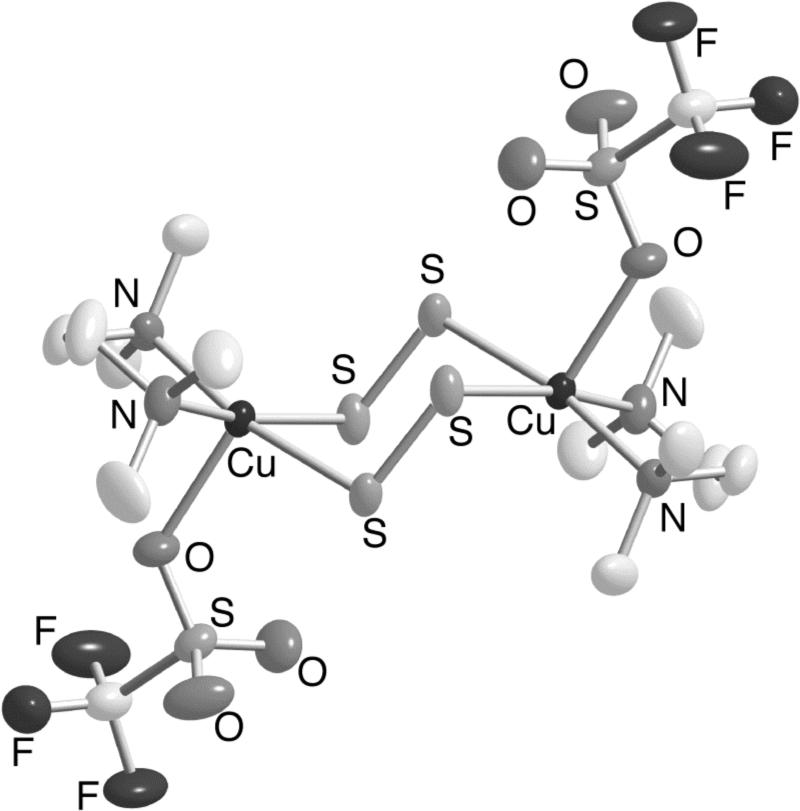
X-ray structure of 7.
3.2 Trinuclear Complexes
Clusters 8a-c were isolated from reactions of [LCu(CH3CN)]X (L = Me4eda, X = SbF6; L = Me4chd, X = PF6) [35] with S8 or through mixing Me4chd, Cu(O3SCF3)2, and Li2S or Na2S2 [31] (Figure 10). These clusters are structural models of CuZ insofar as their Cu3(μ-S) “half” mimics the subcluster of CuZ defined by CuII, CuIII, CuIV, and S (Figure 1). X-ray crystallography results for 8a-c show them to exist as 3+ cations consistent with the formal oxidation state assignment [CuIIICuII2(μ-S2−)2]3+. A similar overall charge and formal oxidation state assignment was identified previously for the oxygen analog [(Me4chd)3Cu3(μ-O)2]3+ (9) [36]. However, while the latter was shown to adopt a valence localized structure with structurally distinct CuIII and CuII sites, for 8a-c structural, spectroscopic, and theoretical data support a fully valence delocalized core. Thus, the metal-ligand distances in each of 8a-c are essentially identical and EPR data at room temperature and at 10 K indicate an S = 1 ground state with equivalent hyperfine coupling to the three Cu ions. While deeper insights await a more complete spectroscopic comparison of 8 and 9, DFT calculations point to an interesting reversal of the energies of the frontier e” and a2” orbitals arising from ‘cross-cluster’ S-S interactions that destabilizes the a2” orbital as the reason for the shift from a Jahn-Teller distorted localized structure in 9 to a symmetric delocalized configuration in 8 (Figure 10) [35].
Figure 10.
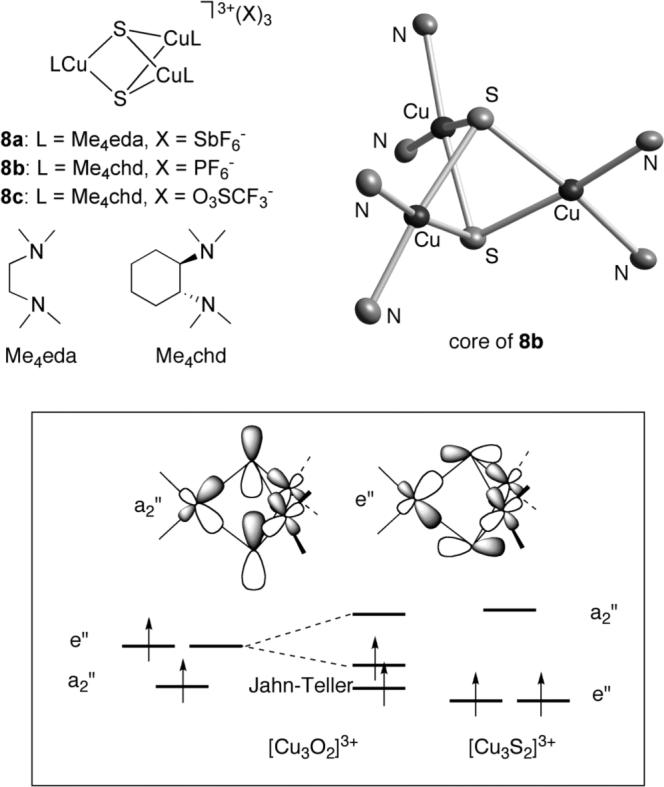
Drawing of clusters 8a-c, the X-ray structure of the tricationic core portion of 8b (C and H atoms omitted), and (in the box) a partial orbital energy level diagram comparing the ordering of the frontier molecular orbitals for 8 and its oxygen analog 9.
3.3 High Nuclearity Complexes
An X-ray crystal structure of an N-donor ligated cluster with bridging μ4-sulfido and μ3-thiolato ions between 13 copper(I) ions has been reported (Figure 11) [37]. This cluster, [(LS−)6CuI13(S2−)2]3+ (10), was isolated during attempted crystallization of [(LSH)CuI]ClO4, with the sulfido groups apparently being derived from decomposition of the supporting thiolate ligand. As noted by the authors, the arrangement of four copper(I) ions about each sulfido group resembles that in the CuZ core, and is also related to the core geometry of 1 (Figure 2).
Figure 11.

X-ray structure of [(LS−)6CuI13(S2−)2]3+ (10) with (left) and without (right) the supporting N-donor ligand (LS−) shown.
Recently, tetra- and hexacopper clusters with bindentate N-donors and bridging disulfido ligands were isolated and structurally defined (Figure 12) [31]. The complex [(Me4chd)4Cu6(μ-S2)4Cl4] (11) was obtained from a mixture of Me4chd, CuCl2, and Li2S, along with a copper(I) coproduct, (Me4chd)CuICl, apparently resulting from oxidation of S2− to S22− by CuII. Subsequent reaction of 11 with [[(Me4chd)CuI(MeCN)]SbF6 led to [(Me4chd)4Cu4(μ-S2)2Cl2](SbF6)2 (12). The μ4-S22− coordination in 11 and 12 has been seen previously in transition metal complexes [38], but to our knowledge it is new for copper [39]. It is notable that with one specific supporting N-donor ligand, Me4chd, compounds with core stoichiometries and redox levels ranging from [Cu2(μ-S2)2]2+ (7b), [Cu3(μ-S)2]3+ (8b,c), [Cu4(μ-S2)2]4+ (12), to [Cu6(μ-S2)4]4+ (11) have been isolated, depending on the particular reaction conditions.
Figure 12.
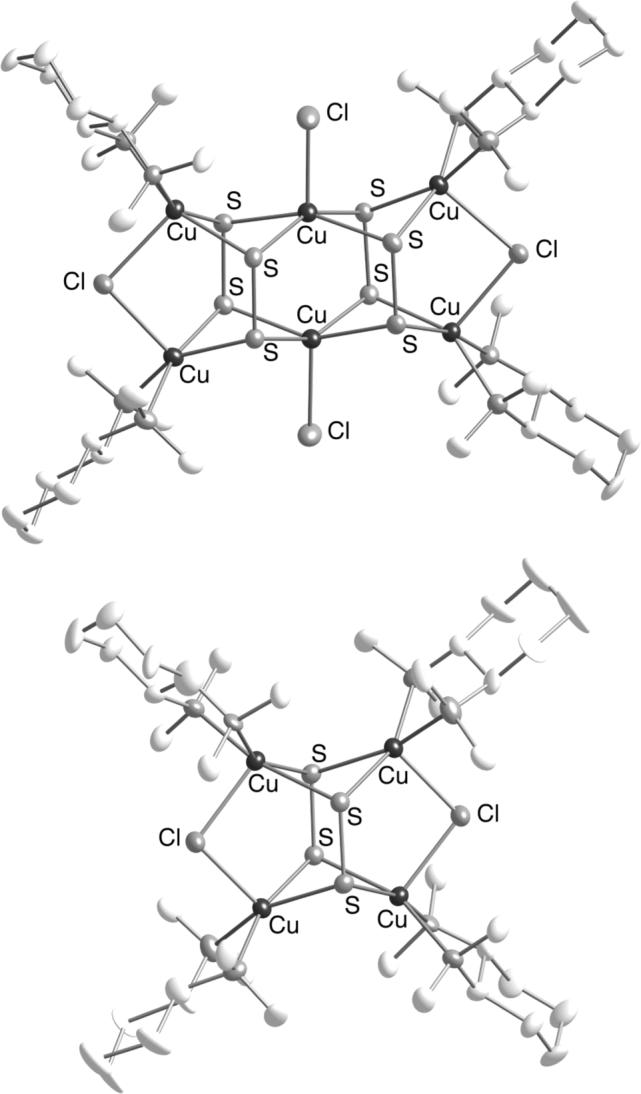
X-ray structures of [(Me4chd)4Cu6(μ-S2)4Cl4] (11, top) and the cationic portion of [(Me4chd)4Cu4(μ-S2)2Cl2](SbF6)2 (12, bottom).
4. Concluding Comments
In efforts to prepare copper-sulfide complexes with N-donor ligands, novel structures with a variety of copper nuclearities and sulfur oxidation levels have been isolated. The nature of the clusters prepared via these approaches is sensitive to the specific N-donor ligand, sulfur source, copper salt, and reaction conditions used. For instance, with one specific supporting N-donor ligand (Me4chd) compounds with core stoichiometries and redox levels ranging from [Cu2(μ-S2)2]2+ (7b), [Cu3(μ-S)2]3+ (8b,c) [Cu4(μ-S2)2]4+ (12) to [Cu6(μ-S2)4]4+ (11) were isolated. Spectroscopic and theoretical data have provided insight into the nature of the Cu-S bonding interactions in these compounds, key findings being the particularly high degrees of S-S bond activation in disulfido-dicopper complexes supported by β-diketiminate and anilido-imine ligands and a novel delocalized mixed-valent structure in [Cu3(μ-S)2]3+ cores. From the comparison with structurally similar oxygen analogs, Cu-S bonding is stronger and more covalent, with possible implications for enhanced charge and/or spin delocalization mediated by the bridging sulfur in the CuZ cluster. While new insights into fundamental copper-sulfur chemistry have been obtained, much remains to be accomplished in efforts to model CuZ, including the development of new synthetic protocols for the synthesis of high nuclearity copper(I)-sulfur clusters supported by low denticity N-donor ligands that would enable studies of biomimetic reactivity with N2O.
5. Acknowledgements
We thank the NIH (GM47365) for financial support for the research performed in our laboratory.
Biography
 Left: William B. Tolman was an undergraduate student at Wesleyan University, Middletown, CT, and received his Ph.D. in 1987 from the University of California, Berkeley, under the tutelage of K. Peter C. Vollhardt. After a postdoctoral period at the Massachussetts Institute of Technology with Stephen J. Lippard, he began his independent career at the University of Minnesota in 1990, where he is currently a Distinguished McKnight University and Lee Irvin Smith Professor of Chemistry.
Left: William B. Tolman was an undergraduate student at Wesleyan University, Middletown, CT, and received his Ph.D. in 1987 from the University of California, Berkeley, under the tutelage of K. Peter C. Vollhardt. After a postdoctoral period at the Massachussetts Institute of Technology with Stephen J. Lippard, he began his independent career at the University of Minnesota in 1990, where he is currently a Distinguished McKnight University and Lee Irvin Smith Professor of Chemistry.
Middle: Itsik Bar-Nahum was born in Jerusalem, Israel, in 1972. He received his B.Sc. and M.Sc. in chemistry from the Hebrew University of Jerusalem, Israel, in 1999, and completed his Ph.D. on the synthesis and catalytic applications of polyoxometalate – organometallic hybrid compounds in 2005, under the supervision of Ronny Neumann at The Weizmann Institute of Science, Rehovot, Israel. Since then he has been a postdoctoral associate in the laboratory of W. Tolman at the University of Minnesota.
Right: John T. York received his B.S. in Chemical Engineering from North Carolina State University in 1995 and his B.A. in Secondary Science Education from the University of Wyoming in 2001. He recently completed his Ph.D. research on synthetic copper-sulfur and copper-dioxygen systems in the laboratory of W. Tolman at the University of Minnesota, and is beginning his independent career as Assistant Professor of chemistry at Stetson University in DeLand, Florida.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References
- 1.Trogler WC. Coord. Chem. Rev. 1999;187:303. [Google Scholar]
- 2.Zhu S, Wang X, Wang A, Cong Y, Zhang T. Chem. Commun. 2007:1695. doi: 10.1039/b702502e. For example. references cited therein. [DOI] [PubMed] [Google Scholar]
- 3.Lee D-H, Mondal B, Karlin KD. In: Activation of Small Molecules: Organometallic and Bioinorganic Perspectives. Tolman WB, editor. Wiley-VCH; Weinheim: 2006. pp. 43–79. [Google Scholar]
- 4.a Costas M, Mehn MP, Jensen MP, Que JL. Chem. Rev. 2005;104:939. doi: 10.1021/cr020628n. Selected recent reviews: [DOI] [PubMed] [Google Scholar]; b Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]; c Lewis EA, Tolman WB. Chem. Rev. 2004;104:1047. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]; d Tshuva EY, Lippard SJ. Chem. Rev. 2004;104:987. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]; e Bois JD, Mizoguchi TJ, Lippard SJ. Coord. Chem. Rev. 2000;200−202:443. [Google Scholar]; f Borovik AS, Zinn PJ, Zart MK. In: Activation of Small Molecules: Organometallic and Bioinorganic Perspectives. Tolman WB, editor. Wiley-VCH; Weinheim: 2006. pp. 187–234. Editon edn. [Google Scholar]
- 5.Paulat F, Kuschel T, Nather C, Praneeth VKK, Sander O, Lehnert N. Inorganic Chemistry. 2004;43:6979. doi: 10.1021/ic049302i. references cited therein. [DOI] [PubMed] [Google Scholar]
- 6.a Zumft WG, Kroneck PMH. Adv. Microb. Phys. 2007;52:107. doi: 10.1016/S0065-2911(06)52003-X. [DOI] [PubMed] [Google Scholar]; b Zumft WG. Microbiol. Rev. 1997;61:533. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Brown K, Tegon M, Prudencio M, Pereira AS, Besson S, Moura JJ, Moura I, Cambillau C. Nat. Struct. Biol. 2000;7:191. doi: 10.1038/73288. [DOI] [PubMed] [Google Scholar]; b Haltia T, Brown K, Tegoni M, Cambillau C, Saraste M, Mattila K, Djinovic-Carugo K. Biochem. J. 2003;369:77. doi: 10.1042/BJ20020782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Rasmussen T, Berks BC, Sanders-Loehr J, Dooley DM, Zumft WG, Thomson AJ. Biochemistry. 2000;39:12753. doi: 10.1021/bi001811i. [DOI] [PubMed] [Google Scholar]; b Alvarez ML, Ai J, Zumft W, Sanders-Loehr J, Dooley DM. J. Am. Chem. Soc. 2001;123:576. doi: 10.1021/ja994322i. [DOI] [PubMed] [Google Scholar]; c Oganesyan VS, Rasmussen T, Fairhurst S, Thomson AJ. Dalton Trans. 2004:996. doi: 10.1039/b313913a. [DOI] [PubMed] [Google Scholar]
- 9.a Chen P, Gorelsky SI, Ghosh S, Solomon EI. Angew. Chem. Int. Ed. 2004;43:4132. doi: 10.1002/anie.200301734. [DOI] [PubMed] [Google Scholar]; b Chen P, DeBeer George S, Cabrito I, Antholine WE, Moura JJG, Moura I, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 2002;124:744. doi: 10.1021/ja0169623. [DOI] [PubMed] [Google Scholar]; c Chen P, Cabrito I, Moura JJG, Moura I, Solomon EI. J. Am. Chem. Soc. 2002;124:10497. doi: 10.1021/ja0205028. [DOI] [PubMed] [Google Scholar]; d Ghosh S, Gorelsky SI, Chen P, Cabrito I, Moura JJG, Moura I, Solomon EI. J. Am. Chem. Soc. 2003;125:15708. doi: 10.1021/ja038344n. [DOI] [PubMed] [Google Scholar]; e Ghosh S, Gorelsky SI, DeBeerGeorge S, Chan JM, Cabrito I, Dooley DM, Moura JJG, Moura I, Solomon EI. J. Am. Chem. Soc. 2007;129:3955. doi: 10.1021/ja068059e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a Kroneck PMH, Antholine WA, Riester J, Zumft WG. FEBS Letters. 1989;248:212. doi: 10.1016/0014-5793(89)80464-8. [DOI] [PubMed] [Google Scholar]; b Farrar JA, Neese F, Lappalainen P, Kroneck PMH, Saraste M, Zumft WG, Thomson AJ. J. Am. Chem. Soc. 1996;118:11501. [Google Scholar]; c Randall DW, Gamelin DR, LaCroix LB, Solomon EI. J. Biol. Inorg. Chem. 2000;5:16. doi: 10.1007/s007750050003. [DOI] [PubMed] [Google Scholar]
- 11.a González-Duarte P. In: Comprehensive Coordination Chemistry II. McCleverty JA, Meyer TJ, editors. Vol. 8. Elsevier; Amsterdam: 2004. pp. 213–228. We distinguish clusters with copper and inorganic sulfur (e.g. S2− or S2n−, n = 1 or 2) from the relatively more common copper(I)-thiolate assemblies in metallotheineins [11a] and synthetic molecules [11b]. [Google Scholar]; b Henkel G, Krebs B. Chem. Rev. 2004;104:801. doi: 10.1021/cr020620d. [DOI] [PubMed] [Google Scholar]
- 12.Solomon E, Sarangi R, Woertink J, Augustine AJ, Yoon J, Ghosh S. Acc. Chem. Res. 2007;40 doi: 10.1021/ar600060t. ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a Ibers JA, Holm RH. Science. 1980;209:223. doi: 10.1126/science.7384796. [DOI] [PubMed] [Google Scholar]; b Karlin KD. Science. 1993;261:701. doi: 10.1126/science.7688141. [DOI] [PubMed] [Google Scholar]; c Chem. Rev 2004104347.(issue on Biomimetic Inorganic Chemistry)14871127 [Google Scholar]
- 14.CSD Version 5.28. Nov, 2006.
- 15.Guo J, Sheng T, Zhang W, Wu X, Lin P, Wang Q, Lu J. Inorg. Chem. 1998;37:3689. doi: 10.1021/ic970900p. [DOI] [PubMed] [Google Scholar]
- 16.Lang JP, Tatsumi K. Inorg. Chem. 1998;37:160. doi: 10.1021/ic970894x. [DOI] [PubMed] [Google Scholar]
- 17.Dehnen S, Eichhöfer A, Fenske D. Eur. J. Inorg. Chem. 2002:279. [Google Scholar]
- 18.a Yang R-N, Sun Y-A, Hou Y-M, Hu X-Y, Jin D-M. Inorg. Chim. Acta. 2000;304:1. [Google Scholar]; b Yam VW-W, Lee W-K, T.-Fai Chem. Commun. 1993:1571. [Google Scholar]
- 19.Hathaway BJ. In: Comprehensive Coordination Chemistry I. Gillard RD, McCleverty JA, editors. Vol. 5. Pergamon; Oxford: 1987. pp. 533–774. [Google Scholar]
- 20.Fujisawa K, Moro-oka Y, Kitajima N. J. Chem. Soc., Chem. Commun. 1994:623. [Google Scholar]
- 21.Chen P, Fujisawa K, Helton ME, Karlin KD, Solomon EI. J. Am. Chem . Soc. 2003;125:6394. doi: 10.1021/ja0214678. [DOI] [PubMed] [Google Scholar]
- 22.Helton ME, Chen P, Paul PP, Tyeklar Z, Sommer RD, Zakharov LN, Rheingold AL, Solomon EI, Karlin KD. J. Am. Chem. Soc. 2003;125:1160. doi: 10.1021/ja027574j. [DOI] [PubMed] [Google Scholar]
- 23.a Jacobson RR, Tyeklár Z, Farooq A, Karlin KD, Liu S, Zubieta J. J. Am. Chem. Soc. 1988;110:3690. {[(TMPA)Cu]2(O2)}2+: [Google Scholar]; b Tyeklár Z, Jacobson RR, Wei N, Murthy NN, Zubieta J, Karlin KD. J. Am. Chem. Soc. 1993;115:2677. [Google Scholar]
- 24.Kitajima N, Fujisawa K, Fujimoto C, Moro-oka Y, Hashimoto S, Kitagawa T, Toriumi K, Tatsumi K, Nakamura A. J. Am. Chem. Soc. 1992;114:1277. (TpiPr2Cu)2(O2) [Google Scholar]
- 25.Helton ME, Maiti D, Zakharov LN, Rheingold AL, Porco J. John A., Karlin KD. Angew. Chem. Int. Ed. 2006;45:1138. doi: 10.1002/anie.200503216. [DOI] [PubMed] [Google Scholar]
- 26.Brown EC, Aboelella NW, Reynolds AM, Aullón G, Alvarez S, Tolman WB. Inorg. Chem. 2004;43:3335. doi: 10.1021/ic049811k. [DOI] [PubMed] [Google Scholar]
- 27.Brown EC, Bar-Nahum I, York JT, Aboelella NW, Tolman WB. Inorg. Chem. 2007;46:486. doi: 10.1021/ic061589r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badger RM. J. Chem. Phys. 1935;3:710. [Google Scholar]
- 29.Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Jr., Que L, Jr., Zuberbühler AD, Tolman WB. Science. 1996;271:1397. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- 30.York JT, Brown EC, Tolman WB. Angew. Chem. Int. Ed. 2005;44:7745. doi: 10.1002/anie.200503134. [DOI] [PubMed] [Google Scholar]
- 31.York JT, Bar-Nahum I, Tolman WB. submitted for publication. [Google Scholar]
- 32.Steudel R. Angew. Chem. Int. Ed. 1975;14:655. [Google Scholar]
- 33.Yee KK, Barrow RF, Rogstad A. J. Chem. Soc., Faraday Trans. 1972;2(68):1808. [Google Scholar]
- 34.a Treichel PM, Crane RA, Haller KJ. Polyhedron. 1990;9:1893. Selected examples: [Google Scholar]; b Matsumoto K, Sugiyama H. Acc. Chem. Res. 2002;35:915. doi: 10.1021/ar000103m. [DOI] [PubMed] [Google Scholar]; c Schneider R, Wieghardt K, Nuber B. Inorg. Chem. 1993;32:4935. [Google Scholar]
- 35.Brown EC, York JT, Antholine WE, Ruiz E, Alvarez S, Tolman WB. J. Am. Chem . Soc. 2005;127:13752. doi: 10.1021/ja053971t. [DOI] [PubMed] [Google Scholar]
- 36.a Cole AP, Root DE, Mukherjee P, Solomon EI, Stack TDP. Science. 1996;273:1848. doi: 10.1126/science.273.5283.1848. [DOI] [PubMed] [Google Scholar]; b Root DE, Henson MJ, Machonkin T, Mukherjee P, Stack TDP, Solomon EI. J. Am. Chem. Soc. 1998;120:4982. [Google Scholar]
- 37.Lee Y, Sarjeant AAN, Karlin KD. Chem. Commun. 2006:621. doi: 10.1039/b513768c. [DOI] [PubMed] [Google Scholar]
- 38.a Stevenson DL, Magnuson VR, Dahl LF. J. Am. Chem. Soc. 1967;89:3727. Two examples: [Google Scholar]; b Kuhlman ML, Rauchfuss TB. Angew. Chem. Int. Ed. 2004;43:6742. doi: 10.1002/anie.200461264. [DOI] [PubMed] [Google Scholar]
- 39.Lin P, Wu X, Huang Q, Wang Q, Sheng T, Zhang W, Guo J, Lu J. Inorg. Chem. 1998;37:5672. doi: 10.1021/ic9704142. A report of a [CuI6(μ-S22−)]4+ core within a larger MoCu cluster has appeared. [DOI] [PubMed] [Google Scholar]