

Abstract
Solar ultraviolet (UV) irradiation is an important carcinogen that leads to the development of skin cancer, which is the most common human cancer. However, the receptors that mediate UV-induced skin carcinogenesis have not yet been unequivocally identified. Here we showed that UV irradiation directly activates the cannabinoid receptors 1 and 2 (CB1/2). Notably, our data indicated that the absence of the CB1/2 receptors in mice results in a dramatic resistance to UVB-induced inflammation and a marked decrease in UVB-induced skin carcinogenesis. A marked attenuation of UVB-induced activation of mitogen-activated protein (MAP) kinases and nuclear factor-kappaB (NF-κB) was associated with CB1/2 deficiency. These data provide direct evidence indicating that the CB1/2 receptors play a key role in UV-induced inflammation and skin cancer development.
Keywords: cannabinoid receptors, UVB, inflammation, skin cancer
Introduction
Skin cancer is the most common type of human cancer and is associated with excessive exposure to ultraviolet (UV) solar irradiation. Exposure to UV irradiation can activate various oncogenes and inactivate many tumor suppressor genes. The net result is abnormal proliferation of keratinocytes that might harbor DNA damage leading to the onset of skin cancer (1). Exposure to UVB irradiation has been shown to induce activation, aggregation, and internalization of cell surface receptors for epidermal growth factor (EGF), tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1) (2, 3).
Manipulation of the cannabinoid receptors has been useful in the management of pain, treatment of osteoporosis, inflammation, and cancer (4), but the mechanisms of these effects are still not fully understood. Two cannabinoid receptors from mammalian tissues have been cloned and characterized (5, 6). Cannabinoid receptor 1 (CB1) is highly expressed in the brain, whereas the CB2 receptor is found mainly in the immune system (5, 6). However, no direct experimental evidence has confirmed that these membrane-bound receptors are required for UV-induced skin carcinogenesis.
CB1 and CB2 are members of a superfamily of seven-transmembrane-spanning (7-TM) receptors, which have a protein structure defined by an array of seven membrane-spanning helices with intervening intracellular loops and a C-terminal domain that can associate with G proteins. A CB1 (7) and CB2 homology model (8) was constructed using the published x-ray crystal structure of bovine rhodopsin (9), a photosensitive G-protein coupled receptor (GPCR) found in the retina. Arrestin-1 (or visual arrestin) and arrestin-4 (or cone arrestin) modulate the action of rhodopsin (10, 11) and arrestin-2 and arrestin-3 modulate non-retinal GPCRs (12, 13). Recently, desensitization of the CB1 receptor was reported to be mediated by the interaction of the C-terminal residues (419–439) of CB1 with arrestin-2 (14), indicating that CB1 and CB2 might act as a photoreceptor in non-retinal tissues. However, a role for CB1 and CB2 in skin cancer has not been reported. The identification of the “UV-receptor” associated with skin cancer is still to be elucidated and the CB1/2 receptors are potential candidates.
The function of CB1 and CB2 in cancer is controversial. The expression levels of both CB1 and CB2 are significantly higher in CA-human papillomavirus-10 cells, which are virally transformed cells derived from human prostate adenocarcinoma, and also in other human prostate cancer cell lines, including LNCaP, DUI45, PC3, and CWR22Rnu1, compared to human prostate epithelial and PZ-HPV-7 cells that are virally transformed cells derived from normal human prostate tissues (15). Moreover, the CB1 antagonist, Rimonabant or SR141716, inhibited human breast cancer cell proliferation and was more effective in highly invasive metastatic MDA-MB-231 cells than in the less invasive T47D or MCF-7 cells (16). In leukemic precursor cells, the CB2 receptor is an oncoprotein that blocks neutrophilic differentiation when overexpressed in myeloid precursor cells (17). However, treatment of LNCaP cells with WIN-55,212-2, a mixed CB1/CB2 agonist, resulted in inhibition of cell growth and induction of apoptosis (15, 18). Furthermore, cannabinoids have been shown to suppress the growth of several models of tumor xenografts in rats and mice (19). CB1 and CB2 receptors are expressed in normal skin and skin tumors and local activation of cannabinoid receptors induced the apoptotic death of tumorigenic epidermal cells, suppressed proliferation of melanoma cells and inhibited the growth and angiogenesis of skin tumor xenografts in nude mice (20, 21). But cannabinoids may inhibit human keratinocyte proliferation through a non-CB1/CB2 mechanism (22). Therefore, although CB1 and CB2 are physiologically linked with cancer, the functional role of these receptors in cancer is not clear.
Here, we used wildtype CB1/2 (CB1/2+/+) and CB1/2 deficient (CB1/2−/−) mice and respective mouse embryonic fibroblasts (MEFs) to elucidate the function of endogenous CB1 and CB2 in UVB-induced inflammation and skin cancer development.
Materials and Methods
Plasmids and chemicals
The mouse CB1 plasmid (pcDNA3-CB1) was kindly provided by Dr. Beat Lutz (Group Molecular Genetics of Behavior Max-Planck-Institute of Psychiatry, Kraepelinstr. 2–10 D-80804 Munich, Germany) and the mouse CB2 plasmid (pcDNA3-CB2) was kindly provided by Dr. Ruud Delwel (Erasmus MC, Department of Hematology, Dr Molewaterplein 50, 3015GE Rotterdam, The Netherlands). Chemical reagents, including (R)-(+)-WIN55212-2, PTX (pertussis toxin), Tris, NaCl and SDS for molecular biology and buffer preparation were purchased from Sigma-Aldrich Corp. (St. Louis, MO). The luciferase assay substrate was from Promega (Madison, WI). Restriction enzymes were purchased from Roche Diagnostics Corp. (Indianapolis, IN) and Taq DNA polymerase was from Qiagen, Inc. (Valencia, CA). The DNA ligation kit (version 2.0) was purchased from TAKARA Bio, Inc. (Otsu, Shiga, Japan). Cell culture media and other supplements were purchased from Life Technologies, Inc. (Rockville, MD). [3H] CP55940 and membrane fractions, which were isolated from HEK293 EBNA cells overexpressing human CB1 or CB2 receptors were obtained from Perkin Elmer Corp. (Boston, MA). The HisG antibody was from Invitrogen Corp. (Carlsbad, CA) and antibodies against phosphorylated (p)-JNKs, total JNKs, p-p38, total p38, p-ERKs and total ERKs were from Cell Signaling Technology, Inc. (Beverly, MA). The antibody to detect phosphorylated serine residues was from Abcam, Inc. (Cambridge, MA) and the antibody against TNF-α was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA).
Mice
The CB1/2 deficient and wildtype mice were provided by Dr. Andreas Zimmer from the Laboratory of Molecular Biology Clinic of Psychiatry, University Hospital (Bonn, Germany).
In vivo mouse studies
Age- and gender-matched CB1/2+/+ and CB1/2−/− mice were divided into groups and initiated by topical application of 200 nmol of 7,12-dimethyl benz(a)anthracene (DMBA) and then 2 wk later, 1 group each of CB1/2+/+ and CB2−/− mice were treated with increasing doses (increase of 1.5 kJ/m2/per week) of UVB 3 times per week as follows: Week 1: 3 min for 1.5 kJ/m2 (3 doses); Week 2: 6 min for 3.0 kJ/m2 (3 doses); Week 3: 9 min for 4.5 kJ/m2 (3 doses); Week 4: 12 min for 6.0 kJ/m2 (3 doses); Week 5: 15 min for 7.5 kJ/m2 (3 doses); Week 6: 18 min for 9.0 kJ/m2 (3 doses); Week 7 through week 34 continued at 9.0 kJ/m2 (3 doses/week). Groups 1 (CB1/2+/+; n = 30) and 2 (CB1/2−/−, n = 30) were untreated and groups 3 (CB1/2+/+; n = 30) and 4 (CB1/2−/−, n = 30) were treated with UVB only as indicated above. Mice were weighed, photographed, and tumors counted and measured once a week beginning when the first measurable tumors (1 mm3) were observed.
Histopathology and immunohistochemistry
Depilated adult CB1/2+/+ and CB1/2−/− mice (6–8 wk old, 6 per group) were irradiated with 4 kJ/m2 UVB. Dorsal trunk skin was harvested 24 h after irradiation, immediately fixed in 10% neutral buffered formalin and processed for hematoxylin–eosin staining and immunohistochemistry staining with an antibody against TNF-α.
Binding analysis
The ready-to-use membrane fractions were purchased and the overexpression of CB1 or CB2 was confirmed using a [3H] CP55940 binding assay following the manufacturer’s (Perkin Elmer Corp.) suggested protocol. In a competition binding assay, 5 µg of membrane fractions were mixed with 0.5 nM [3H] CP55940 and challenged with increasing concentrations (0.1 to 10 µM) of unlabeled ligand (R)-(+)-WIN55212-2. These fractions were or were not exposed to UVA (60 or 120 kJ/m2) or UVB (9kJ/m2) and incubated at 30 °C for 90 min. Nonspecific binding was assessed using 10 µM unlabeled ligand. Reactions were terminated by filtration through Whatman GF/C paper and washed with ice-cold incubation buffer (50 mM Tris-HCl pH 7.5, 2.5 mM EGTA, 5 mM MgCl2, 1.0 mg/ml BSA). Bound radioactivity was then determined by liquid scintillation counting. Results were analyzed using GraphPad Prism (Version 4, GraphPad software, Inc).
Cell culture and transfections
CB1/2+/+ and CB1/2−/− mouse embryonic fibroblasts (MEFs) were isolated as previously described for similar cell lines (1). HEK293 cells and CB1/2+/+ and CB1/2−/− MEFs were cultured at 37 °C in a 5% CO2 incubator in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). When cells reached 50–60% confluence, transfection of the expression vectors was performed using the Lipofectamine™ Reagent (Invitrogen Corp.) following the manufacturer’s suggested protocol.
Construction of His-fusion proteins
The His-fusion proteins for the CB1 and CB2 receptors were constructed with PCR-amplified open reading frame (ORF) DNA fragments and the pcDNA4/HisMax vector (Invitrogen Corp.). The reading frame of each plasmid construct was confirmed by restriction mapping and DNA sequencing.
Extraction of membrane and cytosolic protein
HEK293 cells were harvested, suspended in cell lysis buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl) and disrupted by freezing and thawing. The supernatant fraction including cytosolic and nuclear soluble proteins was recovered by centrifugation at 15,000 g for 10 min at 4°C and the pellet was resuspended with cell suspension buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% Triton-100). The cell suspension was stirred on ice for 1 h, centrifuged at 15,000 g for 10 min at 4°C and membrane proteins were recovered in the supernatant fraction.
Immunoprecipitation and Western blotting
Immunoblotting was performed following the instructions from Cell Signaling Technology Inc. In brief, HEK293 cells or CB1/2+/+ or CB1/2−/− MEFs were starved for 24 h in DMEM supplemented with 0.1% FBS. Cells were then treated with UVB (4 kJ/m2) and harvested in cell lysis or suspension buffer. Samples were equalized for protein and analyzed by Western blotting. To analyze CB1 or CB2 phosphorylation, a HisG antibody was used for immunoprecipitation with membrane proteins (500 µg). The antibody binding was carried out at 4°C overnight and proteins were visualized by Western blotting using an antibody against phosphorylation of serine residues.
NF-κB activity assay
The NF-κB luciferase reporter plasmid (pGL2-NF-κB-Luc) was transfected into CB1/2+/+ or CB1/2−/− MEFs together with the phRL-SV40 plasmid. The MEFs were cultured for an additional 36 h, starved for 24 h and then exposed to UVB (4 kJ/m2). The cells were disrupted with lysis buffer 12 or 18 h later, and luciferase activity was measured.
Results
CB1 and CB2 are activated by UVA or UVB
The CB1/2 receptors are seven transmembrane helix receptors that belong to the superfamily of G-protein coupled receptors (GPCRs) (5, 6). A GPCR isomerizes between two different states, an inactive (R) and an active (R*) conformation (23) and the R* state is appropriate for functional coupling to G proteins. Because an agonist has a high relative affinity for R* forms compared with the inactive form, receptor binding of the agonist results in a shift of the equilibrium toward the active state and G protein coupling can occur virtually concomitantly (23). Activation of the CB1/2 receptors by UV treatment produced a similar shift in affinity for the agonist, (R)-(+)-WIN55212-2, compared to the untreated control, which was detected by an agonist-specific increase in binding affinity. Competition binding experiments using membrane fractions from cells overexpressing CB1 and exposed to UVB (9 kJ/m2) or UVA (60 or 120 kJ/m2) showed a 1.9-, 2.1-, or 2.9-fold lower Ki value, respectively, compared to untreated control membrane fractions (Table 1). Similar increases in affinity were observed in membrane fractions from cells overexpressing the CB2 receptor– a 1.4 fold lower Ki value with UVB (9 kJ/m2) and a 2.6-fold lower Ki value with UVA (60 kJ/m2) (Table 1). These findings suggested that the CB1 and CB2 receptors are activated by UVA or UVB treatment.
Table 1.
Effect of UV radiation in CB1 or CB2 on ligand binding affinities
| vs [3H] CP55940 |
|||
|---|---|---|---|
| Receptor | UV radiation | Dose(kJ/m2) | Control:UV treated Ki ratio |
| CB1 | UVB | 9 | 1.9:1 |
| CB1 | UVA | 60 | 2.1:1 |
| CB1 | UVA | 120 | 2.9:1 |
| CB2 | UVB | 9 | 1.4:1 |
| CB2 | UVA | 60 | 2.6:1 |
Competition binding studies, using [3H] CP55940 as a tracer, were conducted with membrane preparations isolated from cells overexpressing CB1 or CB2 (Perkin Elmer Corp.)
CB1/2 double knockout mice show resistance to UV-induced skin inflammation
Because of the potential role of cannabinoids in cell-growth inhibition and inflammation (24), we investigated whether CB1/2−/− mouse skin would exhibit an altered susceptibility to UVB-induced inflammation. At the established peak response time of 24 h after irradiation (25), CB1/2+/+ and CB1/2−/− mouse skin was harvested and analyzed. Histological analysis indicated that UVB-treated CB1/2+/+ mouse skin showed a pronounced inflammation with the emergence of subcorneal pustules and epidermal erosion (Fig. 1A and B), whereas the epidermis of CB1/2−/− mice was far more resistant to UVB-induced inflammation (Fig. 1C and D).
Figure 1.
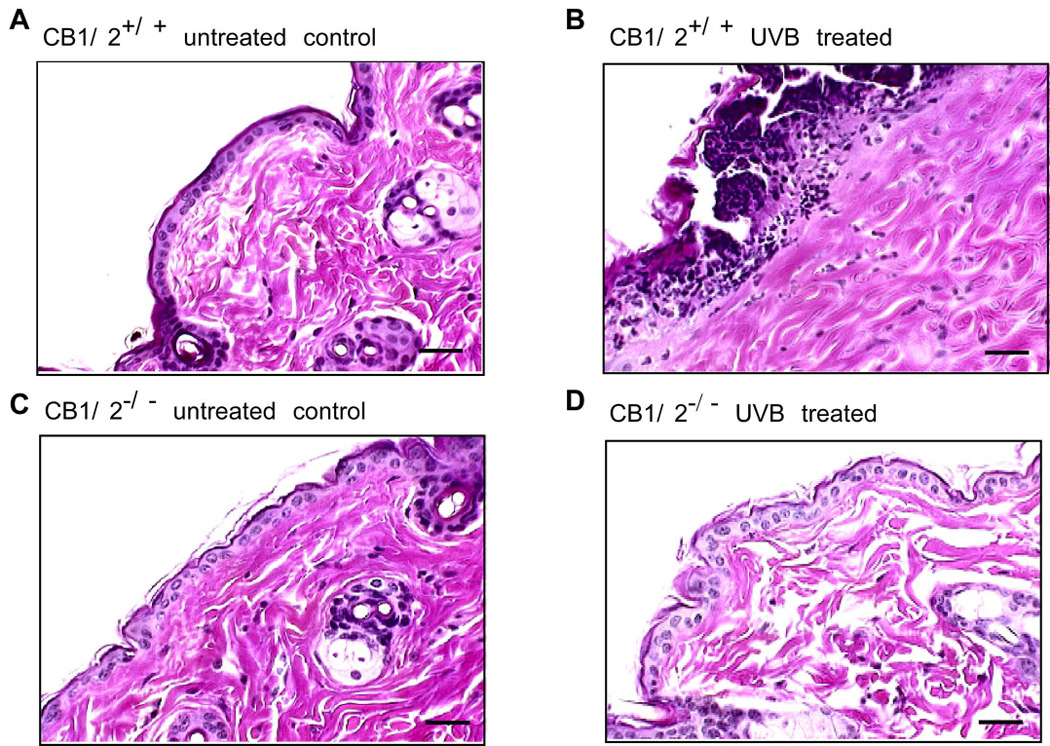
CB1/2 deficiency reduces UV-induced skin inflammation. CB1/2+/+ (A and B) or CB1/2−/− (C and D) mice were or were not irradiated with UVB (4 kJ/m2). Dorsal skin from irradiated (B and D) or non-irradiated (A and C) mice was harvested 24 h after UVB exposure. For assessment of inflammation, tissues were fixed in 10% neutral-buffered formalin and stained with H&E (hematoxylin and eosin). All UV irradiation experiments were performed in triplicate for each condition and representative images are shown. Scale bar indicates 25 µm at X200 magnification.
CB1/2 receptors are phosphorylated in response to UV treatment
Current theories for GPCR regulation predict that the active state conformation is the target for phosphorylation, internalization and desensitization (26). Phosphorylation hinders further association of the receptor with G proteins, thereby terminating the signaling pathways (27). In the continued presence of agonist, receptors are targeted to lysosomes for degradation (28) and agonist treatment has been shown to result in rapid phosphorylation of CB1 and CB2 receptors in transfected cell systems (29, 30). The quick internalization of CB1 and CB2 receptors after agonist exposure has been reported (30, 31). Activation of the CB1/2 receptors by UV irradiation might produce a similar rapid phosphorylation and internalization of these receptors. To test this hypothesis, we transfected His-epitope-tagged CB1 or CB2 into HEK293 cells and treated the cells with UVB followed by immunoprecipitation and Western blotting. Results indicated that treatment with UVB induced a rapid phosphorylation of the CB1 and CB2 receptors, which was observed at 5 minutes and remained at this level until 30 minutes after UVB treatment (Fig. 2A and B). Treatment with UVB also induced rapid internalization of CB1 and CB2 as indicated by the time-dependent decrease in protein level of CB1 and CB2 in membrane fractions after UVB treatment (Fig. 2C and D). These results indicated that the CB1 and CB2 receptors are phosphorylated and internalized in response to UVB treatment.
Figure 2.
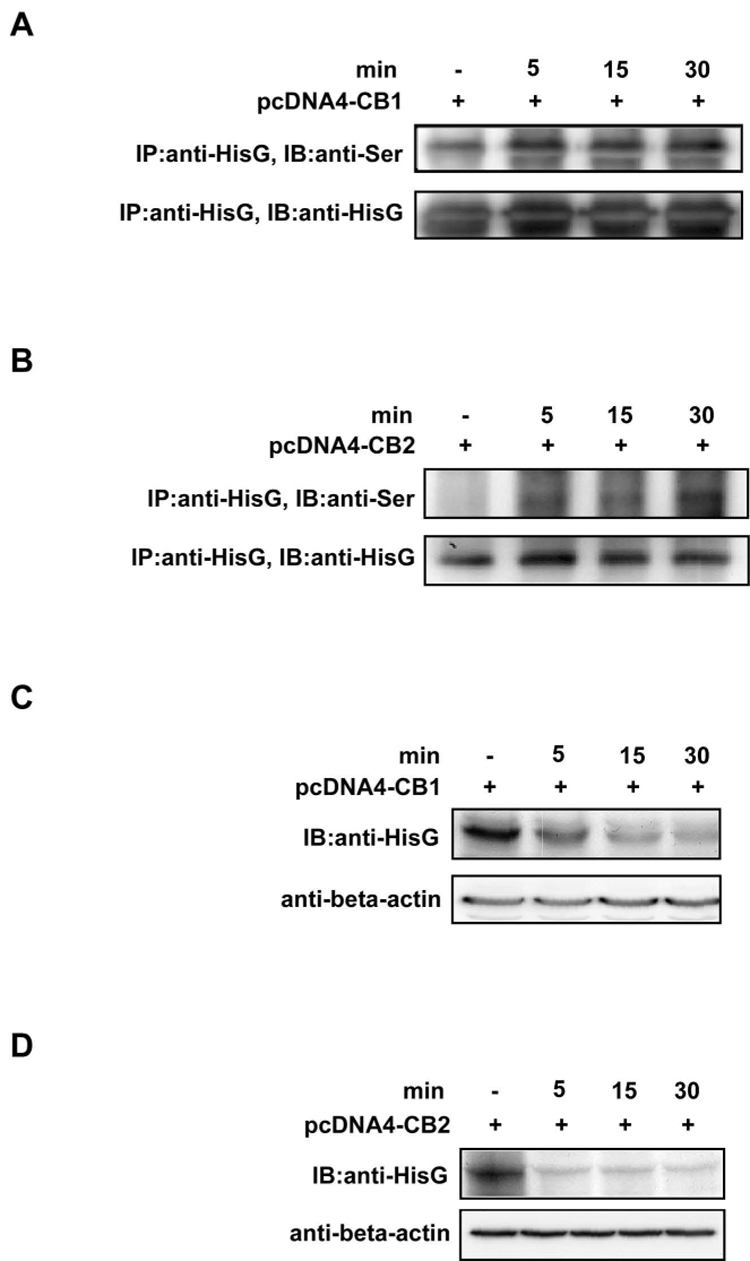
UVB induces CB1 or CB2 phosphorylation and internalization. HEK293 cells transfected with (A) pcDNA4-HisG-CB1 or (B) pcDNA4-HisG-CB2 were irradiated with UVB (4 kJ/m2) followed by incubation at 37°C in a 5% CO2 incubator for 5, 15, or 30 min. The transfected cells not exposed to UVB were used as a negative control. Cells were harvested at the indicated time and the membrane protein was extracted and subjected to immunoprecipitation with a HisG monoclonal antibody. CB1 or CB2 serine phosphorylation was detected by Western blotting with a phospho-serine antibody. The total protein levels of (C) CB1 or (D) CB2 using the same sample as in (A) and (B), respectively, were determined by Western blotting with the HisG monoclonal antibody to show the localization of the CB1 or CB2 receptor in the membrane fraction after UVB treatment.
CB1/2 modulate JNKs and ERKs signaling cascades in response to UV treatment
The GPCRs can stimulate the MAP kinase cascades, which have prominent roles in the control of cell growth, differentiation, and proliferation (32). To demonstrate a potential role for the cannabinoid receptors in UV-induced signal transduction, we examined the phosphorylation of ERKs, JNKs, and p38 in CB1/2+/+ and CB1/2−/− MEFs following UVB irradiation (4 kJ/m2). Results of Western blot analysis indicated that phosphorylation of ERKs, JNKs and p38 was induced in a time-dependent fashion after exposure of CB1/2+/+ MEFs to UVB (Fig. 3A). In marked contrast, UVB-induced-phosphorylation of ERKs and JNKs in CB1/2−/− cells was almost abolished (Fig. 3A). Although p38 phosphorylation was somewhat attenuated, the decrease was not as substantial as the decrease in ERKs and JNKs phosphorylation (Fig. 3A). These data provide direct evidence supporting the involvement of the CB1/2 receptors in mediating the activation of the MAP kinase pathways in the cellular response to UVB.
Figure 3.
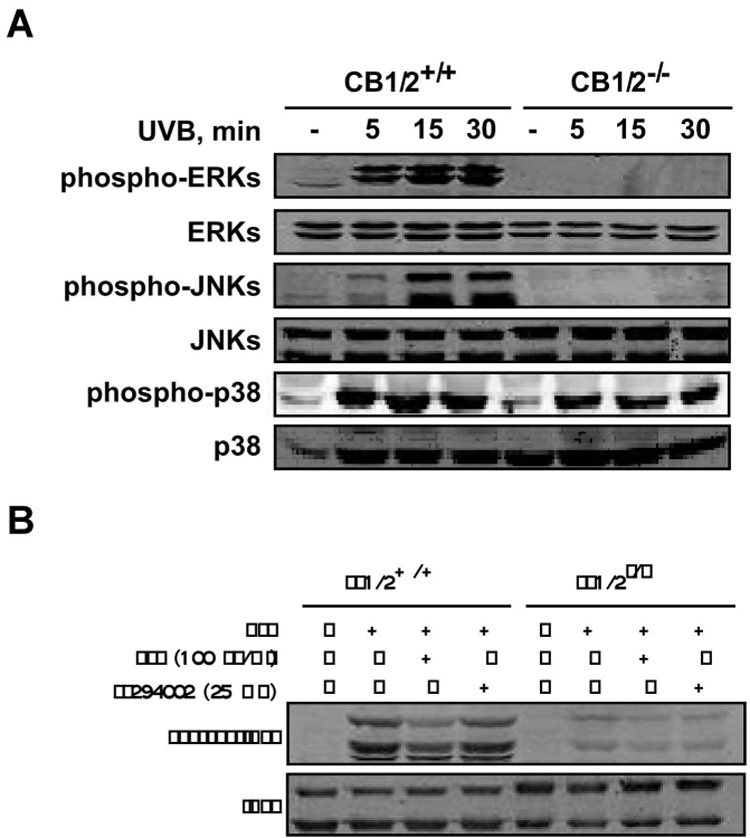
UVB-induced MAP kinase activation is markedly attenuated in cannabinoid receptor (CB) 1/2 deficient (CB1/2−/−) mouse embryonic fibroblasts (MEFs). (A) CB1/2 wildtype (CB1/2+/+) or CB1/2−/− MEFs were irradiated with UVB (4 kJ/m2) and then incubated at 37°C for 5, 15, or 30 min. The cells not exposed to UVB were used as a negative control. Cells were then harvested and phosphorylation of ERKs, JNKs and p38 was detected by immunoblotting using the respective, specific phospho-antibodies. Total protein levels of ERKs, JNKs and p38 were used as loading controls. (B) CB1/2+/+ and CB1/2−/− MEFs were treated with PTX (100 ng/ ml) or LY294002 (25 µM) prior to UVB (4 kJ/m2) exposure. Cells were then incubated at 37°C for 15 min and harvested. Phosphorylation of JNKs was detected by immunoblotting using a specific phospho-antibody. Total protein levels of JNKs were used as a loading control.
Both CB1 and CB2 receptors are coupled to Gi/o proteins, through which they stimulate the activity of the MAP kinases (33). The involvement of Gi/o proteins in coupling the CB1 and/or CB2 receptor to JNKs activation in this system was assessed using PTX (pertussis toxin; 100 ng/ ml), a compound that has been widely used as a reagent to characterize the involvement of heterotrimeric G-proteins in signaling (34). PTX catalyses the ADP-ribosylation of specific G-protein α subunits of the Gi family, which prevents the occurrence of the receptor–G-protein interaction (34). PTX pretreatment prior to UVB treatment partially prevented UVB-induced JNKs activation in CB1/2+/+ cells, indicating the involvement of Gi/o-proteins in UVB-induced JNKs activation (Fig. 3B). Cannabinoid receptors are also known to stimulate the phosphatidylinositol 3-kinase (PI3-K)/Akt survival pathway (35), although the negative coupling of cannabinoid receptors to Akt has also been reported (36). In CB1/2+/+ and CB1/2−/− cells, LY294002, a PI3-K inhibitor, was used to determine whether the PI3-K/Akt survival pathway is involved in the UVB-induced JNKs activation. Results indicated that UVB-induced JNKs phosphorylation is not mediated through PI3-K (Fig. 3B). Notably, neither compound affected JNKs phosphorylation in CB1/2−/− cells, which was still markedly decreased compared to wildtype cells.
CB1/2 is involved in TNF-α mediated NF-κB activation
Manipulation of the cannabinoid receptors has been useful in the management of pain, treatment of osteoporosis, inflammation, and cancer (4). Human keratinocytes constitutively secrete only a small amount of TNF-α and other cytokines, including IL-1, IL-6, IL-8, colony-stimulating factor, transforming growth factors, and platelet-derived growth factor. However, UV exposure significantly induces the production of cytokines (37, 38). TNF-α is an important member of the cytokine cascade and might be involved in the mediation of the inflammatory response to UV exposure, either directly or through the induction of other cytokines (39). We therefore examined the effect of UVB on the expression of TNF-α in CB1/2+/+ and CB1/2−/− mouse skin. Interestingly, compared to adult CB1/2−/− mice, adult CB1/2+/+ mouse skin demonstrated a more elevated inflammatory cytokine TNF-α level (Fig. 4A and B) following UVB treatment, corresponding with the observed elevated inflammatory response (Fig. 1A–D). Thus, the CB1/2 receptors are involved in orchestrating events leading to the inflammation observed in UVB-treated mouse skin. Furthermore, CB1 or CB2 activation has been reported to result in enhanced NF-κB transactivation (40). To test whether CB1/2 deficiency is associated with suppression of NF-κB activity, we transfected an NF-κB-linked luciferase promoter plasmid into CB1/2+/+ or CB1/2−/− MEFs and stimulated the cells with UVB (4 kJ/m2). The results indicated that NF-κB activation was induced in CB1/2+/+ MEFs, but not in CB1/2−/− MEFs (Fig. 4C), suggesting that CB1/2 receptors are involved in UVB-induced NF-κB activation.
Figure 4.
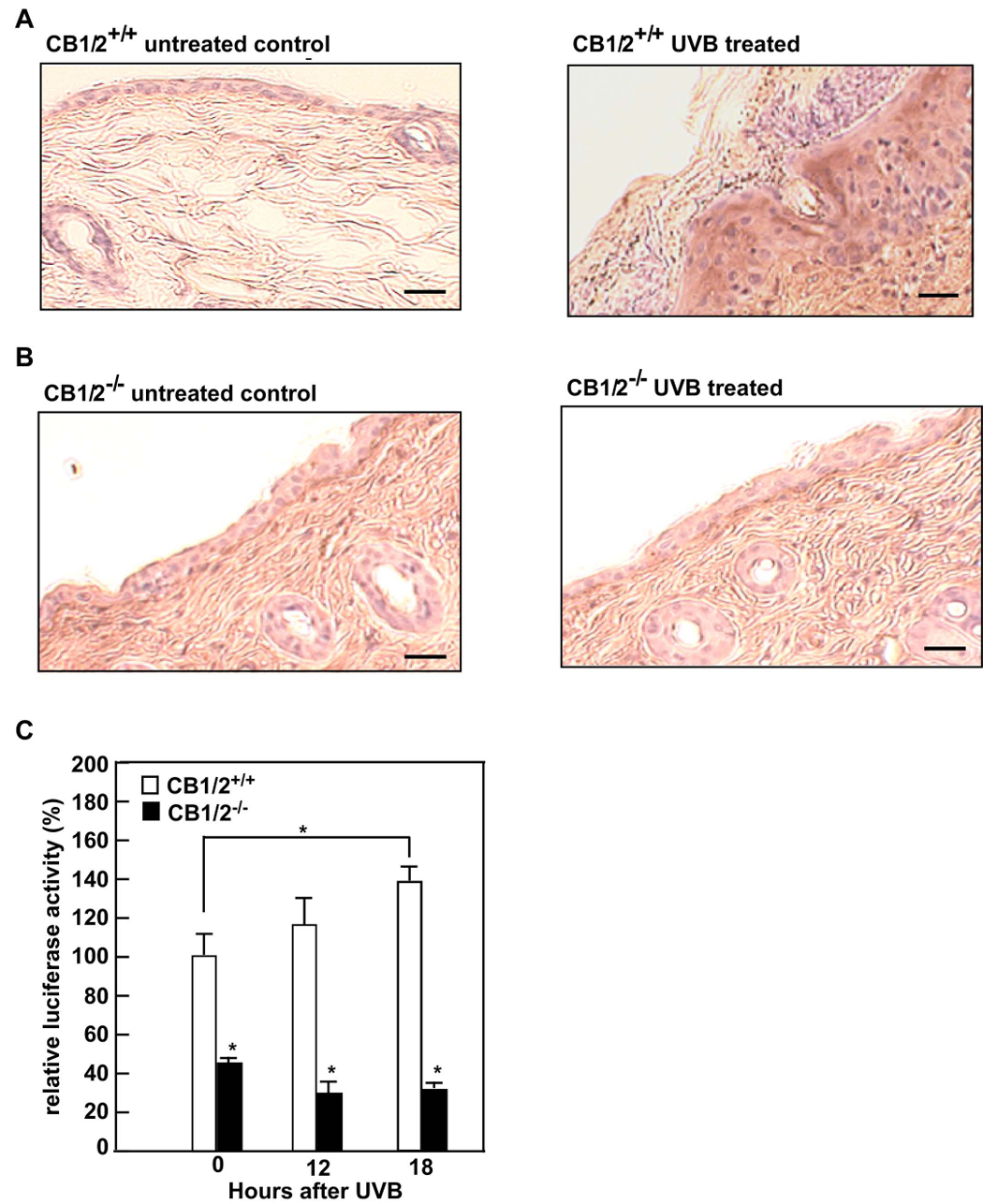
CB1/2 deficiency suppresses UV-induced NF-κB activation. CB1/2+/+ (A) or CB1/2−/− (B) mice were or were not irradiated with UVB (4 kJ/m2). Dorsal skin from irradiated (right panel of A and B) or non-irradiated (left panel of A and B) mice was harvested 24 h after UVB exposure. Tumor necrosis factor-α (TNF-α) was detected in formalin-fixed and paraffin-embedded sections of skin using the ABC (avidin-biotin complexes) immunohistochemistry staining method. TNF-α is visualized as the brown DAB (3, 3′-diaminobenzidine) staining. All UV irradiation experiments were performed in triplicate for each condition and representative images are shown. The scale bar indicates 25 µm at X200 magnification. (C) CB1/2+/+ or CB1/2−/− MEFs were transiently transfected with 2 µg of NF-κB luciferase reporter plasmid together with 0.1 µg of the pRL-SV-40 plasmid. At 24 h after transfection, cells were starved in 0.1% FBS/DMEM for another 24 h, and then were or were not exposed to UVB (4 kJ/m2) and incubated at 37°C. After 12 or 18 h, the firefly luciferase activity was determined in cell lysates and normalized against Renilla luciferase activity. The relative activity is shown as compared to the activity of CB1/2+/+ MEFs without UVB exposure. Data are represented as means ± S.D. of values from triplicate experiments. Significant differences were evaluated using the Student’s t-test (*, p< 0.05)
CB1/2 deficiency reduces UV-induced skin papilloma formation
Studies in CB1−/−, CB2−/− or double-knockout mice have revealed responses to cannabinoids that are not mediated by the CB1/2 receptors (41). Candidates for the observed responses included other membrane-bound receptors (42) and the PPAR (peroxisome-proliferator-activated receptor) family of nuclear receptor transcription factors (43). These results emphasize the importance of identifying the function of the CB1/2 receptors in carcinogenesis. To elucidate the function of the CB1/2 receptors in skin cancer development, we exposed mice to UVB in the two-stage skin carcinogenesis mouse model. Mice were initiated with 7, 12-dimethyl benz[a]anthracene (DMBA) and then treated with increasing doses of UVB beginning 2 weeks after initiation. Notably, CB1/2+/+ mice showed substantially greater overall papilloma development compared to CB1/2−/− mice. Forty percent of wildtype mice developed papillomas compared to less than 10% for CB1/2−/− mice over the same period of time (Fig. 5A). Furthermore, the papillomas found on CB1/2+/+ mice were more numerous (Fig. 5B) and larger (Fig. 5C) compared to CB1/2−/− mice (Fig. 5D). DMBA alone had no effect on tumor development in either wildtype or CB1/2 deficient mice (data not shown). These results suggested that CB1/2 receptors might have an enhancing effect on DMBA/UVB-induced skin tumor development.
Figure 5.
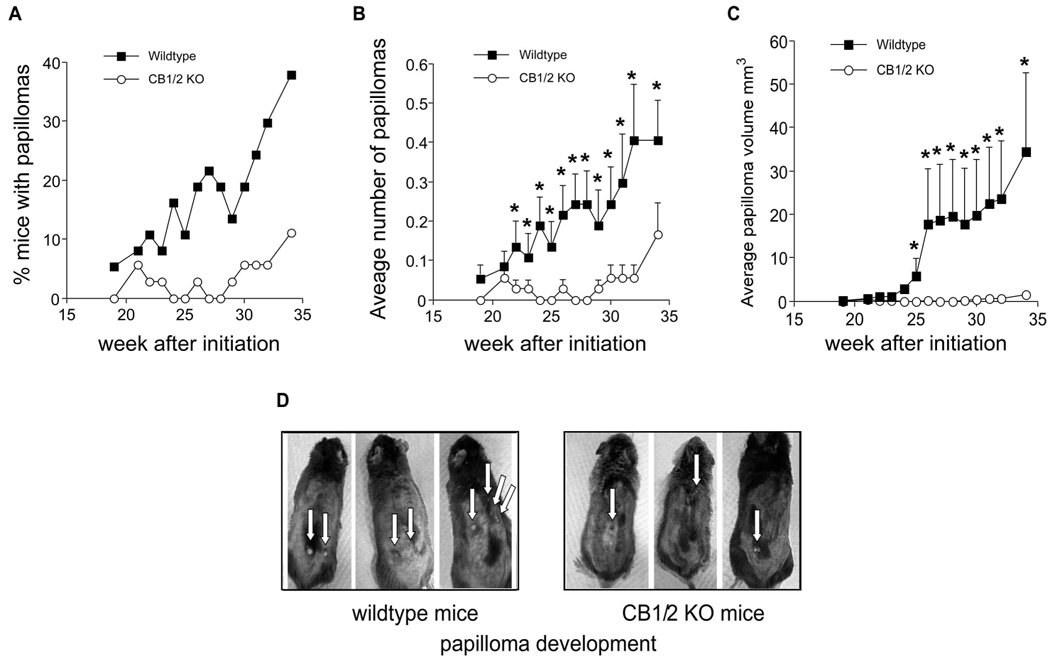
CB1/2 deficiency suppresses skin papilloma formation in the two-stage skin carcinogenesis mouse model. (A) Percentage of mice developing papillomas over a total of 34 wk. (B) Average number of papillomas per mouse. (C) Average papilloma volume (mm3) per mouse as measured by caliper and calculated according to the following formula: [(short diameter2/long diameter)/2]. For B and C, data are represented as means ± S.D. and the asterisk (*) indicates a significantly greater (p < 0.05) multiplicity (B) or volume (C) of papillomas in wildtype mice compared to CB1/2 knockout (KO) mice. (D) Representative photographs of UVB-treated CB1/2+/+ and CB1/2−/− mice.
Discussion
Solar radiation induces acute and chronic reactions in human and animal skin. Whereas acute UV irradiation causes apoptosis, long-term exposure to UV irradiation induces activation of oncogenes and inactivation of tumor suppressor genes resulting in abnormal proliferation of keratinocytes that may contain DNA damage leading to the onset of skin cancer (1, 44). In addition, UV irradiation stimulates cell proliferation through signal transduction initiated on the cell membrane. The activation, clustering and internalization of cell surface receptors for EGF, TNF-α and IL-1 after exposure to UVB has been reported (2, 3). The effects of UV may be produced by a perturbation of membrane structure or a conformational change in membrane proteins resulting from energy absorption (3). Whether CB1 and CB2 receptors are activated by UV radiation is not clear. Our in vitro binding studies indicated that UVB or UVA increased the affinity of CB1 and CB2 receptors for a selected agonist; and UVB also induced CB1 and CB2 phosphorylation and internalization in the membrane fragments of cells overexpressing these proteins. These data indicated that UV activates CB1 and CB2 receptors directly. Furthermore, from our in vivo studies, we found that tumorigenesis was significantly increased in wildtype (CB1/2+/+) mice compared with mice lacking the CB1/2 receptors (CB1/2−/−) after chronic or long-term exposure to UVB. These data showed that, besides the cell surface receptors for EGF, TNF-α or IL-1, the CB1/2 receptors are also the target of UV irradiation and have a marked effect on UV-induced skin carcinogenesis.
Our results also indicated that after acute exposure to UVB, CB1/2+/+ mice displayed a much higher incidence of inflammation compared with CB1/2−/− mice, coinciding with an increased inflammatory cytokine TNF-α level. These are known effects of acute UV radiation (1). Cannabinoids have been reported to suppress the production of cytokines in animal models and in human cell culture, but they have also been shown to increase the production of cytokines, including TNF, IL-1, IL-6, and IL-10, when they are administered alone or together with bacteria or other antigens (45). Thus, cannabinoids might either suppress or enhance the production of these pro-inflammatory agents, depending on either the nature of the pro-inflammatory stimulus or the type of cannabinoids used. Our results showed that activation of the CB1/2 receptors by UV irradiation enhanced the production of TNF-α and inflammation in CB1/2 wildtype mouse skin. One of the crucial mediators of inflammation-induced tumor growth and progression is activation of NF-κB by a classical pathway, which may promote tumor development (46, 47). Our data also indicated that NF-κB activation was decreased in CB1/2−/− mice compared with CB1/2+/+ mice. Overall, these observations suggested that the CB1/2 receptors are required in the induction of the pro-inflammatory cascade-dependent tumor development in response to UVB.
Acknowledgements
We would like to thank Dr. Andreas Zimmer (Laboratory of Molecular Biology Clinic of Psychiatry, University Hospital, Bonn, Germany) for the CB1/2 deficient mice; Dr. Beat Lutz (Group Molecular Genetics of Behavior Max-Planck-Institute of Psychiatry, Kraepelinstr. 2-10D-80804 Munich, Germany) for the mouse CB1 plasmid; Dr. Ruud Felwel (Erasmus MC, Department of Hematology, Molewaterplein 50, 3015GE Rotterdam, Netherlands) for the mouse CB2 plasmid.
Grant support: This work is supported in part by The Hormel Foundation, National Institutes of Health grants CA77646, CA27502, and CA111356.
References
- 1.Melnikova VO, Ananthaswamy HN. Cellular and molecular events leading to the development of skin cancer. Mutat Res. 2005;571:91–106. doi: 10.1016/j.mrfmmm.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 1.Sachsenmaier C, Radler-Pohl A, Zinck R, Nordheim A, Herrlich P, Rahmsdorf HJ. Involvement of growth factor receptors in the mammalian UVC response. Cell. 1994;78:963–972. doi: 10.1016/0092-8674(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 3.Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 4.Marx J. Drug development. Drugs inspired by a drug. Science. 2006;311:322–325. doi: 10.1126/science.311.5759.322. [DOI] [PubMed] [Google Scholar]
- 5.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 6.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365:61–65. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 7.Shim JY, Welsh WJ, Howlett AC. Homology model of the CB1 cannabinoid receptor: sites critical for nonclassical cannabinoid agonist interaction. Biopolymers. 2003;71:169–189. doi: 10.1002/bip.10424. [DOI] [PubMed] [Google Scholar]
- 8.Xie XQ, Chen JZ, Billings EM. 3D structural model of the G-protein-coupled cannabinoid CB2 receptor. Proteins. 2003;53:307–319. doi: 10.1002/prot.10511. [DOI] [PubMed] [Google Scholar]
- 9.Palczewski K, Kumasaka T, Hori T, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 10.Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 A crystal structure of visual arrestin: a model for arrestin's regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 11.Craft CM, Whitmore DH, Wiechmann AF. Cone arrestin identified by targeting expression of a functional family. J Biol Chem. 1994;269:4613–4619. [PubMed] [Google Scholar]
- 12.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 13.Attramadal H, Arriza JL, Aoki C, et al. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem. 1992;267:17882–17890. [PubMed] [Google Scholar]
- 14.Bakshi K, Mercier RW, Pavlopoulos S. Interaction of a fragment of the cannabinoid CB1 receptor C-terminus with arrestin-2. FEBS Lett. 2007;581:5009–5016. doi: 10.1016/j.febslet.2007.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarfaraz S, Afaq F, Adhami VM, Mukhtar H. Cannabinoid receptor as a novel target for the treatment of prostate cancer. Cancer Res. 2005;65:1635–1641. doi: 10.1158/0008-5472.CAN-04-3410. [DOI] [PubMed] [Google Scholar]
- 16.Sarnataro D, Pisanti S, Santoro A, et al. The cannabinoid CB1 receptor antagonist rimonabant (SR141716) inhibits human breast cancer cell proliferation through a lipid raft-mediated mechanism. Mol Pharmacol. 2006;70:1298–1306. doi: 10.1124/mol.106.025601. [DOI] [PubMed] [Google Scholar]
- 17.Jorda MA, Lowenberg B, Delwel R. The peripheral cannabinoid receptor Cb2, a novel oncoprotein, induces a reversible block in neutrophilic differentiation. Blood. 2003;101:1336–1343. doi: 10.1182/blood-2002-07-2034. [DOI] [PubMed] [Google Scholar]
- 18.Sarfaraz S, Afaq F, Adhami VM, Malik A, Mukhtar H. Cannabinoid receptor agonist-induced apoptosis of human prostate cancer cells LNCaP proceeds through sustained activation of ERK1/2 leading to G1 cell cycle arrest. J Biol Chem. 2006;281:39480–39491. doi: 10.1074/jbc.M603495200. [DOI] [PubMed] [Google Scholar]
- 19.Guzman M. Cannabinoids: potential anticancer agents. Nature reviews. 2003;3:745–755. doi: 10.1038/nrc1188. [DOI] [PubMed] [Google Scholar]
- 20.Casanova ML, Blazquez C, Martinez-Palacio J, et al. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. The Journal of clinical investigation. 2003;111:43–50. doi: 10.1172/JCI16116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blazquez C, Carracedo A, Barrado L, et al. Cannabinoid receptors as novel targets for the treatment of melanoma. Faseb J. 2006;20:2633–2635. doi: 10.1096/fj.06-6638fje. [DOI] [PubMed] [Google Scholar]
- 22.Wilkinson JD, Williamson EM. Cannabinoids inhibit human keratinocyte proliferation through a non-CB1/CB2 mechanism and have a potential therapeutic value in the treatment of psoriasis. Journal of dermatological science. 2007;45:87–92. doi: 10.1016/j.jdermsci.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 23.D'Antona AM, Ahn KH, Kendall DA. Mutations of CB1 T210 produce active and inactive receptor forms: correlations with ligand affinity, receptor stability, and cellular localization. Biochemistry. 2006;45:5606–5617. doi: 10.1021/bi060067k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guzman M. Effects on cell viability. Handb Exp Pharmacol. 2005:627–642. doi: 10.1007/3-540-26573-2_21. [DOI] [PubMed] [Google Scholar]
- 25.Schwarz A, Mahnke K, Luger TA, Schwarz T. Pentoxifylline reduces the formation of sunburn cells. Exp Dermatol. 1997;6:1–5. doi: 10.1111/j.1600-0625.1997.tb00138.x. [DOI] [PubMed] [Google Scholar]
- 26.Leurs R, Smit MJ, Alewijnse AE, Timmerman H. Agonist-independent regulation of constitutively active G-protein-coupled receptors. Trends Biochem Sci. 1998;23:418–422. doi: 10.1016/s0968-0004(98)01287-0. [DOI] [PubMed] [Google Scholar]
- 27.Abood ME. Molecular biology of cannabinoid receptors. Handb Exp Pharmacol. 2005:81–115. doi: 10.1007/3-540-26573-2_3. [DOI] [PubMed] [Google Scholar]
- 28.von Zastrow M, Kobilka BK. Ligand-regulated internalization and recycling of human beta 2-adrenergic receptors between the plasma membrane and endosomes containing transferrin receptors. J Biol Chem. 1992;267:3530–3538. [PubMed] [Google Scholar]
- 29.Garcia DE, Brown S, Hille B, Mackie K. Protein kinase C disrupts cannabinoid actions by phosphorylation of the CB1 cannabinoid receptor. J Neurosci. 1998;18:2834–2841. doi: 10.1523/JNEUROSCI.18-08-02834.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouaboula M, Dussossoy D, Casellas P. Regulation of peripheral cannabinoid receptor CB2 phosphorylation by the inverse agonist SR 144528. Implications for receptor biological responses. J Biol Chem. 1999;274:20397–20405. doi: 10.1074/jbc.274.29.20397. [DOI] [PubMed] [Google Scholar]
- 31.Rinaldi-Carmona M, Le Duigou A, Oustric D, et al. Modulation of CB1 cannabinoid receptor functions after a long-term exposure to agonist or inverse agonist in the Chinese hamster ovary cell expression system. J Pharmacol Exp Ther. 1998;287:1038–1047. [PubMed] [Google Scholar]
- 32.Howlett AC. Cannabinoid receptor signaling. Handb Exp Pharmacol. 2005:53–79. doi: 10.1007/3-540-26573-2_2. [DOI] [PubMed] [Google Scholar]
- 33.Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- 34.Fields TA, Casey PJ. Signalling functions and biochemical properties of pertussis toxin-resistant G-proteins. Biochem J. 1997;321(Pt 3):561–571. doi: 10.1042/bj3210561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanchez MG, Ruiz-Llorente L, Sanchez AM, Diaz-Laviada I. Activation of phosphoinositide 3-kinase/PKB pathway by CB(1) and CB(2) cannabinoid receptors expressed in prostate PC-3 cells. Involvement in Raf-1 stimulation and NGF induction. Cell Signal. 2003;15:851–859. doi: 10.1016/s0898-6568(03)00036-6. [DOI] [PubMed] [Google Scholar]
- 36.Gomez del Pulgar T, Velasco G, Sanchez C, Haro A, Guzman M. De novo-synthesized ceramide is involved in cannabinoid-induced apoptosis. Biochem J. 2002;363:183–188. doi: 10.1042/0264-6021:3630183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kock A, Schwarz T, Kirnbauer R, et al. Human keratinocytes are a source for tumor necrosis factor alpha: evidence for synthesis and release upon stimulation with endotoxin or ultraviolet light. J Exp Med. 1990;172:1609–1614. doi: 10.1084/jem.172.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oxholm A, Oxholm P, Staberg B, Bendtzen K. Immunohistological detection of interleukin I-like molecules and tumour necrosis factor in human epidermis before and after UVB-irradiation in vivo. Br J Dermatol. 1988;118:369–376. doi: 10.1111/j.1365-2133.1988.tb02430.x. [DOI] [PubMed] [Google Scholar]
- 39.Munker R, Gasson J, Ogawa M, Koeffler HP. Recombinant human TNF induces production of granulocyte-monocyte colony-stimulating factor. Nature. 1986;323:79–82. doi: 10.1038/323079a0. [DOI] [PubMed] [Google Scholar]
- 40.Daaka Y, Zhu W, Friedman H, Klein TW. Induction of interleukin-2 receptor alpha gene by delta9-tetrahydrocannabinol is mediated by nuclear factor kappaB and CB1 cannabinoid receptor. DNA Cell Biol. 1997;16:301–309. doi: 10.1089/dna.1997.16.301. [DOI] [PubMed] [Google Scholar]
- 41.Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez C, Galve-Roperh I, Canova C, Brachet P, Guzman M. Delta9-tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett. 1998;436:6–10. doi: 10.1016/s0014-5793(98)01085-0. [DOI] [PubMed] [Google Scholar]
- 43.Sun Y, Alexander SP, Kendall DA, Bennett AJ. Cannabinoids and PPARalpha signalling. Biochem Soc Trans. 2006;34:1095–1097. doi: 10.1042/BST0341095. [DOI] [PubMed] [Google Scholar]
- 44.Ichihashi M, Ueda M, Budiyanto A, et al. UV-induced skin damage. Toxicology. 2003;189:21–39. doi: 10.1016/s0300-483x(03)00150-1. [DOI] [PubMed] [Google Scholar]
- 45.Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5:400–411. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- 46.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 47.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]