

Abstract
Fluorescent molecules that emit in the near infra-red are potentially useful as probes for biotechnology. A relatively under-explored design for probes of this type are the aza-BODIPY dyes; this study was performed to enhance our understanding of these materials and ways in which they may be used in dye cassette systems. Thus, the aza-BODIPY dyes 1a – g were prepared. An advanced intermediate towards an eighth compound in the series, 6h, was made but it could not be complexed with boron effectively to give 1h. Spectroscopic properties of these compounds were recorded, and correlations between substituent effects, UV absorbance, fluorescence emissions, and quantum yields were made. Compound 1a was coupled with a fluorescein-alkyne derivative to give the energy transfer cassettes 2 and 3. Both these compounds gave poor energy transfer, and the possible reasons for this were discussed.
Keywords: fluorescent compounds, biochemical markers, heterocyclic compounds
1. Introduction
There is considerable interest in preparing new fluorescent dyes that emit towards the near-IR region.1–4 Labeled-biomolecules are more easily observed in vivo, for instance, when the probes used emit in this region.5 Unfortunately, there are a very limited number of organic molecules that emit in this range and could be adapted to form probes for biomolecules. Probably, the most widely used are the cyanine dyes,6–10 and few other choices are commercially available.
BODIPY (difluoroboradiaza-s-indacene) dyes A are highly fluorescent, stable, and insensitive to the solvents’ polarity and pH. BODIPYs are unusual in that they are relatively non-polar and are electronically neutral. They have found widespread applications as laser dyes, sensors, and molecular probes that emit in the region around 520 – 600 nm.11
Modifications to the BODIPY core can lead to dyes that emit above 600 nm. Such modifications include appending strong electron-donating groups, rigidifying substituents around the core, and extending the conjugation of the system. The strategy of attaching electron-donating substituents, however, has limitations. For instance, amine groups make the probes sensitive to quenching via electron transfer to the excited state, and the fluorescence becomes pH sensitive.
There is a subset of BODIPY dyes that are related to the parent systems by N-for-C substitution at the C8-position. These are commonly called “aza-BODIPY” dyes, and almost all the known compounds have 3,3,5,5-aryl substituents, of which the tetraphenyl system B is the most widely studied. The parent heterocycle without a boron atom has been known since 1943,12–15 but it was not until 1994 that a B-derivative was mentioned in the literature and its remarkable fluorescent properties were noted.16 Specifically, the tetraphenyl compound B was shown to emit at 695 nm (in 1,2-Me2C6H4, Φ = 0.77), i. e a marked red-shift relative to other BODIPY dyes.

To date, there have been only two deliberate attempts to further extend the emission of aza-BODIPY dyes into the near IR-region. In the first, Carreira and co-workers used extended heterocycles to prepare constrained systems such as C; this gave molecules that emitted up to 815 nm with enhanced quantum yields.17,18 Second, work from our laboratory focused on using ortho-oxygens on the 3,3-aryl substituents to constrain them giving systems D; this approach also shifts the fluorescence emissions to the red and increased the quantum yields.19 Finally, when designing pH sensitive probes, O’Shea found that when 4-N,N-dimethylaminobenzene substituents were created at the 3,3-aza-BODIPY sites in molecule E, then an emission at 823 nm was observed under conditions where the amine groups were not protonated.20 This was reported while the work described in this manuscript was in progress. Compound E has limited value as a potential core-structure for biomolecular probes unless protonated, in which case the red-shift is not observed. Nevertheless, that observation supported the hypothesis we were exploring, i. e. that strongly electron-withdrawing or -donating aryl-substituents could significantly shift the emissions of aza-BODIPY dyes. A second goal of this study was to prepare aza-BODIPY systems with halide substituents that could be substituted with “donor” fragments that absorb considerably shorter UV radiation. In the event, both these goals were realized, and the data are reported here. Specifically, a series of aza-BODIPY dyes 1a – g, having a range of different substituted-aryl groups were prepared. One of these potential “acceptor” molecules, compound 1a was elaborated into the donor-acceptor cassette21–23 systems 2 and 3. Important spectral parameters for all these dyes are reported here.
 |
cpd | R1 | R2 | R3 |
| a | OMe | I | H | |
| b | Br | OMe | H | |
| c | H | H | OMe | |
| d | Br | H | OMe | |
| e | I | H | OMe | |
| f | Br | OMe | OMe | |
| g | Br | NMe2 | OMe | |
| ha | Br | I | H |
compound 1h could not be obtained. Compound 6h could however be obtained.

2. Results and Discussion
2.1 Synthesis of Aza-BODIPYS 1 a – g
Synthesis of dyes 1 began with diaryl α,β-unsaturated ketones 4 (chalcones; readily prepared by an aldol/dehydration reaction of the corresponding benzaldehyde and acetophenone derivatives). O’Shea’s modification 24 of an older Rogers’ procedure was then used.12 Thus, Michael addition of the anion from nitromethane to the chalcones 4 gave the 1,3-diaryl-4-nitrobutan-1-ones 5 in essentially quantitative yields after aqueous work-up; these were then used without further purification. Condensation with ammonium acetate in refluxing butanol gave the azadipyrromethenes 6 via a cascade of events (in situ formation of the pyrrole and corresponding nitroso-pyrrole, and subsequent condensation of those two entities). Finally, complexation of the azadipyrromethenes with boron trifluoride gave the azaBODIPYs 1 in excellent yields.
Using the procedure outlined in Scheme 1, dipyrromethene intermediate 6h was only obtained in low yield (8%, crude) and in an impure form, so other conditions were developed (Scheme 2). Variations of solvent (butanol, pentanol, hexanol, neat), ammonia source (HCONH2 instead of NH4OAc) and heating conditions (conventional heating or microwave radiation) to make the reaction viable were unsuccessful. However, 6h•HCl was obtained from the γ-ketonitrile 7 (made from the corresponding chalcone and acetone cyanohydrin) via condensation with hydroxylamine hydrochloride in methanol; this is one of the procedures developed by Rogers’ in 1944. 13,25 Surprisingly, complexation of 6h with boron trifluoride etherate did not give the desired product, but instead led to a compound with two BF2 units (19F); the structure of this adduct was tentatively assigned as 8. Presumably steric and/or electronic interactions within 6h prevented formation of the desired aza-BODIPY 1h, an observation supported by the fact that when the iodo groups were substituted (via Sonogashira for example) prior to complexation, formation of the aza-BODIPY proceeds easily and in good to excellent yield (data not shown).
Scheme 1.


Syntheses of aza-BODIPY 1a – g.
Scheme 2.

Modified Route for the Preparation of aza-BODIPY 1h.
Compounds 1a – g have good solubilities in most organic solvents (eg chloroform, toluene, tetrahydrofuran). An X-ray structure of compound 1a is shown in Figure 1. Compound 1a crystallized in the triclinic space group P-1 with two molecules in the asymmetric unit. The overall conjugated nature of the chromophore was confirmed from the analysis of the crystal structure with comparable bond lengths.
Figure 1.

X-ray structure of 1a. The molecule exists in two O-Me conformations in the crystal lattice.
2.2 Spectroscopic Properties of Aza-BODIPY Derivatives 1 a – g
Compounds 1 were prepared mainly to facilitate syntheses of more elaborate molecules containing donor groups (eg 2 and 3, vide infra), consequently most of them contain halogen atoms. However, this series of compounds does provide some non-systematic basis for comparing absorption and fluorescence properties.
Absorption spectra of compounds 1 show strong S0→S1 transitions with absorbance maxima between 650 and 798 nm (Table 1 and Figure 2a). Entry 1 and 2 in Table 1 are literature data24 for the tetra-aryl substituted aza-BODIPYs B and F.
Table 1.
Key Spectroscopic Data for the aza-BODIPY Derivatives 1a–g
 | ||||||
|---|---|---|---|---|---|---|
| entry | dye | λabs (nm) | ε (M−1cm−1) | λem. (nm) | fwhma (nm) | Φb |
| 1 | B | 655 | 79 000 | 676 | - | 0.34 |
| 2 | F | 693 | 85 000 | 717 | - | 0.36 |
| 3 | 1a | 680 | 109 000 | 711 | 47 | 0.18 ± 0.01 |
| 4 | 1b | 702 | 84 120 | 731 | 30 | 0.42 ± 0.03 |
| 5 | 1c | 640 | 73 850 | 688 | 47 | 0.07 ± 0.01 |
| 6 | 1d | 650 | 55 000 | 694 | 47 | 0.10 ± 0.01 |
| 7 | 1e | 653 | 71 600 | 701 | 35 | 0.10 ± 0.01 |
| 8 | 1f | 692 | 66 200 | 738 | 50 | 0.20 ± 0.01 |
| 9 | 1g | 798 | 68 610 | 830 | 30 | 0.07 ± 0.01 |
Full width at half maximum peak height.
Measured in 1% pyridine/toluene, Zn-phthalocyanine standard (Φ = 0.30 in the same solvent).
Figure 2.

a Normalized absorption and b fluorescence spectra of compounds 1a – g in toluene at 5 µM.

Using these as references, the data collected on compounds 1 indicate that introduction of electron-donating groups onto the aryl substituents results in significant bathochromic shifts. The largest red-shift in the series was observed for compound 1g (entry 9) which has two strongly electron donating groups attached to the 3-aryl substituents in ortho- and para-positions. This is consistent with the idea that the HOMO/LUMO energy levels of the aza-BODIPY core are influenced in such a way that fluorescence is enhanced by the electron rich aryl substituents. Comparison of molecules 1c – e with the rest of those in the series implies that the presence of an ortho-methoxy group on the 3-aryl substituent correlates with a blue-shift in the absorbance, but this may be overridden by a strongly electron donating group in the para-position of that same aromatic ring. Throughout, the extinction coefficients for these new dyes are high, in the range of 55 000 to 109 000 M−1cm−1, characteristic of aza-BODIPY compounds.
Fluorescence properties of the new dyes are also shown in Table 1, and the spectra are shown in Figure 2b. Their emission maxima range from 676 to 830 nm in toluene. Quantum yields for compounds in this series range from 0.07 to 0.41 in toluene. Interestingly, the fluorescence quantum yields of the bromoaryl-substituted derivatives were not significantly altered, while the fluorescence quantum yields of the iodoaryl-substituted derivatives show a significant decrease. There has been a tendency to over-generalize on the influence of some substituents on fluorescence, and the so-called “heavy atom effect” is a prime example of this. For compounds 1, bromine-containing aryl-substituents do not reduce the quantum yields to trivial values (eg 1b, entry 4). The only iodinated analogs in the series, 1a and 1e, also have reasonable quantum yields (0.18 and 0.10, respectively, entries 3 and 7). Most probably, the origins of the heavy atom effect are electron donation or acceptance from excited states of the chromophore, which often, but not always, occurs when heavy atoms are present. For the aza-BODIPY dyes 1 the halo-substituents are isolated from the chromophore by the aryl twist. Consequently, the main parameter influencing the fluoresence properties of the core is the oxidation potentials of those aryl substituents. This more sophisticated argument, rather than generalizations about “heavy atom effects”, is one advocated in other situations in several papers by Nagano and co-workers.26–32
2.3 Synthesis of Through-bond Energy Transfer Cassettes 2 and 3
Scheme 3 shows syntheses of molecules 2 and 3 that have donor fluorescein-derived entities attached to BODIPY cores. These molecules were made to test their efficacy as energy transfer cassettes. The difference between compounds 2 and 3 is only a carboxylic acid linker in the acceptor part. Throughout this donor fragment was derived from the ethynyl fluorescein diacetate 923,33 which served as a Sonogashira34 coupling partner for the aza-BODIPYs 1a or 10, respectively. Compound 10 was obtained via a one-pot, two step procedure involving deprotection and alkylation of 1a. In the synthesis of compound 10, demethylation of the aza-BODIPY 1a gave an intermediate with two phenolic-OH groups. This was unstable to air, hence it was alkylated without isolation at that stage. The final step in both syntheses was removal of the acetate groups from the fluorescein parts (potassium carbonate or TMSOK35); in the synthesis of compound 3 this also hydrolyzed the methyl ester functionality. Overall, the synthesis of cassette 2 was reasonably straightforward. However, cassette 3 was practically more difficult to prepare because of the instability of the intermediate mentioned above, and the need to isolate the final product via RP-HPLC. We were unable to fully and properly characterize compound 3 by NMR (1H and 13C) in most organic solvents, including CD3CO2D and D2O, because the compound aggregated. However, compound 12 was fully characterized. Synthesis of compound 3 was supported by mass spectrometry analysis, and UV-fluorescence properties.
Scheme 3.




Syntheses of a, through-bond energy transfer system 2, and b, system 3.
2.4 Spectroscopic Studies of Cassette 2
The absorption and emission spectra of cassettes 2 are solvent and pH dependent, reflecting the characteristics of the fluorescein part. This is consistent with the properties of fluorescein itself.36–38 Fluorescein tends to close to its lactone form at pH values of somewhat less than 6.5, and it is not particularly fluorescent in that state (Figure 3). Such pH-dependencies are undesirable if these cassettes were to be used as probes for imaging biomolecules.
Figure 3.

pH dependency of fluorescein fluorescence quantum yield.
Preliminary investigations of the spectroscopic properties of compound 2 showed poor energy transfer from the donor to the acceptor in a 1:1 mixture of THF:buffer pH 7.4 (Figure 4a) i. e. weak fluorescence from the aza-BODIPY (eg at 730 nm) was observed, and mostly fluorescence emission from the fluorescein was seen upon excitation of the donors at 488 nm,. To test if aggregation was responsible for this poor energy transfer, a concentration study was performed. No significant shift could be seen for both the absorption maxima of the donor and acceptor at 502 and 702 nm, respectively (Figure 4b). Further, the relationship between fluorescence intensity and concentration was essentially linear (Figure 4c). However, increasing concentrations of the non-ionic detergent Triton X-100 had significant effect on the system. The absorption peak of the fluorescein diminished with increased concentration of Triton X-100 (Figure 4d), and ultimately no peak was observed. The fluorescence emission of the donor showed the same tendency; no emission was observed at high concentration of Triton X-100 (Figure 4e). On the other hand, the fluorescence emission from the aza-BODIPY increased with increasing concentration of Triton X-100 (Figure 4f). It appears that in presence of the non-ionic detergent Triton X-100, the fluorescein is converted to its non-fluorescent lactone form. This change (from the quinoid to the lactone form) can occur due to the presence of the polyoxyethylene group in Triton X-100. It is well established that fluorescein dye retains its lactone form in presence of solvents having oxygen groups.39,40
Figure 4.


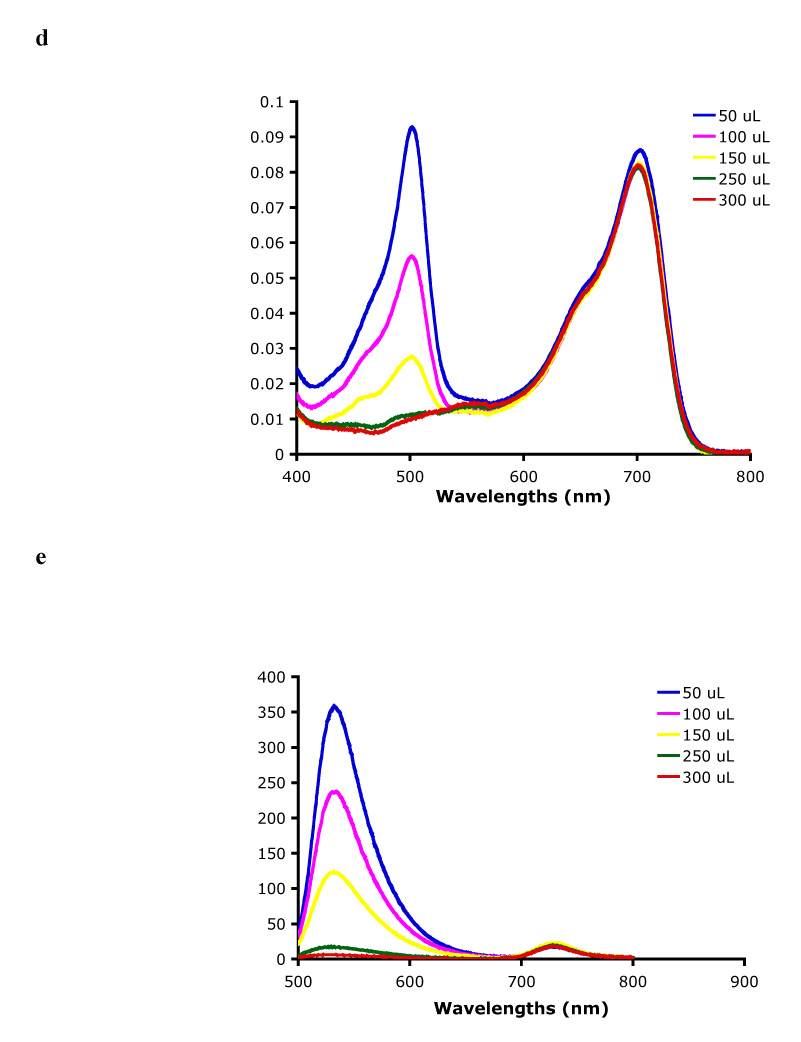

Spectra of: a UV absorption and corrected fluorescence spectra (excitation at 488 and 702 nm) of 2 in 1:1 THF:buffer pH 7.4; b UV absorption of 2 with increasing concentration of 2 in 1:1 THF:buffer pH 7.4; c uncorrected fluorescence intensities (at 530 and 730 nm) versus concentration of compound 2 in 1:1 THF:buffer pH 7.4; d UV absorption of compounds 2 in 1:1 THF:buffer pH 7.4 with increasing amount of Triton-X 100; e uncorrected fluorescence intensity of compound 2 upon excitation at 488 nm with increasing amount of Triton-X 100; f uncorrected fluorescence intensity of compound 2 upon excitation at 702 nm with increasing amount of Triton-X 100.
3. Conclusions
These studies show that the fluorescence emissions observed from aza-BODIPY dyes can be manipulated by altering the electronic substituents on the aryl-substituents. Red-shifts in the fluorescence tend to correspond to strongly electron donating para-groups, at least for the 3-aryl substituent. A series of seven functionalized aza-BODIPY dyes were prepared. Of these, compound 1a proved to be a useful starting material for attachment of alkyne-based energy-trasnfer-donor entities. In this study, the donor was a fluorescein-derived alkyne. Cassettes 2 and 3 were prepared from this coupling procedure exhibited absorption and fluorescence characteristics that were highly dependent on the pH and solvent media. These observations correlate with equilibria at the fluorescein part corresponding to lactone formation, and to solvent polarity effects that are influenced by the addition of the non-ionic detergent Triton X-100. Overall, we conclude that aza-BODIPY dyes have great potential application as biomolecular probes if they can be modified to increase solubility in aqueous media. Fluorescein is probably not a good donor for through-bond energy transfer cassettes designed for applications around physiological pH values, but the potential for using the aza-BODIPY acceptor fragment in conjunction with other donor groups is apparent.
4. Experimental Section
General Experimental Procedures
All chemicals were obtained from commercial suppliers and used without further purification. Chromatography on silica gel was performed using a forced flow of the indicated solvent on EM reagents silica gel 60 (230–400 mesh). Dichloromethane was dried/degassed by passing it down an alumina column. 1H and 13C NMR spectra were recorded on an Inova Instrument at 500 MHz (1H), 125 MHz (13C). 11B and 19F NMR spectra were recorded on a Inova 400 Broad Band instrument at 128 MHz (11B) and 376 MHz (19F). NMR chemical shifts are expressed in ppm relative to internal solvent peaks, and coupling constants were measured in Hz. For 11B NMR, BF3.OEt2 has been used as an external reference; similarly, CFCl3 was used as an external standard for the 19F spectra.
1-(4-Iodophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (4a)
41,42 4-Methoxybenzaldehyde (5.53 g, 0.04 mol), 4-iodoacetophenone (10.00 g, 0.04 mol) and potassium hydroxide (0.16 g, 4.06 mmol) were dissolved in methanol / H2O (1:1 v/v, 120 mL) and stirred under reflux for 12 h. During the course of the reaction, the product precipitated from the reaction mixture. After cooling, the reaction mixture was filtered and washed with methanol to give the product as a white solid (11.35 g, 77%), m.p. (non corrected) 162.2–163.3 °C. δH (500 MHz, CDCl3): 7.84 (d, 2H, J= 8.1 Hz), 7.78 (d, 1H, J= 15.6 Hz), 7.71 (d, 2H, J= 8.1 Hz), 7.59 (d, 2H, J= 8.3 Hz), 7.33 (d, 1H, J= 15.6 Hz), 6.93 (d, 2H, J= 8.3 Hz), 3.85 (s, 3H); δC (125 MHz, CDCl3): 189.6, 161.8, 145.2, 137.8, 137.7, 130.3, 129.8, 127.3, 118.9, 114.4, 100.3, 55.4; m/z (ESI): theoretical mass (M+H)+: 365.00; found : 365.01.
3-(4-Bromophenyl)-1-(4-methoxyphenyl)prop-2-en-1-one (4b)
43 4-Bromobenzaldehyde (5.00 g, 0.027 mol), 4-methoxyacetophenone (3.22 g, 0.027 mol) and potassium hydroxide (0.108 g, 2.7 mmol) were dissolved in ethanol (30 mL) and stirred at room temperature for 12 h. The product which precipitated upon formation was filtered and washed with ethanol and water. The titled product was obtained as a white solid (7.3 g, 85 %), m.p. (non corrected) 149–150 ° C. δH (500 MHz, CDCl3): 8.04 (d, 2H, J= 8.8 Hz), 7.73 (d, 1H, J= 15.6 Hz), 7.49–7.55 (m, 5 H), 6.99 (d, 2H, J= 8.8 Hz), 3.89 (s, 3H); δC (125 MHz, CDCl3): 188.3, 163.5, 142.4, 133.9, 132.1, 130.8, 130.7, 129.7, 124.5, 122.3, 113.8, 55.5; m/z (ESI): theoretical mass (M+H)+: 317.01; found : 317.02–319.02 (Br isotope).
1-(2-Methoxyphenyl)-3-phenyl-2-propene-1-one (4c)
was prepared according to the reported procedure.44–46 The chalcone was obtained as a yellow oil (2.9 g, 60% yield). δH (500 MHz, CDCl3): 7.66–7.63 (m, 2H), 7.57–7.59 (m, 2H), 7.47 (td, 1H, J= 7.3 Hz, J= 1.7 Hz), 7.42–7.38 (m, 4H), 7.04 (t, 1H, J= 7.6 Hz), 6.99 (d, 1H, J= 8.3 Hz ), 3.89 (s, 3H); δC(125 MHz, CDCl3): 192.7, 157.9, 142.9, 134.9, 132.7, 130.1, 130.1, 129.0, 128.7, 128.2, 126.8, 120.5, 111.4, 55.5; m/z (HR-ESI): theoretical mass (M+H)+: 239.1072; found : 239.1139.
3-(4-Bromophenyl)-1-(2-methoxyphenyl)prop-2-en-1-one (4d)
A solution of acetophenone (3.20 g, 0.027 mol) in ethanol (27 mL) was added gradually to an aqueous solution of 10% KOH (80 mL) at 0 °C. After stirring for 30 min, 4-bromobenzaldehyde (5.00 g, 0.027 mol) was added to the solution and stirred at 0 °C for an additional 15 min. The reaction mixture was then allowed to warm up to room temperature and stirred for 12h. The product precipitated from the reaction mixture over the course of the reaction. It was filtered and washed with water to afford the desired product as a white solid (8.05 g, 94 %). Both isomers (E) and (Z) were obtained. δH (500 MHz, CDCl3): 7.64 (dd, 1H, J= 7.6 Hz, J= 1.9 Hz), 7.58–7.45 (m, 6H), 7.38 (d, 1H, J= 15.8 Hz), 7.05 (td, 1H, J= 7.6 Hz. J= 0.9 Hz), 7.01 (d, 1H, J= 8.3 Hz), 3.91 (s, 3H); δC (125 MHz, CDCl3): 192.6, 158.1, 142.1, 141.6, 134.1, 133.1, 132.2, 132.1, 130.4, 129.7, 129.0, 127.5, 125.7, 124.4, 120.8, 111.6, 55.57; m/z (HR-ESI): theoretical mass (M+H)+: 317.0099; found : 317.0231.
(E)-3-(4-iodophenyl)-1-(2-methoxyphenyl)prop-2-en-1-one (4e)
A solution of 2-methoxyacetophenone (1.43 g, 0.011 mol) in ethanol (11 mL) was added gradually to an aqueous solution of 10% KOH (32 mL) at 0 °C. After stirring for 30 min, 4-iodobenzaldehyde (2.0 g, 0.009 mol) was added to the solution and stirred at 0 °C for an additional 15 min. The reaction mixture was then allowed to warm up to room temperature and stirred for 12h. The product precipitated from the reaction mixture over the course of the reaction. It was filtered and washed with water to afford the desired product as a white solid (2.8 g, 85%). δH (500MHz, CDCl3): 7.74 (d, 2H, J = 8.3 Hz), 7.64 (dd, 1H, J = 7.6 Hz, J = 1.7 Hz), 7.56 – 7.37 (m, 3H), 7.32 (d, 2H, J = 8.3 Hz ), 7.05 (td, 1H, J = 7.6 Hz, J = 1 Hz ), 7.01 (d, 1H, J = 8.3 Hz), 3.91 (s, 3H); δC (125MHz, CDCl3): 192.5, 158.1, 141.6, 138.0, 134.6, 133.1, 130.4, 129.8, 129.0, 127.8, 120.8, 111.6, 96.4, 55.7; m/z (HR-ESI): theoretical mass (M+H)+: 365.0038; found : 365.0098.
3-(4-Bromophenyl)-1-(2,4-dimethoxyphenyl)prop-2-en-1-one (4f)
47,48 A solution of acetophenone (1.95 g, 0.011 mol) in ethanol (11 mL) was added gradually to an aqueous solution of 10% KOH (32 mL) at 0 °C. After stirring for 30 min, 4-bromobenzaldehyde (2.00 g, 0.011 mol) was added to the solution and stirred at 0 °C for an additional 15 min. The reaction mixture was then allowed to warm up to room temperature and stirred for 12h. The product precipitated from the reaction mixture over the course of the reaction. It was filtered and washed with water to afford the desired product as a white solid (2.8 g, 73%). δH (500 MHz, CDCl3): 7.77 (d, 1H, J= 8.8 Hz), 7.44–7.62 (m, 6H), 6.57 (dd, 1H, J= 8.8 Hz, J= 2.4 Hz), 6.50 (d, 1H, J= 2.2 Hz), 3.88 (s, 3H), 3.91 (s, 3H); δC (125 MHz, CDCl3): 190.0, 164.3, 160.4, 140.3, 134.4, 132.9, 131.9, 129.6, 127.7, 124.0, 121.9, 105.2, 98.6, 55.7, 55.5; m/z (HR-ESI): theoretical mass (M+Li)+: 353.0365; found : 353.0384–355.0366 (Br isotope).
(E)-3-(4-Bromophenyl)-1-(4-(dimethylamino)-2-methoxyphenyl)prop-2-en-1-one (4g)
NaOH (776mg, 19.4mmol) was added to a solution of bromobenzaldehyde (1.44g, 7.8mmol) and 1-(4-dimethylamino-2-methoxyphenyl)ethanone (1.5g, 7.8mmol) in 20 mL MeOH. The reaction mixture was stirred at room temperature for 24h. The solid was collected on a filter and washed with cold MeOH to afford the title compound (2.5 g, 89%) as a light yellow solid. δH (500MHz, CDCl3): 7.81 (d, 1H, J = 8.9 Hz), 7.62 (m, 2H), 7.45 (m, 4H), 6.32 (dd, 1H, J = 8.9 Hz, J= 2.4 Hz ), 6.09 (d, 1H, J = 2.4 Hz ), 3.91 (s, 3H), 3.05 (s, 6H); δC (125MHz, CDCl3): 188.4, 161.3, 154.8, 139.0, 135.0, 133.3, 131.9, 129.5, 128.4, 123.5, 116.8, 104.7, 94.0, 55.5, 40.1; m/z (HR-ESI) theoretical mass (M+Li)+ C18H18BrLiNO2 366.0681; found 366.0584.
(E)-3-(4-bromophenyl)-1-(4-iodophenyl)prop-2-en-1-one (4h)
49 4-Bromobenzaldehyde (20.0 g, 0.11 mol), 4-iodoacetophenone (26.6 g, 0.11 mol) and sodium hydroxide (0.432 g, 0.01 mol) were dissolved in a 1:1 mixture of methanol:water (400 mL) and stirred under reflux for 12 h. After cooling to room temperature, the product was filtered and washed with methanol. The title product was obtained as a white solid (42.7 g, 96%). δH (500 MHz, CDCl3): 7.88 (d, 2H, J= 8.6 Hz), 7.75 (d, 2H, J= 15 Hz), 7.73 (d, 2H, J= 8.6 Hz), 7.51 (d, 2H, J= 8.6 Hz), 7.45 (d, 2H, J= 15 Hz); δC (125 MHz, CDCl3): 189.4, 143.9, 137.9, 137.2, 133.6, 132.3, 129.9, 129.8, 125.1, 121.9, 100.8; m/z (APCI) theoretical mass (M)+ 411.9; found 413.09.
General Procedure for the Synthesis of Michael Adduct 5 a – g
A solution of chalcone (1 mmol), nitromethane (20 mmol) and KOH (0.2 mmol) in ethanol (1 mL) was heated at 60 °C for 12 h. After cooling to room temperature, the solvent was removed in vacuo and the oily residue obtained was dissolved in ethyl acetate and washed with water (3 × 50 mL). The combined organic layers were washed with brine, dried over sodium sulfate and concentrated to give the target compound as a yellow/off white solid or oily residue (that solidifies upon standing overtime) in nearly quantitative yield. This product was used in the next without further purification.
1-(4-Iodophenyl)-3-(4-methoxyphenyl)-4-nitrobutan-1-one (5a)
δH (500 MHz, CDCl3): 7.82 (d, 2H, J= 8.3 Hz), 7.61 (d, 2H, J= 8.3 Hz), 7.18 (d, 2H, J= 8.8 Hz), 6.86 (d, 2H, J= 8.8 Hz), 4.78 (ddAB system, 1H, J= 12.5 Hz, J= 6.8 Hz), 4.64 (ddAB system, 1H, J=12.5 Hz, J=7.8 Hz), 4.15 (apparent quint, 1H, J=7.1 Hz), 3.77 (s, 3H), 3.37 (ddAB system, 2H, J= 7.3 Hz, J= 1.2 Hz); δC (125 MHz, CDCl3):196.3, 159.1, 138.0, 135.6, 130.7, 129.3, 128.4, 114.4, 101.6, 79.7, 55.2, 41.5, 38.5; m/z (APCI): theoretical mass (M)+: 425.01; found : 425.1.
3-(4-Bromophenyl)-1-(4-methoxyphenyl)-4-nitrobutan-1-one (5b)
δH (500 MHz, CDCl3): 7.89 (d, 2H, J= 8.8 Hz), 7.45 (d, 2H, J= 8.5 Hz), 7.17 (d, 2H, J= 8.5 Hz), 6.93 (d, 2H, J=8.8 Hz), 4.8 (ddAB system, 1H, J= 12.5 Hz, J= 6.1 Hz), 4.65 (ddAB system, 1H, J= 12.5 Hz, J= 8.3 Hz), 4.19 (apparent quint, 1H, J= 7.1 Hz), 3.87 (s, 3H), 3.37 (2 coalesced ddAB system, 2H, J= 17.6 Hz, J= 6.6 Hz); δC (125 MHz, CDCl3):194.9, 163.9, 138.2, 132.1, 130.3, 129.2, 129.2, 121.7, 113.9, 79.3, 55.5, 40.8, 38.8; m/z (APCI): theoretical mass (M)+: 377.02; found : 377.9-379.9 (Br isotope)
1-(2-Methoxyphenyl)-4-nitro-3-phenylbutan-1-one (5c)
50,45,46 δH (500 MHz, CDCl3): 7.62 (dd, 1H, J= 7.8 Hz, J= 1.9 Hz), 7.48 (td, 1H, J= 7.3 Hz, J= 1.9 Hz), 7.30–7.34 (m, 2H), 7.25–7.24 (m, 3H), 7.00–6.96 (m, 2H), 4.8 (ddAB system, 1H, J= 7.6 Hz, J= 1.2 Hz), 4.65 (ddAB system, 1H, J= 8.3 Hz, J= 1.2 Hz), 4.18 (apparent quint, 1H, J= 7.1 Hz), 3.89 (s, 3H), 3.46 (ddAB system, 2H, J= 2.9 Hz, J= 3.7 Hz); δC (125 MHz, CDCl3): 198.9, 158.6, 139.4, 134.0, 130.4, 128.8, 127.6, 127.5, 127.4, 120.8, 111.5, 79.9, 55.5, 46.7, 39.7; m/z (HR-ESI): theoretical mass (M+H)+: 300.1236; found : 300.1237.
3-(4-Bromophenyl)-1-(2-methoxyphenyl)-4-nitrobutan-1-one (5d)
δH (500 MHz, CDCl3): 7.64 (dd, 1H, J= 7.6 Hz, J= 1.7 Hz), 7.49 (td, 1H, J= 7.6 Hz, J= 1.7 Hz), 7.45 (d, 2H, J= 8.5 Hz), 7.14 (d, 2H, J= 8.5 Hz), 7.00–6.97 (m, 2H), 4.78 (ddAB system, 1H, J= 12.5 Hz, J= 6.3 Hz), 4.62 (ddAB system, 1H, J= 12.5 Hz, J= 8.3 Hz), 4.16 (apparent quint, 1H, J= 6.8 Hz), 3.91 (s, 3H), 3.42 (dd, 2H, J= 6.8 Hz, J= 1.7 Hz); δC (125 MHz, CDCl3): 158.5, 138.4, 134.1, 131.8, 130.3, 129.2, 127.0, 121.3, 120.7, 111.4, 79.4, 55.3, 46.4, 39.0 (signal for CO missing); m/z (HR-ESI): theoretical mass (M+Li)+: 384.0423; found : 384.0429-386.0451 (Br isotope).
3-(4-Iodophenyl)-1-(2-methoxyphenyl)-4-nitrobutan-1-one (5e)
δH (500MHz, CDCl3): 7.66 – 7.63 (m, 3H), 7.50 (2 touching dd, 1H, J = 7.3 Hz, J = 1.9 Hz ), 7.02 (d, 2H, J = 8.3 Hz), 6.98 (t, 2H, J = 8.3 Hz), 4.78 (m, 1H), 4.62 (m, 1H), 4.12 (m, 1H), 3.90 (s, 3H), 3.42 (m, 2H); δC (125MHz, CDCl3): 158.6, 139.2, 137.9, 134.2, 130.5, 129.5, 127.1, 120.8, 111.5, 93.1, 79.5, 55.5, 46.5, 39.3 (C for carbonyl group not seen); m/z (APCI): theoretical mass (M)+: 425.01; found : 425.1.
3-(4-Bromophenyl)-1-(2,4-dimethoxyphenyl)-4-nitrobutan-1-one (5f)
δH (500 MHz, CDCl3): 7.76 (d, 1H, J= 8.8 Hz), 7.44 (d, 2H, J= 8.5 Hz), 7.14 (d, 2H, J= 8.5 Hz), 6.51 (dd, 1H, J= 8.8 Hz, J= 2.4 Hz), 6.45 (d, 1H, J= 2.2 Hz), 4.77 (ddAB system, 1H, J= 12.4 Hz, J= 6.3 Hz), 4.61 (ddAB system, 1H, J= 12.4 Hz, J= 8.8 Hz), 4.13 (apparent quint, 1H, J= 7.1 Hz), 3.87 (s, 3H), 3.85 (s, 3H), 3.39 (ddAB system, 1H, J= 17.6 Hz, J= 6.6 Hz), 3.34 (ddAB system, 1H, J= 17.6 Hz, J= 7.6 Hz) ; δC (125 MHz, CDCl3): 196.0, 164.9, 160.8, 138.8, 132.9, 131.9, 129.2, 121.4, 119.9, 105.4, 98.2, 79.6, 55.5, 55.4, 46.4, 39.2; m/z (HR-ESI): theoretical mass (M+Li)+: 414.0528; found : 414.0537–416.0497 (Br isotope).
3-(4-bromophenyl)-1-(4-(dimethylamino)-2-methoxyphenyl)-4-nitrobutan-1-one (5g)
δH (500MHz, CDCl3): 7.75 (d, 1H, J = 8.9 Hz), 7.41 (m, 2H), 7.13 (m, 2H), 6.26 (dd, 1H, J = 8.9, J= 2.3 Hz), 6.00 (d, 1H, J = 2.3 Hz), 4.78 (ddAB system, 1H, J= 12.7 Hz, J= 5.8 Hz), 4.58 (ddAB system, 1H, J= 12.7 Hz, J= 9.1 Hz), 4.12 (m, 1H), 3.86 (s, 3H), 3.34 (ddAB system, 1H, J= 17.0 Hz, J= 6.3 Hz), 3.26 (ddAB system, 1H, J= 17.0 Hz, J= 8.0 Hz), 3.03 (s, 6H); δC (125MHz, CDCl3): 194.7, 161.5, 155.1, 139.3, 132.8, 131.9, 129.3, 121.2, 114.8, 104.6, 93.3, 79.7, 55.1, 46.2, 40.1, 39.4; m/z (APCI): theoretical mass (M+H)+: 421.06; found : 421.14.
2-(4-Bromophenyl)-4-(4-iodophenyl)-4-oxobutanenitrile (7)
6 mL of a solution of 10% sodium carbonate was added to a solution of (E)-3-(4-bromophenyl)-1-(4-iodophenyl)prop-2-en-1-one 4h (1.0 g, 2.42 mmol) and acetone cyanohydrin (0.5 g, 6.05 mmol) in acetone (20 mL). After refluxing for 12h, the reaction mixture allowed to cool to room temperature. The content of the flask was poured into an Erlenmeyer, and water was added to induce the separation of the product. Compound 7 was obtained as a yellow powder in 75 % yield (0.805 g). δH (500 MHz, CDCl3): 7.82 (d, 2H, J= 8.6 Hz), 7.59 (d, 2H, J= 8.6 Hz), 7.50 (d, 2H, J= 8.3 Hz), 7.30 (d, 2H, J= 8.3 Hz), 4.49 (apparent t, 1H, J= 7.6 Hz, J= 6.3 Hz), 3.65 (ddAB system, 1H, J= 18.1 Hz, J= 7.6 Hz), 3.44 (ddAB system, 1H, J= 18.1 Hz, J= 6.3 Hz); δC (125 MHz, CDCl3): 193.7, 138.1, 134.5, 133.9, 132.3, 129.6, 129.2, 122.4, 119.9, 98.8, 43.9, 31.2; m/z (APCI): theoretical mass (M)+: 440.07; found: 440.08.
General Procedure for the Synthesis of Azadipyrromethene 6 a – g
A 100 mL round-bottomed flask was charged with 5 (1 eq.), ammonium acetate (35 eq.), and butanol and heated under reflux for 24h. After cooling to room temperature, the solvent was concentrated to a quarter of its original volume, filtered, and the isolated solid was washed with ethanol to yield the desired product as a dark blue-black solid. The crude product was used in the next step without any further purification.
Azadipyrromethene 6a
δH (500 MHz, CDCl3): 7.70 (d, 4H, J=7.8 Hz), 7.48 (d, 4H, J=8.3 Hz), 7.26 (d, 2H, J=7.8 Hz), 7.04 (s, 2H), 6.92 (d, 4H, J=8.3 Hz), 6.77 (s, 2H), 3.96 (s, 6H); m/z (APCI): theoretical mass (M)+: 762.01; found: 762.10.
Azadipyrromethene 6b
m/z (HR-ESI): theoretical mass (M+H)+: 666.0392; found: 666.0399–668.0366 (Br isotope). Attempt to take nmr in CDCl3only gave broad signals probably because of aggregation.
Azadipyrromethene 6c
δH (500 MHz, CDCl3): 8.13 (d, 2H, J= 7.8 Hz), 8.07 (d, 4H, J= 7.3 Hz), 7.44–7.33 (m, 10H), 7.08 (t, 2H, J= 7.3 Hz), 7.04 (d, 2H, J= 8.3 Hz), 3.96 (s, 6H); δC (125 MHz, CDCl3): 158.0, 153.5, 148.5, 141.0, 134.1, 130.9, 129.1, 128.1, 127.9, 127.5, 121.4, 121.0, 117.8, 111.8, 55.9; m/z (HR-ESI): theoretical mass (M+H)+: 510.2182; found: 510.2174.
Azadipyrromethene 6d
δH (500 MHz, CDCl3): 8.08 (d, 2H, J= 7.3 Hz), 7.87 (d, 4H, J= 8.5 Hz), 7.51 (d, 4H, J= 8.8 Hz), 7.41 (dt, 2H, J= 7.3 Hz, J= 1.7 Hz), 7.28 (s, 2H), 7.09–7.02 (m, 4H), 3.95 (s, 6H); δC (125 MHz, CDCl3): 157.9, 153.5, 148.2, 139.5, 132.9, 131.1, 130.4, 129.1, 121.7, 121.1, 121.0, 117.9, 117.8, 111.7, 55.9; m/z (HR-ESI): theoretical mass (M+H)+: 666.0392; found: 666.0399–668.0366 (Br isotope).
Azadipyrromethene 6e
δH (500 MHz, CDCl3): 8.09 (dd, 2H, J= 7.6 Hz, J= 1.7 Hz), 7.75 (apparent doublet of quart, 8H, J= 8.5 Hz, J= 2.2 Hz), 7.41 (dt, 2H, J= 7.3 Hz, J= 1.7 Hz), 7.30 (s, 2H), 7.08 (dt, 2H, J= 7.6 Hz, J= 1.0 Hz), 7.04 (d, 2H, J= 8.5 Hz), 3.95 (s, 6H); δC (125 MHz, CDCl3): 158.1, 153.7, 148.4, 139.8, 137.3, 133.5, 131.3, 130.7, 129.2, 121.2, 121.1, 117.9, 111.8, 93.6, 55.9; m/z (APCI): theoretical mass (M)+: 762.01; found: 762.10.
Azadipyrromethene 6f
δH (500 MHz, CDCl3): 8.03 (d, 2H, J=8.5 Hz), 7.88 (d, 4H, J=8.3 Hz), 7.51 (d, 4H, J=8.3 Hz), 7.21 (s, 2H), 6.60 (dd, 2H, J=8.5 Hz, J=2.4 Hz), 6.56 (d, 2H, J=2.4 Hz), 3.95 (s, 6H), 3.90 (s, 6H); δC (125 MHz, CDCl3): 162.5, 159.4, 152.9, 148.1, 139.3, 133.2, 131.2, 130.5, 130.3, 121.6, 117.2, 114.5, 105.9, 98.8, 56.0, 55.6; m/z (HR-ESI): theoretical mass (M+H)+: 726.0603; found: 726.0587–728.0555 (Br isotope).
Azadipyrromethene 6g
Not isolated, used in the next step without purification and characterization; m/z (APCI) : theoretical mass (M+H)+: 752.12; found: 754.33
Azadipyrromethene 6h
A 100 mL round-bottomed flask was charged with 7 (3.0 g, 6.2 mmol.), hydroxylamine hydrochloride (15.0 g, 217 mmol), and methanol (30 mL) and heated at 100 ° for 3 d. After cooling to room temperature, the solvent was concentrated to a quarter of its original volume, filtered, and the isolated solid was washed with water then methanol to yield the desired product as a light blue-green solid (1.68 g, 32 %). δH (500 MHz, CDCl3): 7.88 (d, 4H, J= 8.6 Hz), 7.72 (d, 4H, J= 8.6 Hz), 7.64 (d, 4H, J= 8.6 Hz), 7.54 (d, 4H, J= 8.6 Hz), 7.49 (s, 2H), 3.96 (s, 6H); δC (125 MHz, CDCl3): 162.9, 154.2, 138.7, 135.4, 132.4, 131.3, 130.7, 130.5, 128.3, 126.1, 125.9, 98.3; m/z (APCI) : theoretical mass (M+H)+: 859.13; found: 859.98
General Procedure for the Synthesis of AzaBODIPY 1 a – g
A flame dried Schlenk flask was charged with the azapyrromethene (1 eq.) and flushed with nitrogen. Dry dichloromethane and dry diisopropylethylamine (11 eq.) were then added. The solution was stirred at 25 °C for 15 min, then distilled BF3•OEt2 (15.6 eq.) was added. After stirring at 25 °C for 24h, the mixture was washed with water, and the organic layer dried over sodium sulfate and concentrated in vacuo to give the target compound.
AzaBODIPY 1a
δH (500 MHz, CDCl3): 8.04 (d, 4H, J=8.8 Hz), 7.83 (d, 4H, J=8.5 Hz), 7.75 (d, 4H, J=8.8 Hz), 7.00 (d, 4H, J=8.5 Hz), 6.89 (s, 2H), 3.91 (s, 6H); δC (125 MHz, CDCl3): 161.1, 157.9, 145.6, 144.1, 137.8, 131.1, 130.9, 130.8, 125.1, 117.2, 114.3, 98.0, 55.4; δB (128 MHz, CDCl3): 0.84 (t, 1B, J B–F = 31 Hz); δF (376 MHz, CDCl3): −134.89 (q, 2F, J B–F = 31 Hz); MS (HR-maldi) m/z calcd for (M+H)+ C34H25BI2F2N3O2:810.0097; found :810.0071; λmax abs (PhMe)/nm 680 (ε/dm3mol−1cm−1 108 996); λmax emis (PhMe)/nm 711; Φ = 0.18 in 1% pyridine in toluene.
AzaBODIPY 1b
δH (500 MHz, CDCl3): 8.08 (d, 4H, J=8.8 Hz), 7.91 (d, 4H, J=8.5 Hz), 7.60 (d, 4H, J=8.5 Hz), 7.04 (s, 2H), 7.02 (d, 4H, J=8.8 Hz), 3.90 (s, 6H); δC not taken due to poor solubility of compound; δB (128 MHz, CDCl3): 1.05 (t, 1B, J B–F = 32 Hz); δF (376 MHz, CDCl3): −132.36 (q, 2F, J B–F = 32 Hz); MS (HR-MALDI) m/z calcd for (M)+ C34H24BBr2F2N3O2: 715.0278; found: 715.0285; λmax abs (PhMe)/nm 702 (ε/dm3mol−1cm−1 84 118); λmax emis (PhMe)/nm 731; Φ = 0.42 in 1% pyridine in toluene.
AzaBODIPY 1c
δH (500 MHz, CDCl3): 8.07–8.08 (m, 4H), 7.89 (dd, 2H, J= 7.8 Hz, J= 1.7 Hz), 7.48 – 7.38 (m, 8H), 7.04 (dd, 2H, J= 15.1 Hz, J= 0.9 Hz), 7.03 (s, 2H), 6.97 (d, 2H, J= 8.3 Hz), 3.80 (s, 6H); δC (125 MHz, CDCl3): 158.0, 156.7, 144.9, 142.8, 132.6, 131.7, 129.3, 129.2, 129.1, 128.5, 121.3, 121.1, 120.5, 111.1, 55.9; δB (128 MHz, CDCl3): 0.558 (t, 1B, J B–F = 30 Hz); δF (376 MHz, CDCl3): −133.37 (q, 2F, J B–F = 30 Hz); MS (HR-ESI) m/z calcd (M+H)+ C34H27BF2N3O2: 558.2164; found : 558.2166; λmax abs (PhMe)/nm 640 (ε = 73 858 dm3mol−1cm−1); λmax emis (PhMe)/nm 688; Φ = 0.07 in 1% pyridine in toluene.
AzaBODIPY 1d
δH (500 MHz, CDCl3): 7.90 (d, 4H, J= 8.8 Hz), 7.87 (dd, 2H, J= 7.6 Hz, J= 1.7 Hz), 7.60 (d, 4H, J= 8.8 Hz), 7.41 (dt, 2H, J= 7.6 Hz, J= 1.7 Hz), 7.05–7.01 (m, 4H), 6.97 (d, 2H, J= 7.8 Hz), 3.86 (s, 6H); δC (125 MHz, CDCl3): 158.0, 157.0, 144.7, 141.4, 131.9, 131.8, 131.7, 131.4, 130.6, 123.7, 121.4, 120.7, 120.5, 111.0, 55.8; δB (128 MHz, CDCl3): 0.51 (t, 1B, J B–F = 30 Hz); δF (376 MHz, CDCl3): −137.46 (q, 2F, J B–F = 30 Hz); MS (HR-MALDI) m/z calcd for (M+H)+ C34H25BBr2F2N3O2: 714.0375; found : 714.0363–715.0370 (Br isotope); λmax abs (PhMe)/nm 650 (ε = 55 014 dm3mol−1cm−1); λmax emis (PhMe)/nm 694; Φ = 0.10 in 1% pyridine in toluene.
AzaBODIPY 1e
δH (500 MHz, CDCl3): 7.86 (dd, 2H, J = 7.8 Hz, J = 1.7 Hz), 7.79 (apparent quart, 8H, J = 8.3 Hz), 7.40 (td, 2H, J = 9 Hz, J = 1.7 Hz), 7.03 (td, 2H, J = 7.8 Hz, J = 1 Hz), 7.02 (s, 2H), 6.97 (d, 2H, J = 8.3 Hz), 3.85 (s, 6H); δC (125MHz, CDCl3): 158.3, 157.3, 141.9, 138.0, 132.3, 132.2, 131.9, 130.9, 121.7, 121.6, 121.0, 120.8, 111.3, 96.1, 56.2; δF (376MHz, CDCl3): 45.45 (q, 2F, J = 31.5 Hz). MS (HR-ESI) m/z calcd for (M+Li)+ C34H24BI2F2N3O2 816.0179; found 816.0019; λmax abs (PhMe)/nm 653 (ε/dm3mol−1cm−1 71 600); λmax emis (PhMe)/nm 701; Φ = 0.10 in 1% pyridine in toluene.
AzaBODIPY 1f
δH (500 MHz, CDCl3): 7.96 (d, 2H, J=8.8 Hz), 7.90 (dd, 4H, J=1.8 Hz, J= 6.7 Hz), 7.58 (dd, 4H, J=1.8 Hz, J= 6.7 Hz), 7.06 (s, 2H), 6.58 (dd, 2H, J=8.8 Hz, J=2.4 Hz), 6.51 (d, 2H, J=2.4 Hz), 3.86 (s, 6H), 3.85 (s, 6H); δC (125 MHz, CDCl3): 163.1, 159.8, 155.9, 144.6, 140.6, 133.2, 131.7, 131.6, 130.5, 123.4, 121.5, 113.8, 104.8, 98.8, 55.8, 55.5; δB (128 MHz, CDCl3): 0.71 (t, 1B, J B–F = 30 Hz); δF (376 MHz, CDCl3): −170.14 (q, 2F, J B–F = 30 Hz); MS (HR-ESI) m/z calcd for (M+H)+ C36H29BBr2F2N3O4 774.0586; found : 774.0573–776.0550 (Br isotope); λmax abs (PhMe)/nm 692 (ε/dm3mol−1cm−1 66 200); λmax emis (PhMe)/nm 738; Φ = 0.20 in 1% pyridine in toluene.
AzaBODIPY 1g
(from butyrophenone 5g ). NH4OAc (17.4g, 0.23mol) was added to the solution of 5g (2.73g, 6.5mmol) in 50mL n-BuOH. The reaction mixture was heated at reflux for 24h. After cooling to room temperature, the solution was concentrated to half its original volume. The solid was filtered and washed with cold EtOH to afford 6g (1.0 g, 41%) as a green solid which was used in the next step without further purification. DIEA (1.8 ml, 10.3 mmol) was added to the solution of 6g (970mg, 1.3 mmol) in 30mL dry DCM. The solution was stirred at room temperature for 15min and BF3•OEt2 (1.6 ml, 12.9mmol) was added. After stirring at room temperature for 24h, the mixture was washed with water (1 × 30 mL) and brine (1 × 30 mL). The organic layer was dried over Na2SO4 and concentrated under reduced pressure. The crude product was purified by flash chromatography on silica gel eluting with 2 : 3 EtOAc-hexane to afford 1g (867mg, 84%) as a purple solid. δH (500MHz, CDCl3): 8.03 (d, 2H, J = 9.2 Hz), 7.90 (d, 4H, J = 8.5 Hz), 7.53 (d, 4H, J = 8.5 Hz), 7.14 (s, 2H), 6.34 (d, 2H, J = 9.1 Hz), 6.13 (d, 2H, J = 2.3 Hz), 3.84 (s, 6H), 3.01 (s, 12H); δC (125MHz, CDCl3): 160.2, 154.5, 153.2, 144.4, 138.5, 133.4, 132.2, 131.4, 130.4, 122.6, 121.5, 109.3, 104.8, 94.8, 55.6, 40.1; δF (376MHz, CDCl3): 45.45 (q, 2F, J = 31.5 Hz). MS (HR-ESI) m/z calcd for (M+H)+ C38H35BBr2F2N5O2 800.1219; found 800.1275; λmax abs (PhMe)/ nm 798 (ε/dm3mol−1cm−1 68 610); λmax emis (PhMe)/ nm 830; Φ = 0.07 in 1% pyridine in toluene.
Compound 11
A flame dried Schlenk tube was charged with ethynyl fluorescein diacetate 923,33 (0.24 g, 0.54 mmol), aza-BODIPY 1a (0.2 g, 0.25 mmol), Pd (PPh3)4 (17 mg, 0.025 mmol), CuI (2.4 mg, 0.012 mmol), THF (15 mL) and triethylamine (0.4 mL, 2.5 mmol). The reaction mixture was deoxygenated 3 times using the freeze-pump-thaw technique. It was then stirred at room temperature for 28h. The solvent was then removed in vacuo, and the residue purified by flash chromatography (dry loading). The column was first eluted with 50% Hexanes:CH2Cl2 to 100% CH2Cl2, then 2% ethyl acetate:CH2Cl2to get the mono-coupling product and finally with 5% ethyl acetate:CH2Cl2 to get the bis-coupling product. The bis-coupling product coeluted with the ethynyl fluorescein bis acetate homo-coupling product and is obtained in a pure form after a second flash chromatography eluted with 50% ethyl acetate:hexanes (107 mg, 30%). δH (500 MHz, CDCl3): 8.18 (s, 2H), 8.10 (d, 4H, J= 8.1 Hz), 8.06 (d, 4H, J= 8.3 Hz), 7.83 (d, 2H, J= 8.1 Hz), 7.66 (d, 4H, J= 8.3 Hz), 7.19 (d, 2H, J= 8.1 Hz), 7.11 (d, 4H, J= 2.2 Hz), 7.02 – 6.99 (m, 6H), 6.88 – 6.83 (m, 8H), 3.91 (s, 6H), 2.32 (s, 12H); δC (125 MHz, CDCl3): 168.8, 168.2, 161.1, 157.7, 152.2, 152.1, 151.5, 145.9, 143.9, 138.3, 131.9 (2C), 130.9, 129.6, 128.9, 128.2, 126.6, 125.5, 125.1, 124.6, 124.2, 117.8, 117.6, 115.9, 114.2, 110.5, 91.7, 89.9, 81.8, 55.4, 21.1; δB (128 MHz, CDCl3): 0.97 (t, 1B, J B–F = 31 Hz); δF (376 MHz, CDCl3): −130.72 (q, 2F, J B–F = 31 Hz); m/z (HR-MALDI) calculated mass for C86H54BF2N3O16 (M+H)+: 1434.3643; found: 1434.3638.
Compound 2
A solution of 11 in a mixture of methanol and THF (1:1) was treated with baked potassium carbonate. The reaction mixture was stirred at room temperature for 2 h. When the reaction was complete according to TLC, the solvent was removed in vacuo to afford the pure product. δH (500 MHz, CD3OD): 8.30 (d, 4H, J=8.5 Hz), 8.18 (d, 2H, J=1.7 Hz), 8.15 (d, 4H, J=8.8 Hz), 7.72 (dd, 2H, J=1.7 Hz, J=7.8 Hz), 7.66 (d, 4H, J=8.5 Hz), 7.26 (s, 2H), 7.24 (d, 2H, J=7.8 Hz), 7.06 (d, 4H, J=9.3 Hz), 7.05 (d, 4H, J=8.8 Hz), 6.54 (dd, 4H, J=2.2 Hz, J= 9.3 Hz), 6.49 (d, 4H, J=2.2 Hz), 3.90 (s, 6H); δC (125 MHz, CD3OD): 182.6, 173.2, 162.6, 160.3, 159.9, 148.3, 144.6, 142.3, 134.9, 133.7, 133.6, 132.8, 132.5, 132.2, 132.1, 131.4, 131.3, 126.6, 126.2, 125.1, 124.1, 119.4, 115.3, 114.8, 113.1, 104.4, 92.1, 91.4, 55.9; δB (128 MHz, CD3OD): 5.34; δF (376 MHz, CD3OD): −187.46; m/z (HR-MALDI) calculated for C78H46BF2N3O12: 1265.3143; found: 1267.3880; λmax abs (50% EtOH:H2O)/nm 500 (fluorescein) and 698 (AzaBODIPY); λmax emis (50% EtOH:H2O)/nm 530 and 733 when excited at 498 nm; λmax emis (50% EtOH:H2O)/nm 733 when excited at 690 nm.
Compound 10
BBr3 (1.0 mol solution in hexane) (4.9 mmol, 1.9 mL) was added to a solution of iodoaza-BODIPY 1a (0.49 mmol, 0.4 g) in dry CH2Cl2 (200 mL) at 0 °C and stirred at 25 °C for 2h. Dichloromethane was removed afterwards EtOAc and H2O (1:1, 300 mL) were added. The organic layer was separated, dried over anhydrous MgSO4, filtered and finally THF (10 X 50 mL) was used to dissolve sticky material during filtration. The filtrate was concentrated and dried under vacuum. The crude product was used in next step without further purification.
NaH (60% suspension in paraffin oil) (4.9 mmol, 290 mg) was added to a solution of the crude unprotected aza-BODIPY [(49.0 mmol, 383 mg)(assumed quantity)] and methyl bromoacetate (1.86 mmol, 1.47 mL) in dry THF (150 mL). After stirring for 12 h at 25 °C, flash silica gel was added to quench the NaH as well as to make slurry for column chromatography. The solvents were removed and chromatographed (SiO2; 50 % hexanes/EtOAc) to afford 10 (79 mg) in 17 % yield as dark blue amorphous solid: Rf = 0.30 (50 % hexanes/EtOAc); 1H NMR (500 MHz, CDCl3) δ 7.93 (d, 4H, J = 9.0 Hz), 7.77 (d, 4H, J = 9.0 Hz), 7.68 (d, 4H, J = 9.0 Hz), 6.92 (d, 4H, J = 9.0 Hz), 6.82 (s, 2H), 4.66 (s, 2H), 3.85 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 169.2, 159.2, 158.1, 143.7, 138.1, 131.1(3C), 126.2, 117.7, 115.0, 98.4, 65.4, 52.6; MS (HR-MALDI) calcd for C38H28BF2I2N3O6 925.0129 found 925.6324.
Compound 12
Ethynyl fluorescein diacetate 9 (0.43 mmol, 0.187 g) was added to a solution of aza-BODIPY 10 (0.085 mmol, 0.79 g), PdCl2 (PPh3)2 (25 mol %, 15 mg), CuI (5 mol %, 1 mg) and Et3N (1 mL) in dry THF (30 mL). The resulting solutition was freeze thawed (−78 °C) for three times (every time purged with Ar). After stirring for 18 h at 40 °C, the solvents were removed and chromatographed (SiO2; 12% EtOAc/CH2Cl2) to afford 12 (53 mg) in 40 % yield as dark brown solid; Rf = 0.30 (6% EtOAc/CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 8.18 (s, 2H), 8.08 (d, 4H, J = 8.5 Hz), 8.02 (d, 4H, J = 8.5 Hz), 7.84–7.80 (m, 2H), 7.65 (d, 4H, J = 8.5 Hz), 7.18 (d, 2H, J = 7.5 Hz), 7.13 (d, 4H, J = 2.0 Hz), 6.99–7.01 (m, 6H), 6.84–6.85 (m, 8H), 4.73 (s, 4H), 3.85 (s, 6H), 2.31 (s, 12H); 13C NMR (125 MHz, CDCl3) δ 179.3, 169.1, 168.5, 159.45, 158.2, 152.5 (2C), 151.8, 146.2, 143.9, 138.7, 132.3, 132.1, 131.3, 129.3 (2C), 129.3, 128.5, 126.9, 126.4, 125.8, 125.1, 124.6, 118.2, 116.3, 115.2, 110.8, 92.0, 90.4, 82.1, 65.5, 52.7, 21.4; IR (neat); ν (cm−1); 1684; MS (HR-MALDI) calcd for C90H58BF2N2O20 1549.3637 found (M-F) 1530.1506.
Compound 3
Solid TMSOK (0.37 mmol, 48 mg) was added to a solution of compound 12 (0.031 mmol, 48 mg) in THF (20 mL). After stirring for 2 h 30 min. at 25 °C, the reaction mixture was neutralized with 2N HCl (1 mL) and subsequently added water (5 mL). After stirring for 5 min., THF was removed and aqueous layer was extracted with 25% iPrOH in CH2Cl2 (pH = 2–3). To obtain pure product in 90 % yield (38 mg) solvents were removed and was dried under vacuum. MS (HR-MALDI) calcd for C80H46BF2N3O6 1353.2939 found 1354.10212 (M+H); Pure compound 3 (< 1 mg) was dissolved in 1:1 (CH3CN:H2O) (2 mL) and subjected to reverse phase analytical HPLC {C18, 5:95 (CH3CN:H2O) and 0.1 % TFA , tR= 16.5 min}.
Supplementary Material
Supporting Information Available. General experimental conditions, characterization data for compounds 1 – 12, X-ray crystallography of 1a. This material is available free of charge via the internet at http://pubs.acs.org.
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Weijer CJ. Science. 2003;300:96–100. doi: 10.1126/science.1082830. [DOI] [PubMed] [Google Scholar]
- 2.Stephens DJ, Allan VJ. Science. 2003;300:82–86. doi: 10.1126/science.1082160. [DOI] [PubMed] [Google Scholar]
- 3.Lippincott-Schwartz J, Patterson GH. Science. 2003;300:87–91. doi: 10.1126/science.1082520. [DOI] [PubMed] [Google Scholar]
- 4.Rieder CL, Khodjakov A. Science. 2003;300:91–96. doi: 10.1126/science.1082177. [DOI] [PubMed] [Google Scholar]
- 5.Frangioni JV. Curr. Opin. Chem. Biol. 2003;7:626–634. doi: 10.1016/j.cbpa.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Mishra A, Behera RK, Behera PK, Mishra BK, Behera GB. Chem. Rev. 2000;100:1973–2011. doi: 10.1021/cr990402t. [DOI] [PubMed] [Google Scholar]
- 7.Narayanan N, Little G, Raghavachari R, Patonay G. Proc. SPIE-Int. Soc. Opt. Eng; 1995. pp. 6–15. [Google Scholar]
- 8.McKeown NB. Chem. Ind. 1999:92–98. [Google Scholar]
- 9.Halik M, Hartmann H. Chem. Eur. J. 1999;5:2511–2517. [Google Scholar]
- 10.Kenworthy AK. Methods. 2001;24:289–296. doi: 10.1006/meth.2001.1189. [DOI] [PubMed] [Google Scholar]
- 11.Loudet A, Burgess K. Chem. Rev. 2007;107:4891–4932. doi: 10.1021/cr078381n. [DOI] [PubMed] [Google Scholar]
- 12.Rogers MAT. J. Chem. Soc. 1943:590–596. [Google Scholar]
- 13.Davies WH, Rogers MAT. J. Chem. Soc. 1944:126–131. [Google Scholar]
- 14.Rogers MAT. Nature (London, United Kingdom) 1943;151:504. [Google Scholar]
- 15.Rogers MAT. Azadipyrromethines, A New Class of Dyes. 2382914. US. 1945
- 16.Allik TH, Hermes RE, Sathyamoorthi G, Boyer JH. Proc. SPIE-Int. Soc. Opt. Eng; 1994. pp. 240–248. [Google Scholar]
- 17.Zhao W, Carreira EM. Angew. Chem. Int. Ed. 2005;44:1677–1679. doi: 10.1002/anie.200461868. [DOI] [PubMed] [Google Scholar]
- 18.Zhao W, Carreira EM. Chem. -Eur. J. 2006;12:7254–7263. doi: 10.1002/chem.200600527. [DOI] [PubMed] [Google Scholar]
- 19.Loudet A, Bandichhor R, Burgess K. Angew. Chem. Int. Ed. 2007 submitted. [Google Scholar]
- 20.McDonnell SO, O'Shea DF. Org. Lett. 2006;8:3493–3496. doi: 10.1021/ol061171x. [DOI] [PubMed] [Google Scholar]
- 21.Burgess K, Burghart A, Chen J, Wan C-W. Proc. SPIE-Int. Soc. Opt. Eng. San Jose, CA: 2000. New Chemistry of BODIPY Dyes, and BODIPY Dye Cassettes Featuring Through-Bond Energy Transfer. [Google Scholar]
- 22.Burghart A, Thoresen LH, Chen J, Burgess K, Bergström F, Johansson LB-A. Chem. Commun. 2000:2203–2204. [Google Scholar]
- 23.Jiao G-S, Thoresen LH, Kim TG, Haaland WC, Gao F, Topp MR, Hochstrasser RM, Metzker ML, Burgess K. Chem. Eur. J. 2006;12:7616–7626. doi: 10.1002/chem.200600197. [DOI] [PubMed] [Google Scholar]
- 24.Gorman A, Killoran J, O'Shea C, Kenna T, Gallagher WM, O'Shea DF. J. Am. Chem. Soc. 2004;126:10619–10631. doi: 10.1021/ja047649e. [DOI] [PubMed] [Google Scholar]
- 25.Rio G, Masure D. Bull. Soc. Chim. Fr. 1972:4604–4610. [Google Scholar]
- 26.Miura T, Urano Y, Tanaka K, Nagano T, Ohkubo K, Fukuzumi S. J. Am. Chem. Soc. 2003;125:8666–8671. doi: 10.1021/ja035282s. [DOI] [PubMed] [Google Scholar]
- 27.Gabe Y, Urano Y, Kikuchi K, Kojima H, Nagano T. J. Am. Chem. Soc. 2004;126:3357–3367. doi: 10.1021/ja037944j. [DOI] [PubMed] [Google Scholar]
- 28.Ueno T, Urano Y, Setsukinai K-i, Takakusa H, Kojima H, Kikuchi K, Nagano T. J. Am. Chem. Soc. 2004;126:14079–14085. doi: 10.1021/ja048241k. [DOI] [PubMed] [Google Scholar]
- 29.Urano Y, Kamiya M, Kanda K, Ueno T, Hirose K, Nagano T. J. Am. Chem. Soc. 2005;127:4888–4894. doi: 10.1021/ja043919h. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka K, Miura T, Umezawa N, Urano Y, Kikuchi K, Higuchi T, Nagano T. J. Am. Chem. Soc. 2001;123:2530–2536. doi: 10.1021/ja0035708. [DOI] [PubMed] [Google Scholar]
- 31.Gabe Y, Ueno T, Urano Y, Kojima H, Nagano T. Anal. Biochem. 2006;386:621–626. doi: 10.1007/s00216-006-0587-y. [DOI] [PubMed] [Google Scholar]
- 32.Sunahara H, Urano Y, Kojima H, Nagano T. J. Am. Chem. Soc. 2007;129:5597–5604. doi: 10.1021/ja068551y. [DOI] [PubMed] [Google Scholar]
- 33.Jiao G-S, Han JW, Burgess K. J. Org. Chem. 2003;68:8264–8267. doi: 10.1021/jo034724f. [DOI] [PubMed] [Google Scholar]
- 34.Sonogashira K, Tohda Y, Hagihara N. Tetrahedron Lett. 1975:4467–4470. [Google Scholar]
- 35.Cox RJ, Durston J, Roper DI. J. Chem. Soc., Perkin Trans. 2002;1:1029–1035. [Google Scholar]
- 36.Molecular Probes. Invitrogen Corporation; 2006. http://probes.invitrogen.com. [Google Scholar]
- 37.Zanker V, Peter W. Chem. Ber. 1958;91:572–580. [Google Scholar]
- 38.Klonis N, Sawyer WH. J. Fluoresc. 1996;6:147–157. doi: 10.1007/BF00732054. [DOI] [PubMed] [Google Scholar]
- 39.Mishra VN, Datt N. Indian J. Chem., Sect. A: Inorg., Phys., Theor. Anal. Chem. 1985;24A:597–600. [Google Scholar]
- 40.Mathur AK, Agarwal C, Pangtey BS, Singh A, Gupta BN. Int. J. Cos. Sci. 1988;10:213–218. doi: 10.1111/j.1467-2494.1988.tb00019.x. [DOI] [PubMed] [Google Scholar]
- 41.Buu-Hoi NP, Sy M. Bull. Soc. Chim. Fr. 1958:219–220. [Google Scholar]
- 42.Sherif S. Can. J. Chem. 1961;39:2563–2571. [Google Scholar]
- 43.Barnes RP, Goodwin TC, Jr, Cotten TW., Jr J. Am. Chem. Soc. 1947;69:3135–3138. [Google Scholar]
- 44.Shadakshari U, Nayak SK. Tetrahedron. 2001;57:8185–8188. [Google Scholar]
- 45.Bloom SM, Garcia PP. Novel Dipyrrylmethene Dyes. 3691161. US. 1972
- 46.Bloom SM, Garcia PP. Intermediates for Dipyrrylmethene Dyes. 13691161. US. 1975
- 47.Liu M, Wilairat P, Go M-L. J. Med. Chem. 2001;44:4443–4452. doi: 10.1021/jm0101747. [DOI] [PubMed] [Google Scholar]
- 48.Liu M, Wilairat P, Go M-L. J. Med. Chem. 2002;45:1735. [Google Scholar]
- 49.Southard GE, Murray GM. Process for preparing vinyl-substituted beta-diketones. 20060069288. The Johns Hopkins University; USA: US. 2006
- 50.Dixon DJ, Richardson RD. Synlett. 2006:81–85. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available. General experimental conditions, characterization data for compounds 1 – 12, X-ray crystallography of 1a. This material is available free of charge via the internet at http://pubs.acs.org.