

Abstract
The small molecule PAP-1 (5-(4-phenoxybutoxy)psoralen) is a selective blocker of the voltage-gated potassium channel Kv1.3 that is highly expressed in cell membranes of activated effector memory T-cells (TEM). The blockade of Kv1.3 results in membrane depolarization and inhibition of TEM cell proliferation and function. In this study, the in vitro effects of PAP-1 on rhesus macaques (RM) T cells and the in vivo toxicity and pharmacokinetics (PK) were examined in RM with the ultimate aim of utilizing PAP-1 to define the role of TEM in RM infected with simian immunodeficiency virus (SIV). Electrophysiological studies on RM T-cells revealed a Kv1.3 expression pattern similar to that in human T-cells. Thus, PAP-1 effectively suppressed RM TEM cell proliferation. Intravenously administered PAP-1 showed a half-life of 6.4 hrs and the volume of distribution suggested that it is distributed extensively into extravascular compartments. Orally administered PAP-1 was efficiently absorbed and plasma concentrations in RM undergoing a 30-day chronic dosing study indicated that PAP-1 levels that are suppressive to TEM cells in vitro can be achieved and maintained in vivo at a non-toxic dose. PAP-1 selectively inhibited TEM function in vivo as indicated by a modest reactivation of cytomegalovirus (CMV) replication. Immunization of these chronically-treated RM with the live influenza A/PR8 virus suggest that the development of an in vivo flu-specific central memory response was unaffected by PAP-1. These RM remained disease-free during the entire course of the PAP-1 study. Collectively these data provide a rational basis for future studies with PAP-1 in SIV-infected RM.
Keywords: effector memory T-cells, SIV, PAP-1, K+ channel blockers
INTRODUCTION
Voltage-gated potassium channels (Kv) regulate several physiological functions of lymphocytes including membrane potassium permeability, calcium influx, cytokine production, clonal expansion and cell death (1–4). While Kv1.3 channels are found in all T and B cell subsets in the resting state, their expression is markedly upregulated in activated effector memory T cells (TEMs) and Ig class-switched memory B cells from ~250 to ~1500 channels per cell (5, 6). Naïve and central memory (TCM) T cells as well as IgD+ B cells in contrast increase expression of another potassium channel, the calcium-activated channel KCa3.1 upon cellular activation (1). This differential expression of K+ channels provides an opportunity for selective inhibition of distinct cell subsets. Indeed, the KCa3.1-specific blocker TRAM-34 has been shown to preferentially suppress the proliferation of pre-activated naïve and TCM cells and IgD+ B cells, while Kv1.3 specific blockers suppress the proliferation of activated human TEM cells but not of naïve and TCM cells when stimulated with anti-CD3 monoclonal antibody, antigen or anti-CD40 antibody in vitro (5–8). Manipulation of Kv1.3 expression or function could therefore have therapeutic potential for the treatment of diseases in which TEM cells and/or class-switched memory B cells contribute to pathogenesis. In proof of this hypothesis, recent studies focusing on TEM-mediated autoimmune diseases have shown that Kv1.3-specific peptides such as ShK and its derivative ShK(L5) effectively relieve experimental autoimmune encephalomyelitis and pristane-induced arthritis in rats (9–11). However, since peptides administered in vivo run the risk of inducing an immune response, particularly if chronic dosing is required, it was reasoned that a non-immunogenic small molecule Kv1.3 channel blocker like the recently reported PAP-1 would be more practical.
PAP-1 is a synthetic derivate of the natural product 5-methoxypsoralen and has been shown to exhibit a 23 to 125-fold selectivity for Kv1.3 over other members of the Kv1 family, effectively blocking Kv1.3 with an EC50 of 2 nM and a stoichiometry of 2 inhibitor molecules per channel (7). PAP-1 has been shown to inhibit the in vitro proliferation of human CCR7- TEM cells significantly more potently than the proliferation of naïve and TCM cells and to effectively suppress delayed-type hypersensitivity (DTH) in rats, a response that is mediated primarily by skin-homing CD4+ TEM cells (7). In addition, PAP-1 delayed diabetes onset and reduced diabetes incidence in spontaneously diabetic BB/Worchester rats at doses that did not exhibit any toxicity (11). Thus, the selective effect of PAP-1 for TEM cells does not limit its application to only autoimmune diseases but it also has the potential for the treatment of other ailments in which TEM cells play a major pathogenic role.
Activated memory CD4+ T cells have been shown to be the primary targets for HIV/SIV infection resulting in chronic T cell hyperactivation that contributes to CD4+ T-cell apoptosis, early immune dysfunction and eventual exhaustion of the virus-specific immune response leading to disease and death (12–17). These antigen-specific memory CD4+ T cells are theoretically derived from antigen-activated naïve T cells that differentiate into central and effector T cells (18, 19). SIV infection of non-human primates such as rhesus macaques (RM, Macaca mulatta) result in infection and a disease course remarkably similar to human HIV-1 infection (13, 20), making this species an ideal model for the study of the effect of PAP-1 on SIV pathogenesis and to determine its potential to alter viral replication and disease course in vivo. The question to be asked is whether the administration of PAP-1 in SIV-infected RM prevents or slows disease progression by limiting the expansion of activated memory viral target cells and/or by contributing to the muting of the chronic immune activation response via inhibition of TEM cell function. Conversely, the suppression of a virus-specific effector T-cell response could also have detrimental effects on the control of viral replication and may accelerate the course of infection. Thus, investigating the influence of PAP-1 on SIV pathogenesis will help define cell lineage(s) and/or mechanisms that are involved in the protection or deterioration of the immune system following SIV infection in this species. Before such a task is undertaken, it is vital to first assess both the in vitro and in vivo effects of PAP-1 in RM. The goal of this study was therefore to determine the potential toxicity, if any, and pharmacokinetics (PK) of PAP-1 in healthy uninfected RM in order to determine a therapeutic dose of PAP-1 that could be used in SIV-infected animals in the future. A single dose of PAP-1 was initially administered to RM and upon determination of the safety and PK, a chronic dosing study was initiated to determine the dosage required to maintain a critical trough level of this compound. RM undergoing chronic PAP-1 dosing were also infected intranasally with a live attenuated influenza A/PR8 (flu) virus and the effect of PAP-1 on the primary effector and secondary memory flu-specific responses were assessed in order to confirm the target cell selectivity of PAP-1 in vivo. In parallel with these in vivo studies, in vitro experiments were performed to define the Kv1.3 channel expression profile in RM T-cell subsets and to determine the potential in vitro suppressive effect of PAP-1 on activated RM TEM cells. Results suggest that PAP-1 was both safe and effective in vitro and in vivo, providing a rational and sound foundation for future studies with PAP-1 in SIV-infected RM.
MATERIALS AND METHODS
Animals
Healthy SIV negative RM of comparable age and weight were housed at the Yerkes National Primate Research Center (YNPRC) of Emory University. Their housing, care, diet and maintenance was in conformance to the guidelines of the Committee on the Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council and the Health and Human Services by standard guidelines “Guide for the Care and Use of Laboratory Animals” (21). RM were anesthesized with 10 mg/kg of ketamine hydrochloride that was administered intra-muscularly. For acute PAP-1 dosing experiments that required multiple bleeds at frequent time intervals, anesthesia was administered every 1–2 hr as needed. On days when only a single bleed was required, a single dose of ketamine was administered prior to sample retrieval. RM that were immunized with the influenza virus for the chronic dosing study were anesthesized and intra-nasally administered 1 ml containing 1024 HA units of the virus in a slow drop by drop fashion as described elsewhere (22). The animals were also boosted 4 weeks and 12 weeks post initial immunization with the same viral dose and route of administration.
PAP-1 (4-Phenoxybutoxy)psoralen
PAP-1 was synthesized as previously described (7). For use in in vitro assays, PAP-1 was dissolved in DMSO to a concentration of 10 mM and this stock solution was diluted to desired concentrations using RPMI supplemented with 1% fetal calf serum (FCS), 2 mM glutamine and 50 ug/ml gentamycin. The final DMSO concentration in the culture assays was 0.05%. For intravenous administration, PAP-1 was dissolved at a concentration of 9 mg/ml in a mixture of cremophor (Sigma-Aldrich, St. Louis MO) and 1X PBS (1:4) by heating to ~ 90°C and vigorous stirring. Aliquots of the cooled PAP-1 solution were frozen at −20°C until use. For oral administration PAP-1 was mixed at a final dosage of 25 mg/kg for each animal into ~ 5 g of chocolate paste just prior to administration and the stock stored at 4°C no more than a week before being fed to the animals.
Specimen collection and toxicity analyses
Whole blood was collected from the RM in heparinized tubes for the determination of absolute counts of WBC, platelets and total lymphocyte count using standard methods. In addition, sera were evaluated for the levels of albumin, liver enzymes, bilirubin, blood urea nitrogen/creatinine, and various electrolytes using our standard in vivo toxicology screen. Peripheral blood mononuclear cells (PBMCs) were also isolated from heparinized whole blood by standard Ficoll-Hypaque gradient centrifugation.
Determination of plasma PAP-1 levels by HPLC-MS
PAP-1 was injected intravenously or administered orally to two rhesus macaques (12 and 17 kg respectively). At various time points following administration, approximately 0.5 ml of blood were collected into EDTA blood sample collection tubes. Plasma was separated by centrifugation and stored at −20°C pending analysis. Samples were purified using C18 solid phase extraction (SPE) cartridges. Elution fractions corresponding to PAP-1 were evaporated to dryness under nitrogen and reconstituted in a mixture of acetonitrile (ACN) and water (2:3). LC-ESI-MSMS analysis was performed with a Hewlett-Packard 1100 series HPLC stack equipped with a KGaA RT 250–4 LiChrosorb RP-18 column (Merck, Darmstadt, Germany), an HP1100 variable wavelength detector (VWD), and interfaced to a Finnigan LCQ Classic Mass Spectrometer (MS). The mobile phase consisted of ACN:H2O with 0.2% formic acid. The flow rate was 1.0 mL min-1 and the gradient was ramped from 1:1 to 9:1 ACN:H2O over 20 min. With the column temperature maintained at 24°C, PAP-1 eluted at 14.8 min and was detected with the VWD and the MS in series. Quantitation was by electrospray ionization MS/MS (capillary temp. = 350°C; capillary voltage = 5V; tube lens offset = −25V; positive ion mode; normalized collision energy = 25.7%; parent mass = 351.1 m/z; SRM = 107.0, 149.0, 251.0, 351.2) for samples between 2.5 nM (0.88 ng/mL) and 750 nM (263 ng/mL), and by the VWD (310 nm) for samples with higher concentrations. The related compound PAP-3 (5(4-phenoxypropoxy)psoralen; FW = 336.33; RT = 13.5 min) was used as an internal standard. The pharmacokinetics of the data obtained on samples following i.v injection were plotted as a two-compartment model using both Origin and Winnonlin® software (Pharsight, CA).
Flow cytometry analysis
Aliquots of the PBMCs were subjected to standard immunophenotypic analysis to determine the frequencies of major T-cell subsets including naïve, central memory, effector memory T-cells and regulatory T-cells (Tregs) using the following antibody clones: CD4-PerCP (clone L200), CD8-FITC (clone X), CD28-PE (clone 28.2), CD95-APC or FITC (clone DX-2), all purchased from BD Pharmingen (San Diego, CA), CD25-PE (clone 4E3, Miltenyi Biotec, Auburn, CA), and FoxP3-APC (clone PCH101, eBioscience, San Diego, CA). To phenotype naïve, TCM and TEM subsets, cells were first incubated with 1 μg/ml of anti-FcR antibody (clone 2.4G2, courtesy Dr. R. Mittler, Emory University CFAR) for 15 min at 4°C, washed and then surface-stained for 15 min at 4°C with a pre-determined optimal concentrations of CD4-PerCP, CD8-FITC, CD28-PE and CD95-APC. Cells were washed, re-suspended in freshly prepared 1% paraformaldehyde in PBS pH 7.4 and analyzed. T-cell subsets were defined as naïve (CD28+CD95−), TCM (CD28+CD95+) and TEM (CD28−CD95+) (23). To phenotype Tregs, cells were surface stained in a similar manner with CD4-PerCP and CD25-PE. Fixation and intracellular staining to detect FoxP3 was performed according to E-Biosciences protocol. Appropriate mAb isotype controls were included. Flow cytometric acquisition of at least 100,000 total events from each sample was performed on a FACS Calibur flow cytometer. Data acquisition and analysis was performed using CellQuest (BD Biosciences) and FlowJo (TreeStar, Ashland, OR) software, respectively.
Proliferation assays
PBMCs were isolated from the peripheral blood of healthy RM (n=9) and re-suspended in RPMI at a concentration of 1 million cells/ml. 100 ul containing 100,000 cells was dispensed into each well of a 96-well polystyrene plate that was previously coated overnight with 400 ng/ml of anti-CD3 monoclonal antibody (clone FN-18, Biosource, Invitrogen, Carlsbad, California). PAP-1 was added in a volume of 100 ul of RPMI at increasing concentrations. Cultures were performed in triplicate. For the isolation of CD28− effector memory cells, PBMCs were incubated with anti-CD28-coated magnetic beads (Dynal Cellection Kit, Invitrogen, Carlsbad, CA) for 30 min at 4°C with gentle rotation. CD28+ cells bound to the beads were separated (5 min x2) using a magnet and the supernatant containing CD28− cells was washed twice with 1% RPMI. An aliquot of these isolated cells was subjected to flow analysis and the purity of the CD28− cell population was always > 90%. The negatively isolated CD28− fraction was seeded at 100,000 cells/well in the absence or presence of increasing concentrations of PAP-1. Cultures were incubated at 37°C, 7% CO2 for 3 days and 16 hrs prior to harvest, cells were pulsed with [3H]-thymidine (1 uCi/well). Cells were harvested onto glass-fiber filters and the mean uptake of [3H]-thymidine was determined using a standard β-scintillation counter.
Electrophysiology
Electrophysiological studies were conducted on aliquots of both resting and in vitro activated unfractionated and fractionated T cell subsets. The T-cells were isolated from PBMCs by negative selection using a rhesus macaque-specific T-cell enrichment kit (StemCell Technologies Inc.). Aliquots of the isolated T cells were activated with 10 ug/ml plate-bound anti-CD3 (clone FN-18, BioSource/Invitrogen) for ~ 40 hrs. Both resting and in vitro activated T cells were stained for CD28 and CD95 on ice in RPMI-medium (supplemented with 10% FCS and 0.1% NaN3) with PE-conjugated mouse anti-human CD28 mAb (CD28.2, BD Pharmingen) and FITC-conjugated mouse anti-human CD95 mAb (DX2, BD Pharmingen). Cells were washed, incubated on poly-L-lysine coated cover-slips, and kept in the dark at 4°C for 10–30 min to attach. Naïve (CD28+CD95−), TCM (CD28+CD95+) and TEM (CD28−CD95+) cells were visualized by fluorescence microscopy and studied in the whole-cell configuration of the patch-clamp technique.
Kv1.3 currents were elicited in situ by repeated 200 ms pulses from a holding potential of −80 mV to 40 mV, applied every second to visualize Kv1.3’s characteristic cumulative inactivation, or every 30 sec in experiments to measure the blocking activity by ShK(L5) or PAP-1 on Kv1.3 currents which were recorded in normal Ringer solution with a Ca2+-free pipette solution containing (in mM): 145 KF, 10 HEPES, 10 EGTA, 2 MgCl2, pH 7.2, 300 mOsm. Whole-cell Kv1.3 conductances were calculated from the peak current amplitudes at 40 mV. KCa3.1 currents were elicited with voltage ramps from −120 to 40 mV of 200 ms duration applied every 10 seconds with a pipette solution containing (in mM): 145 K+ aspartate, 2 MgCl2, 10 HEPES, 10 K2EGTA and 8.5 CaCl2 (1 μM free Ca2+), pH 7.2, 290 mOsm. To reduce chloride “leak” currents, we used a Na+ aspartate external solution containing (in mM): 160 Na+ aspartate, 4.5 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES, pH 7.4, 300 mOsm. Kv1.3 and KCa3.1 channel numbers per cell were determined by dividing the whole-cell Kv1.3 or KCa3.1 conductance by the single channel conductance value for each channel - Kv1.3: 12 pS; IKCa1: 11 pS. Cell capacitance, a direct measure of cell surface area, was constantly monitored during recording. Resting (unstimulated) T cells had membrane capacitances < 2 pF (average cell diameter < 7 μM), while activated cells had membrane capacitances > 4 pF (average cell diameter > 11 μM).
Real-time PCR for RhCMV quantification
A real-time PCR assay was performed to quantify RhCMV DNA copy numbers in plasma samples from the RM undergoing the chronic PAP-1 dosing study by measuring levels of the pp65 virus sequence. The template for the reaction was total viral DNA isolated from the plasma samples using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA). Each plasma sample was spiked with an equal amount (10,000 copies) of an unrelated plasmid containing the chloramphenicol acetyl transferase (CAT) sequence to control for the efficiency of DNA extraction. These were later subjected to and quantified by PCR using CAT-specific primers as described before (24, 25) to serve as an internal control to confirm uniformity in DNA isolation from each plasma sample. The forward primer (5′-GTTTAGGGAACCGCCATTCTG-3′) corresponds to residues 719 to 739 and the reverse primer (5′-GTATCCGCGTTCCAATGCA-3′) corresponds to residues 826 to 808 (26). Reactions of a 20 ul total volume containing 1X iQ SYBR Green Supermix (BioRad), 100 ng/ul of each primer, nuclease-free water and the template from each plasma sample were prepared in duplicate and PCR was performed on an iCycler™ (BioRad, Hercules, CA) under the following conditions: 10 min at 95°C followed by 45 cycles of 15s at 95°C and 1 min at 60°C. A standard curve was generated by using 10-fold serial dilutions of a control pGEM-T plasmid containing the pp65 target sequence (106 to 0 copies per reaction). Results were analyzed by using the iCycler iQ Optical System software (BioRad) and the limit of sensitivity was reproducibly between 1–10 copies of the template.
Determination of flu-specific cell-mediated and humoral responses
For determination of CD4+ and CD8+ T-cell responses, aliquots of PBMCs from each monkey were cultured for 2 hrs with whole flu virus (~ 100 HA units/well), ConA (positive control, 7.5 ug/ml) or OVA peptide (negative control, 3 ug/ml) followed by the addition of brefeldin A (6 ug/ml) and incubation overnight at 37°C, 5% CO2. Cells were then washed and surface stained with CD4-PerCP and CD8-FITC (BD Pharmingen) and then fixed/permeabilized and stained for intracellular IFN-γ-APC or IL2-APC and TNF-PE (all purchased from BD Pharmingen). The frequency of cytokine-producing CD4+ or CD8+ cells was determined by standard flow cytometric analysis on the FACS Calibur II. For determination of humoral responses, a standard ELISA to detect flu-specific IgG was performed. Briefly, 96-well plates were coated with poly-L-lysine and whole flu virus dispensed into individual wells overnight. The plates were washed and media containing 2% non-fat milk solution added for blocking non-specific binding. 100 ul of plasma to be assayed diluted to 1/100, 1/1000, 1/5000, 1/10000 and 1/50000 in 10% RPMI were added in triplicate wells. Plates were incubated at 37°C for 2 hrs, washed x4 with PBS-T, and 100 ul of 1:2000 diluted alkaline-phosphatase-conjugated anti-human Ig (Southern Biotech, Birmingham, AL) was added to each well. After one hr incubation at 37°C, wells were washed and the reaction was developed using an alkaline phosphatase substrate kit (BioRad, Hercules, CA). Wells were monitored for color development and absorbance was measured at 405 nM. The cut-off value was determined as the mean of the negative control plus 3 times its standard deviation. Samples with mean absorbance readings exceeding this value were considered positive for the presence of flu-specific IgG and the reciprocal of the highest dilution of plasma giving a positive reaction was considered as the titer in the sample being tested.
Statistical analyses
Data are represented as means +/− SD unless otherwise indicated. Any statistical analyses to determine significant differences were derived using the two-tailed Student’s t-test. A P value of less than 0.05 was considered to be statistically significant.
RESULTS
Kv1.3 expression in RM T cells is comparable to levels present in human T-cells
Prior to investigating the in vivo and in vitro effect of PAP-1 in RM it was essential to first determine if RM resemble humans and express Kv1.3 and KCa3.1 in a T cell subset specific manner. Since there are currently no specific antibodies to Kv1.3 or KCa3.1 suitable for use in flow cytometry a combination of fluorescence microscopy and electrophysiology were utilized to study RM K+ channel expression as was previously done in human T cells (5). CD3+ lymphocytes were isolated by negative selection from PBMCs of healthy RM (n=6), both resting and anti-CD3 activated cells were stained with CD95-FITC and CD28-PE to distinguish among naïve, TCM and TEM cell subsets and their K+ currents were measured in the whole-cell mode of the patch-clamp technique. Each subset was found to express Kv currents, which were confirmed to be Kv1.3 by their biophysical and pharmacological properties. The currents exhibited Kv1.3’s characteristic use-dependence upon rapid pulsing and showed the same inactivation time constant (~200 ms, Fig 1A) and half-activation voltage (V1/2 = −33 mV, Fig. 1B) as the cloned channel (27). Furthermore, the Kv current in all three T cell subsets was inhibited by the Kv1.3-specific blockers ShK(L5) and PAP-1 with IC50s of 72 pM and 2.1 nM (see Fig. 1C and 1D for representative recordings and 1E for the concentration-response curves). The Kv1.1-specific peptide DTX-1 had no effect on the current at a concentration of 100 nM (see Fig. 1C for representative recording from a resting naïve T cell) demonstrating that in contrast to mouse T cells, RM T cells do not express functional Kv1.1 channels. In addition to Kv1.3 currents, we also detected calcium-activated K+ currents exhibiting the biophysical and pharmacological characteristics of KCa3.1 in RM T cells when dialyzing 3 μM of free Ca2+ through the recording pipette. The currents reversed around −80 mV and were sensitive to blockade by the specific KCa3.1 blocker TRAM-34 (Fig. 1F).
Figure 1.

RM T-cells express Kv1.3 and KCa3.1. (A) The Kv current exhibits the characteristic use-dependence of Kv1.3 when currents are elicited with 200-ms pulses every sec. (B) Normalized peak K+ conductance-voltage relationship of Kv current in resting (■, V1/2 = −34 ± 1.4 mV) and activated (△, V1/2 = −33 ± 1.2 mV ) RM T cells (n = 3). (C) The Kv current in a naïve T cell is insensitive to the Kv1.1-blocking peptide toxin DTX-I and sensitive to the Kv1.3-specific blocker ShK(L5). (D) Blockade of the Kv current in an activated TEM cell by PAP-1. (E) Concentration-response curve for Kv current inhibition by ShK(L5) (■, IC50 = 72 ± 8 pM, nH = 1.15) and PAP-1 (○, IC50 = 2.1 ± 0.3 nM, nH = 1.8). (F) KCa current in a naïve T cell elicited by dialysis with 3 μM free Ca2+. The current is blocked by the specific KCa3.1 blocker TRAM-34. Kv1.3 was previously blocked by 1 nM ShK(L5).
In addition, the number of Kv1.3 and KCa3.1 channels in naïve, TCM and TEM cells was also determined by patch-clamping roughly 20 cells from each subset in both the resting and activated state and then dividing the whole-cell Kv1.3 or KCa3.1 conductance by the single channel conductance value for each channel (see Fig. 2A for representative Kv1.3 and KCa3.1 currents). While the number of Kv1.3 channels is generally slightly lower in RM than in human T-cells (5), the expression pattern is very similar. As seen with human naïve and TCM cells, the up-regulation of Kv1.3 channels in these cell subsets in RM was minimal upon activation but as anticipated, activated TEM cells from RM exhibited a significant (~ 8-fold) increase in Kv1.3 channels similar to the level of up-regulation that occurs in activated human TEM cells. In contrast, there was a ~30 to 40-fold increase in the number of KCa3.1 channels in activated naïve and TCM cells with no up-regulation occurring in the TEM subset. The differential up-regulation of these K+ channels among the T cell subsets was reflected by the increase in Kv1.3 and KCa3.1 currents in activated TEM cells and naïve/TCM cells, respectively (Fig. 2A). It should be noted that staining with the CD95 and CD28 antibodies did not interfere with the expression pattern of Kv1.3 and iKCa3.1 currents (data not shown). Taken together, these data indicate that Kv1.3 and KCa3.1 are the only K+ channels expressed in RM T cells and that their expression pattern is similar to human T cells. While KCa3.1 is the dominating channel in activated naïve and TCM cells, activated TEM cells express a Kv1.3high phenotype.
Figure 2.

(A) Representative Kv1.3 and KCa3.1 currents in activated naïve, TCM and TEM RM T cells. (B) Scatterplot of Kv1.3 and KCa3.1 channel numbers per cell in resting (open symbols) and activated (closed symbols) T cells in naïve, TCM and TEM RM T cells. Mean channel numbers (± SD) are shown in the table below the plot.
PAP-1 inhibits the proliferation of RM PBMCs and TEM cells in vitro
Recent studies demonstrated that Kv1.3 expression in human T-cells is low in both CD4+ and CD8+ naïve and TCM cell and high in both activated CD4+ and CD8+ CCR7- TEM cells (5) and as a result PAP-1 effectively suppressed the proliferation of human CCR7- TEM cells in vitro with an IC50 of 10 nM (7). It was reasonable to assume that the same would be true in RM and we therefore evaluated the effect of PAP-1 on total CD28− T-cells since TEM cells in RM are best defined by the phenotype CD28−CD95+ (23). Thus, to determine if a comparable degree of suppression was observed with RM TEM cells an in vitro assay was performed in which CD28− TEM cells were negatively isolated from RM PBMCs (n=9) using anti-CD28-coated magnetic microbeads. Negatively isolated cells were confirmed to be 90% CD28− by flow cytometry. This CD28− fraction, containing primarily the TEM subset, was stimulated in vitro with a low sub-optimal concentration of plate-bound anti-CD3 in order to preferentially activate TEM cells, which exhibit a lower activation threshold than naïve and TCM cells (28). As shown by the representative data in Figure 3, PAP-1 suppressed the proliferation of TEM cells with an IC50 of ~100 nM. The suppressive effect of PAP-1 on bulk PBMCs which predominantly consist of naïve and TCM cells was also tested and was found to be less pronounced with an IC50 of ~1 uM (Fig. 3). Isolated CD28+ T-cells could not be utilized as a pure source of naïve and TCM cells because the T-cells still contained bound anti-CD28 antibody when released from the magnetic beads and this in combination with anti-CD3 resulted in a level of stimulation that abrogated the suppressive effect of PAP-1. This observation is in line with a previous study which reported that T-cell proliferation was unaffected by the K+ channel blocker margatoxin when co-stimulation with anti-CD28 was involved (4). In summary, the results obtained suggest that PAP-1 is an effective inhibitor of RM TEM cell proliferation, which is consistent with previous studies.
Figure 3.
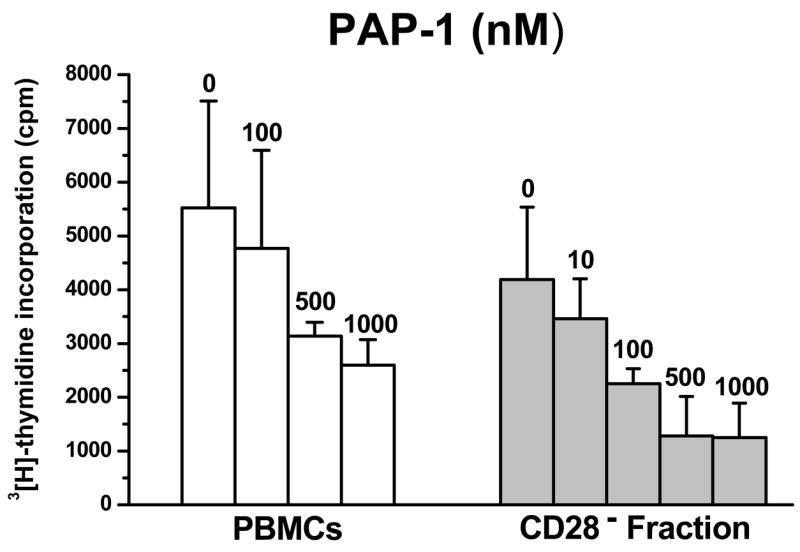
PAP-1 suppresses the proliferation of PBMCs and TEM cells from RM. PBMCs isolated from healthy uninfected RM (n = 9) were seeded at 100,000 cells/well and were stimulated with 400 ng/ml plate-bound anti-CD3 in the absence or presence of increasing concentrations of PAP-1 (nM). To test the effect of PAP-1 on TEM cells, PBMCs were depleted of CD28+ cells using anti-CD28-coated magnetic microbeads and the CD28− fraction was seeded at 100,000 cells/well. Cultures were incubated at 37°C, 7% CO2 for 3 days. Cells were pulsed with 1 uCi/well [3H]-thymidine 16 hrs prior to harvest and thymidine uptake was measured by standard scintillation counting. The mean cpm ± S.D was determined; shown is data representative of three independent assays.
Acute in vivo dosage studies suggest that PAP-1 is non-toxic and its PK is best reflected by a two-compartment model
To determine the in vivo toxicity and PK of PAP-1, two adult RM, RHg7 and RVe7 were administered the compound i.v. at a dose of 3 mg/kg. The i.v. administration of PAP-1 was well tolerated by both animals with no signs of acute side effects. Blood and serum biochemical parameters were determined at 48 hrs and 7 days post-administration and comparison with baseline levels suggest no evidence of PAP-1 related hematological and organ toxicity (Table 1). An increase in the levels of serum creatinine phosphokinase (CPK) from baseline levels was observed in both RM but this was attributed to the effect of the anesthetic ketamine hydrochloride that was used to sedate the animals for blood sample collection. Ketamine anesthesia and the physical restraint associated with its administration have been shown to induce variations in several serum and hematological parameters in non-human primates including RM and cynomologus monkeys (29, 30). Indeed, repeated ketamine administration that was necessary for multiple blood sample withdrawal within the first 48 hours of the acute PAP-1 dosing experiment led to alterations in CPK activity a well fluctuations in other biochemical variables such as glucose and aspartate transaminase (AST) in both RHg7 and RVe7. A decrease in circulating lymphocytes with a coincident increase in neutrophils was also observed as previously described suggesting a redistribution of lymphocytes from the blood to extravascular sites and a mobilization of neutrophils to the peripheral circulation. Further analysis of PBMCs for the frequencies of major T cell subsets including CD4+ and CD8+ naïve, TCM, TEM cells and CD4+ Tregs by flow cytometry also reflected this decrease in lymphocytes at the 8 hr time point (Table 2). However the levels of circulating lymphocytes and serum analytes returned to values near baseline within a week (Tables 1 and 2).
The pharmacokinetics of i.v. administered PAP-1 was also examined in each of the two animals. Plasma samples were collected pre- and at a number of time points post-PAP-1 administration (15 min, 30 min, 1 hr, 2 hrs, 4 hrs, 8 hrs, 24 hrs, 48 hrs and 7 days). PAP-1 plasma concentrations at each time point were determined by HPLC-MS and the data obtained appeared to fit as a two-compartment model and followed second order kinetics (Fig. 4A, left panel). The rate of elimination of PAP-1 was determined to be moderately slow with an in vivo half-life (t1/2) of 6.4 hours. The volume of distribution (Fig 4A, right panel) was between 2–4 L/kg which is similar to the level previously determined in rats although the t1/2 of PAP-1 in these rodents is shorter (3.8 hrs) (11).
Figure 4.

Pharmacokinetics of i.v. and orally administered PAP-1 in RM. (A) Total PAP-1 plasma concentration-time profile in RM after single i.v. injection of 3 mg/kg dose (left) and log plot of averaged data used to determine the volume of distribution (right). (B) Plasma concentration-time profile in RM that were orally administered 25 mg/kg of PAP-1. Concentrations at 48 hours were 20 and 30 nM; concentrations at 7 days 10 and 3 nM.
After a washout period of ~ 2 months and upon confirmation of normal baseline levels of blood/serum chemistry parameters, the safety and efficacy of orally administered PAP-1 (25 mg/kg) was determined in the same 2 RM. As depicted in Fig. 4B, the plasma concentration of PAP-1 peaked within 10 hours of ingestion reaching a maximum of ~1700 nM and ~950 nM in RHg7 and RVe7 respectively, indicating an efficient absorption of PAP-1 via the oral route. There were no signs of toxicity evident in the RM following oral administration of PAP-1. In addition, similar to the i.v. acute dosing results, fluctuations in serum and hematological parameters were observed in the 48 hr blood/serum samples after initial ketamine administration but all values returned to baseline levels within a week (data not shown).
Chronic dosing of PAP-1 in RM immunized with the flu virus does not appear to increase their susceptibility to infection and selectively inhibits TEM function but not the development of a central memory flu-specific response
Since results of the acute dosing studies indicated that there were no detectable toxic side effects at the dose utilized during and in the days following in vivo PAP-1 administration, a 30-day chronic dosing study was initiated in two RM RGq8 and RWk7. The protocol utilized is outlined in Table 3. The purpose of this experiment was threefold. First, it was necessary to assess the safety of chronic administration of PAP-1 and to determine the dose required to maintain a critical trough level of the compound in RM in vivo. Second, given that all TEM cells and not just antigen-specific TEM cells up-regulate Kv1.3 channels upon activation, there is always the possibility that PAP-1 may globally suppress TEM cells and compromise the host’s ability to respond to pathogens. It was therefore critical to ensure that chronic application of PAP-1 does not result in increased susceptibility of the host to opportunistic infection and/or disease. Finally, the main feature of PAP-1 is that it selectively inhibits the function of TEM cells as shown by previous studies and by the in vitro data herein (Fig. 2). It was therefore important to demonstrate that this selectivity holds true in vivo.
To accomplish the aforementioned tasks, a chronic dosing study was therefore initiated by administering PAP-1 orally to the two RM daily for a 30-day period at a dose of 25 mg/kg/day. The oral route was chosen due to logistic limitations of daily IV administration and given the ease of administration and the fact that pharmacologically relevant levels of PAP-1 were readily detectable in the plasma. Since the elimination of PAP-1 in RM was previously determined to be relatively slow with a t1/2 of 6.4 hrs, the dose was administered once daily. Blood samples were collected at baseline and before each feeding at the indicated time points (Table 3) and utilized to determine standard blood/serum chemistry and served as a source of PBMCs for the analysis of major T cell subsets by flow cytometry. As seen with the acute dosing study, chronic oral administration of PAP-1 did not result in any apparent hematological and organ toxicity and no marked fluctuations in the frequencies of major T cell lineages were noted (not shown). Plasma samples were also isolated from the collected blood samples for analysis of PAP-1 concentrations by HPLC-MS. As shown by the results in Table 4, trough levels with concentrations averaging 511 ±113 nM in RGq8 and 77 ± 5 nM in RWk7 were maintained throughout the study suggesting variations in individual uptake levels but also reproducibility within the same individual. After PAP-1 was discontinued, plasma levels decreased according to the established second order kinetics and low detectable levels were still present 17 days after PAP-1 was discontinued.
The selective suppressive effect of PAP-1 on TEM cells in vivo was tested by measuring a potential (but clinically silent) reactivation of cytomegalovirus (CMV) replication by quantifying the levels of CMV DNA in plasma samples from these two RM by real-time PCR. Since CMV levels are believed to be kept in check by TEM cells (26, 31), a loss of TEM cell function was hypothesized to compromise the control of CMV replication. Total viral DNA was isolated from plasma samples from each monkey at various time points before, during and after PAP-1 treatment. Prior to DNA isolation, these plasma samples were also spiked with an equal concentration of a control plasmid containing the CAT sequence that was later quantified by real-time PCR to ensure consistency in DNA isolation from each sample (data not shown). Quantification of RhCMV revealed that at the baseline time point both monkeys exhibited detectable low levels of circulating CMV DNA (~500–1000 copies/ml) which was unexpected (Fig. 5). These RhCMV levels remained relatively near baseline during the first two weeks of treatment but an increase did occur as indicated by the peak on day 21 in both monkeys, with a larger increase being detected in samples from RWk7 (~8800 copies/ml) than RGq8 (~2200 copies/ml). After PAP-1 was discontinued, RhCMV levels rapidly returned to levels near baseline in both animals.
Figure 5.

Effect of PAP-1 on plasma CMV levels in RM undergoing chronic PAP-dosing. Real-time PCR was performed to quantify RhCMV levels in viral DNA extracted from plasma samples that were isolated from the two animals before, during and after PAP-1 application. The last day of PAP-1 treatment (day 30) is indicated.
To further demonstrate the in vivo selectivity of PAP-1, both RM were immunized with the flu virus on day 7 to determine if PAP-1 selectively inhibits the initial flu-specific effector T-cell response but not the development of central memory T cells. In order to determine the functional nature of the TEM response, the cell mediated T-cell immune responses to the flu virus was measured by ICC. The frequency of CD4+ and CD8+ T cells producing the Th1 cytokines IFN-γ, TNF-α and IL-2 in response to whole flu virus were determined. As expected, CD4+ and CD8+ T cells from both animals on day 0 exhibited a minimal response to the OVA peptide (negative control) while ConA induced a substantial increase in the level of all three cytokines tested (Fig. 6A). Similar control data was observed for samples obtained on days 28 and 47 (data not shown). In both RM, background levels of T-cell responses were observed on day 28 (3 weeks after the initial flu immunization) indicating that neither animal exhibited a flu-specific T-cell response while being treated with PAP-1 (Fig. 6B), unlike historical controls in which early flu-specific T-cell responses are clearly evident (32). However on day 47, 10 days after the 1st flu booster and two weeks following the discontinuation of PAP-1 treatment, a substantial and significant increase (p<0.05) in the production of IFN-γ and TNF-α by CD4+ T-cells (~3% and 2.5% of CD4+ T-cells, respectively) was evident in RWk7, the animal which had cleared PAP-1 from the circulation by that time point. An increase in IL-2-producing CD4+ T cells and cytokine producing CD8+ T cells were also noted in this animal. Unlike RWk7, only a modest increase in the flu-specific T-cell response was evident in RGq8 after the 1st flu booster which was administered at a time when this animal still had marked levels of PAP-1 present in its system. The cytokine responses in RWk7 and RGq8 were comparable to, if not above, those noted for historical controls (n=4) that were not treated with PAP-1 at any time and exhibited an ~ two-fold increase in the frequency of IFN-γ producing CD4+ T-cells (1.44% ± 0.35 to 3.25% ±0.35) and CD8+ T-cells (0.42% ±0.13 to 0.98% ±0.10) following a primary flu booster ~ 46 days post initial flu immunization (A. Ansari et al, unpublished data). A second flu boost was administered two months later to RWk7 and RGq8 and PBMC samples were analyzed 10 days thereafter. Similar results were obtained in that a greater flu-specific response was exhibited by RWk7 than RGq8 (data not shown). Next, the humoral flu specific immune response in the plasma from these 2 monkeys was measured using our lab standardized ELISA. Anti-flu IgG levels in a series of diluted plasma samples from both animals were determined at baseline, day 21 (post initial immunization), day 47 (10 days after 1st booster) and day 103 (10 days after 2nd booster). As shown by the Ab titers in Table 5, both animals exhibited a minimal flu-specific Ab response while undergoing PAP-1 treatment. Similar to the pattern observed with ICC results, a response was only evident in RWk7 and not RGq8 after the 1st booster but both animals exhibited a strong humoral response after the 2nd flu booster as indicated by the flu Ab titer of 1:10000. As controls, three additional RM were immunized twice with the flu virus but were not administered PAP-1 at any time. Plasma samples were tested for the presence of flu Ab at time points comparable to those of the PAP-1 treated RM. As shown in Table 5, all three control animals exhibited a flu-specific response after the 1st flu booster but of particular significance is the fact that a response was already readily detectable in all three control RM four weeks after the initial immunization, in contrast to the minimal/lack of response in the PAP1-treated RM. Importantly, neither RGq8 or RWk7 were adversely affected by the live flu immunization and exhibited no other signs of disease during the entire course of the PAP-1 chronic dosing experiment.
Figure 6.

Effect of PAP-1 on the flu virus-specific primary and secondary T-cell responses in RM. PBMCs that were isolated on day 28 (during PAP-1 application, post-initial flu immunization) and day 47 (after PAP-1 application, post-1st flu booster) were cultured in the presence of ConA (positive control), media (background control), Ova (negative control), and whole flu virus in the presence of brefeldin A. Cells were surface stained for CD4 and CD8 and then fixed/permeabilized and stained to detect IFN-γ (clear bars), IL-2 (dark grey bars) and TNF-α (light grey bars). The frequency of cytokine-producing CD4+ and CD8+ T-cells was determined by flow cytometry. Data shown includes (A) the CD4+ and CD8+ T-cell response to positive (ConA) and negative (media, OVA) controls in both RM on day 0 (baseline) and (B) the CD4+ and CD8+ T-cell response (corrected for media background) to the flu virus in RGq8 (left panel) and RWk7 (right panel) on days 28 and 47.
DISCUSSION
In order to delineate the role of different non-human primate lymphoid subsets cells in the pathogenesis of SIV infection our laboratory has been working to identify reagents that can selectively inhibit or deplete certain subsets. Here we evaluated the toxicity and PK of the recently developed small molecule PAP-1 in RM in order to determine a safe and effective dose for future studies aimed at identifying the role of TEM cells in the pathogenesis of SIV infection. By blocking Kv1.3 in TEM cells, PAP-1 and the peptide ShK(L5) have been previously shown to preferentially inhibit human and rat TEM cells in vitro and to effectively suppress or treat delayed-type hypersensitivity, allergic contact dermatitis, EAE, pristane-induced arthritis and type-1 diabetes in rat models (7, 9, 11, 33). Since activated CD4+ TEM cells are the primary targets for SIV viral infection it was reasoned that the use of PAP-1 in SIV-infected RM would shed light on the role of this important cell lineage in disease progression or resistance. However, prior to conducting such intricate studies in SIV-infected RM, a comprehensive set of in vitro and in vivo experiments were first performed to determine the safety and efficacy of PAP-1 in healthy RM. PAP-1 had been previously reported to exhibit a 23–125 fold selectivity over the closely related Kv1-family channels expressed in the heart (Kv1.5 and Kv1.7) and in neurons and a more than 1000-fold selectivity over HERG, Na+, Ca2+ and chloride channels (7). PAP-1 further did not show any acute or chronic toxicity in rats (33). However, PAP-1 had never before been used in primates and we therefore needed to determine if it could be safely used in RM. We further needed to ascertain whether RM express high levels of Kv1.3 in activated TEM cells similar to humans and rats (5, 10). This could not simply be assumed because ion channels often show significant species differences in their expression pattern. For example besides Kv1.3 and KCa3.1 mouse T cells express Kv1.1, Kv1.2 and Kv1.6 in CD4+ T cells and Kv3.1 in CD8+ T cells and do not up-regulate Kv1.3 in TEM cells (34–36). Kv1.3 blockers therefore do not inhibit mouse T cell function in vitro (34) and have no effect in mouse autoimmune disease models (37).
Our patch-clamping experiments clearly demonstrated that the K+ channels in RM T cells are Kv1.3 and KCa3.1. Importantly, of these two channels, Kv1.3 was the one selectively up-regulated in activated TEM cells while KCa3.1 expression was instead increased in activated naïve and TCM subsets, which is in agreement with previous studies involving human T-cells (5), providing the opportunity to selectively manipulate the function of the TEM cell subset using the Kv1.3 channel blocker PAP-1. This compound and the alternative Kv1.3-specific peptide blocker Shk(L5) were shown to inhibit the Kv1.3 current in activated TEM cells and resting naïve T-cells, respectively, which may at first suggest that Kv1.3-specific blockers inhibit the function of all T-cell subsets. However, although Kv1.3 channels dominate in all resting T-cell subsets, KCa3.1 channels are rapidly up-regulated in naïve and TCM cells upon activation and as such, these cells are no longer susceptible to Kv1.3 blockade while the TEM subset remains sensitive to its effects. Additional results from in vitro suppression assays showed that while PAP-1 does suppress the proliferation of bulk RM PBMCs (IC50 = 1 uM), the inhibitory effect on CD28-depleted cells was clearly more potent as indicated by the IC50 of 100 nM which is approximately tenfold higher than that previously reported for human TEM cells (10 nM) (7). Since the assay in the previous study by Schmitz et al had utilized purified effector memory T-cells and not PBMCs depleted of naïve and TCM cells, the effect of PAP-1 on isolated CD28-depleted RM T-cells (as opposed to CD28-depleted PBMCs) was also evaluated to determine if this was the cause of the difference. The IC50 was determined to be ~ 350 nM (data not shown) which is higher than that initially obtained for the CD28-depleted PBMC fraction. This is not unexpected since a given number of purified TEM cells would require a greater concentration of PAP-1 to be suppressed to the same extent as an equivalent number of CD28-depleted PBMC fraction that would contain comparatively fewer TEM cells. An in vivo setting is however likely to be best reflected by the PBMC fraction. Regardless, the in vitro IC50 of PAP-1 for RM TEM cells is still higher than that for isolated human TEM cells and the reason for this difference remains unclear but given that the CD28-depleted cell fraction utilized in the assay was determined to be > 90% pure making purity issues highly unlikely, it is possible that intrinsic differences between RM TEM and human TEM contributed to the observed variation in the response to PAP-1. It is also possible that differences in the stimulus used in the previous and current study contributed to these dissimilarities. Nonetheless, the trend is importantly still maintained, with PAP-1 exerting a more potent suppressive effect on RM TEM cells than naïve and TCM cells.
The Kv1.3 channel expression pattern on RM TEM cells and the in vitro selectivity of PAP-1 for this T cell subset therefore provided a logical basis for pursuing in vivo studies and upon confirmation of the safety and PK of PAP-1, a chronic dosing study was initiated and the moderately long t1/2 of 6.4 hrs allowed for a trough level to be maintained with a single daily dose of 25 mg/kg. Although the trough levels achieved were considerably different in the two RM that were chronically administered PAP-1 (511 nM vs 77 nM), these values are still within the normal range of physiological variability (x5) due to individual differences in absorption and/or rates of metabolism. Importantly, these trough levels indicate that in vivo non-toxic plasma concentrations of PAP-1 were achieved and maintained near to or well above the level that was shown to be suppressive for RM TEM cells in vitro.
In addition to determining trough levels, the main purpose of the PAP-1 chronic dosing study was to demonstrate that PAP-1 selectively suppresses TEM cells in vivo and given that PAP-1 suppresses all TEM cells and not only those specific for a particular antigen, it was also crucial to show that the suppression of this T-cell subset would not increase the host’s susceptibility to opportunistic infection and disease. This was especially important given that RM are carriers of CMV and reactivation of CMV replication due to an immunocompromised system has been documented and this sometimes leads to a serious illness (38–42). Quantification of CMV DNA levels by real-time PCR in plasma samples from the two RM before and during chronic PAP-1 treatment indicated a detectable but low level of CMV in both animals even at baseline, which was unexpected. Elevated stress levels associated with repeated access to these RM for previous experiments and those conducted during the recent studies may be a contributing factor to this observation and may also be the reason for the fluctuations observed in CMV DNA levels during the early phase of PAP-1 application. A control of CMV replication was compromised as indicated by the rise in CMV DNA by day 21. However, the observed increase in CMV DNA copies appears to be well below the levels that have been reported to be associated with CMV-related illness in RM (42, 43). Importantly, CMV DNA copy number decreased to levels near baseline following the discontinuation of PAP-1 treatment and no signs of illness were evident throughout the entire course of the study. These data therefore lend support for the selective suppressive effect of PAP-1 on TEM cells in vivo since this T cell subset has been described to be largely responsibly for the control of CMV replication (26, 31) but the results also highlight the need for caution when pursuing a long-term dosing regimen since the extent of CMV reactivation needs to be taken into account.
In addition to the selective inhibitory effect of PAP-1 on TEM cells, it was also important to demonstrate that the development and/or function of the central memory arm was not affected by PAP-1 in vivo. To achieve this, both RM were intranasally administered live attenuated flu virus during PAP-1 treatment and the primary effector and secondary memory cell mediated and humoral responses were compared before and after PAP-1 application was discontinued. Under normal circumstances of antigenic stimulation the primary immune response typically involves the activation of naïve cells that proliferate to produce cells that are at various stages of differentiation (18, 19). Cells that are at a more terminally differentiated stage exert effector function while those that are at an intermediate level of differentiation persist as TCM after antigen clearance. Thus, following the initial flu immunization naïve cells will develop into flu-specific TEM cells but their further proliferation and function is expected to be inhibited by PAP-1 while the generation of TCM cells should not be affected. Indeed, flow cytometric analyses that were performed 1, 2 and 3 weeks after immunization revealed a modest increase in TEM frequency in both RM (data not shown) and ICC assays also indicated no increase in the production of IFN-γ, IL-2 or TNF-α from CD4+ and CD8+ T-cells in response to the flu virus (Fig. 6B, day 28), suggesting a suppression of the primary effector T cell response. After PAP-1 was discontinued, two flu boosters were administered to both RM to determine if the generation of central memory cells was affected by PAP-1. ICC data indicated that a stronger memory T-cell response was exhibited by RWk7 than RGq8. These cytokine responses were readily detectable and were comparable to, if not above, those exhibited by historical controls that were also administered the flu virus but not treated with PAP-1 suggesting that a functional secondary memory flu-specific response was exhibited by the PAP-1 treated RM. Similarly, results from the ELISA showed a lack of flu-specific IgG production in the PAP-1 treated RM three weeks after the initial immunization in contrast to the untreated RM which clearly exhibited an Ab response but both PAP-1 treated RM exhibited a flu-specific IgG response after administration of a second flu booster suggesting that the generation of central memory B cells was not affected. The secondary IgG response exhibited by RGq8 was again weaker than that of RWk7. Since RGq8 had maintained ~ 7-fold higher trough levels of PAP-1 than RWk7 (Table 4), it is tempting to speculate that this may have been the reason for the minimal flu-specific response in this monkey. However, the response of additional animals to PAP-1 including the parallel use of bona fide controls would need to be examined in order to derive a definite correlation between in vivo PAP-1 plasma concentrations and the suppression of antigen-specific immune responses.
The fact that the IgG responses were also influenced by PAP-1 raises the important point that PAP-1 affects not only a single cell subset i.e. TEM cells but in fact also has an impact on B-cells. Indeed, PAP-1 has been previously shown to inhibit the proliferation and function of class-switched IgD-CD27+ B cells but not activated naïve and IgD+CD27+ B cells (6). This in combination with the suppressive effect of PAP-1 on CD4+ TEM with Th2 function may have therefore contributed to the inhibition of the primary B-cell mediated IgG response. One may then posit how an IgG response was generated after the flu booster given that PAP-1 should have suppressed memory class-switched B-cells. Since PAP-1 does not affect the generation of IgD+CD27+ memory cells, it is possible that this compartment was able to undergo class switching after the booster immunizations. It would be of interest to evaluate the nature of the Ab response elicited when RM are immunized with flu boosters while still being administered PAP-1 to determine if Ab production would be limited to only IgM and not IgG. These observations therefore stress the need for caution when interpreting results from the PAP-1 chronic dosing study since its effect is exerted on not only a single cell subset but in fact influences both the cell-mediated and humoral arms of the immune system. It is also important to bear in mind that the nature and level of Kv1.3 expression in other major cell subsets such as NK/NKT cells has not been elucidated although it has been suggested that components of the innate immune system are unaffected by Kv1.3 blockade (44). A recent study demonstrated the presence of both Kv1.3 and Kv1.5 channels on human peripheral dendritic cells suggesting that PAP-1 may also influence the function of this cell subset. However, the presence of the alternate K+ channel Kv1.5 may offset any inhibitory effect caused by PAP-1. The potential inhibitory effect of PAP-1 on other major cell lineages therefore remains to be clarified and the influence of this on disease outcome cannot be overlooked. However, worthy of mention is that the two RM that were undergoing chronic dosing of PAP-1 were not adversely affected by the live flu virus which normally replicates only in the upper respiratory tract and occasionally produces mild symptoms in healthy monkeys such as transient nasal discharge. Of note, such symptoms were not observed throughout the entire course of the study. Thus, the immune system of the two RM in the study was not significantly compromised by PAP-1 at least during the short-term chronic dosing regimen.
In summary, the results from this study collectively show that the utilization of PAP-1 in RM is both safe and effective opening the door for its use in SIV-infected RM to determine the effect of this small molecule on major cell lineages and to define those that play a major role in SIV pathogenesis. Future studies would therefore include administering PAP-1 to SIV-infected RM during the acute or chronic phase of infection to determine if the inhibition of TEM cell function prevents or slows disease progression to AIDS by limiting the number of viral cell targets and/or by muting the chronic immune hyperactivation exhibited by SIV-infected RM. Although the utilization of PAP-1 in an SIV-infected RM model, particularly during the acute phase of infection, may potentially limit the pool of viral target cells by inhibiting CD4+ TEM cell function and proliferation, the effect on CD8+ TEM cells that assist in VL control cannot be overlooked. It is possible that if a lower VL setting is achieved due to CD4+ TEM inhibition, other players in immune control such as NK cells and B cells may contribute to VL control, possibly by-passing the need for a rigorous CD8+ TEM response. Since the effects of PAP-1 are almost immediately reversible, any detrimental effects on VL control can be relieved by discontinuing treatment or by lowering the dose. One may also test if the PAP-1 dose can be titrated to a concentration that perhaps maintains a balance between CD4+ TEM inhibition and CD8+ TEM function. In addition to the application of PAP-1 in RM, its use can also be applied in SIV-infected sooty mangabeys (SM), a non-human primate that remains disease-free throughout the course of SIV infection (13, 45, 46). The aim would be to determine the cause of disease-resistance in this species and to therefore evaluate if the inhibition of TEM function in SM results in disease development. Preliminary in vitro experiments performed by us have shown that the Kv1.3 channel expression pattern in SM T-cells is very similar to that in RM and human T-cells. In addition, PAP-1 inhibits the Kv1.3 current as shown by patch-clamping experiments and it also effectively suppresses the proliferation of both PBMCs and isolated TEM cells from SM in vitro (data not shown). The potential application for the use of PAP-1 in this non-human primate is therefore a reality and these studies together with those involving RM are sure to further our understanding of the mechanism(s) involved in disease resistance and susceptibility in SIV-infected hosts. Also worthy of note is the observed differential expression of KCa3.1 channels in RM T-cells (Fig 2) and SM T-cells (not shown) and the availability of the KCa3.1-specific blocker TRAM-34 (8) provides the opportunity to manipulate naïve and central memory cell subsets which may prove to be beneficial during acute stages of infection. Further studies involving this additional K+ channel blocker are therefore warranted.
Acknowledgments
The authors would like to thank Daniel Homerick for excellent technical assistance with the HPLC-MS assay, Dr. K. George Chandy for helpful suggestions with the study, and the Yerkes National Primate Research staff for their excellent care of the animals and sample collections.
Footnotes
Supported by NIH RO1 27057, IR24RR16988, RR00165 and RO1 GM076063.
References
- 1.Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freedman BD, Price MA, Deutsch CJ. Evidence for voltage modulation of IL-2 production in mitogen-stimulated human peripheral blood lymphocytes. J Immunol. 1992;149:3784–3794. [PubMed] [Google Scholar]
- 3.Leonard RJ, Garcia ML, Slaughter RS, Reuben JP. Selective blockers of voltage-gated K+ channel depolarize human T lymphocytes: mechanism of the antiproliferative effect of charybdotoxin. Proc Natl Acad Sci USA. 1992;89:10094–10098. doi: 10.1073/pnas.89.21.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin CS, Boltz RC, Blake JT, Nguyen M, Talento A, Fischer PA, Springer MS, Sigal NH, Slaughter RS, Garcia ML, Kaczorowski GJ, Koo GC. Voltage-gated potassium channels regulate calcium-dependent pathways involved in human T lymphocyte activation. J Exp Med. 1993;177:637–645. doi: 10.1084/jem.177.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wulff H, Calabresi PA, Allie R, Yun S, Pennington M, Beeton C, Chandy KG. The voltage-gated Kv1.3 K+ channel in effector memory T cells as a new target for MS. J Clin Invest. 2003;111:1703–1713. doi: 10.1172/JCI16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wulff H, Knaus H-G, Pennington M, Chandy KG. K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J Immunol. 2004;173:776–786. doi: 10.4049/jimmunol.173.2.776. [DOI] [PubMed] [Google Scholar]
- 7.Schmitz A, Sankaranarayanan A, Azam P, Schmidt-Lassen K, Homerick D, Hansel W, Wulff H. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol Pharmacol. 2005;68:1254–1270. doi: 10.1124/mol.105.015669. [DOI] [PubMed] [Google Scholar]
- 8.Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activate K+ channel, IKCa1: A potential immunosuppressant. Proc Natl Acad Sci USA. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beeton C, Pennington MW, Wulff H, Singh S, Nugent D, Crossley G, Khaytin I, Calabresi PA, Chen CY, Gutman GA, Chandy KG. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol Pharmacol. 2005;67:1369–1381. doi: 10.1124/mol.104.008193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beeton C, Wulff H, Barbaria J, Clot-Faybesse O, Pennington M, Bernard D, Cahalan MD, Chandy KG, Beraud E. Selective blockade of T lymphocyte K+ channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc Natl Acad Sci USA. 2001;98:13942–13947. doi: 10.1073/pnas.241497298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beeton C, Wulff H, Standifer NE, Azam P, Mullen KM, Pennington MW, Kolski-Andreaco A, Wei E, Grino A, Counts DR, Wang PH, LeeHealey CJ, Andrews BS, Sankaranarayanan A, Homerick D, Roeck WW, Tehranzadeh J, Stanhope KL, Zimin P, Havel PJ, Griffey S, Knaus H-G, Nepom GT, Gutman GA, Calabresi PA, Chandy KG. Kv1.3 channels are a therapeutic target for T cell-mediated autoimmune diseases. Proc Natl Acad Sci USA. 2006;103:17414–17419. doi: 10.1073/pnas.0605136103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Firpo PP, Axberg I, Scheibel M, Clark EA. Macaque CD4+ T-cell subsets: influence of activation on infection by simian immunodeficiency viruses (SIV) AIDS Res Hum Retroviruses. 1992;8:357–366. doi: 10.1089/aid.1992.8.357. [DOI] [PubMed] [Google Scholar]
- 13.Kaur A, Grant RM, Means RE, McClure H, Feinberg M, Johnson RP. Diverse host responses and outcomes following simian immunodeficiency virus SIVmac239 infection in sooty mangabeys and rhesus macaques. J Virol. 1998;72:9597–9611. doi: 10.1128/jvi.72.12.9597-9611.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Veazey RS, Tham IC, Mansfield KG, DeMaria M, Forand AE, Shvetz DE, Chalifoux LV, Sehgal PK, Lackner AA. Identifying the target cell in primary simian immunodeficiency virus (SIV) infection: highly activated memory CD4(+) T cells are rapidly eliminated in early SIV infection in vivo. J Virol. 2000;74:57–64. doi: 10.1128/jvi.74.1.57-64.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 16.Finzi D, Siliciano RF. Viral dynamics in HIV-1 infection. Cell. 1998;93:6314–6319. doi: 10.1016/s0092-8674(00)81427-0. [DOI] [PubMed] [Google Scholar]
- 17.Hazenberg MD, Otto SA, Benthem BHv, Roos MT, Coutinho RA, Lange JM, Hamann D, Prins M, Miedema F. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS. 2003;17:1881–1888. doi: 10.1097/00002030-200309050-00006. [DOI] [PubMed] [Google Scholar]
- 18.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: Function, Generation, and Maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 19.Song K, Rabin RL, Hill BJ, Rosa SCD, Perfetto SP, Zhang HH, Foley JF, Reiner JS, Liu J, Mattapallil JJ, Douek DC, Roederer M, Farber JM. Characterization of subsets of Cd4+ memory T cells reveals early branched pathways of T cell differntiation in humans. Proc Natl Acad Sci USA. 2005;102:7916–7921. doi: 10.1073/pnas.0409720102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuroda MJ, Schmitz JE, Charini WA, Nickerson CE, Lifton MA, Lord CI, Forman MA, Letvin NL. Emergence of CTL coincides with clearance of virus during primary simian immunodeficiency virus infection in rhesus monkeys. J Immunol. 1999;162:5127–5133. [PubMed] [Google Scholar]
- 21.
- 22.Ansari AA, Bostik P, Mayne AE, Villinger F. Failure to expand influenza and tetanus toxoid memory T cells in vitro correlates with disease course in SIV infected rhesus macaques. Cell Immunol. 2001;210:125–142. doi: 10.1006/cimm.2001.1810. [DOI] [PubMed] [Google Scholar]
- 23.Pitcher CJ, Hagen SI, Walker JM, Lum R, Mitchell BL, Maino VC, Axthelm MK, Picker LJ. Development and homeostasis of T cell memory in rhesus macaque. J Immunol. 2002;168:29–43. doi: 10.4049/jimmunol.168.1.29. [DOI] [PubMed] [Google Scholar]
- 24.Knuchel M, Bednarik DP, Chikkala N, Ansari AA. Biphasic in vitro regulation of retroviral replication by CD8+ cells from non-human primates. J Acquir Immune Defic Syndr. 1994;7:438–446. [PubMed] [Google Scholar]
- 25.Knuchel M, Bednarik DP, Chikkala N, Villinger F, Folks TM, Ansari AA. Development of a novel quantitative assay for the measurement of chloramphenicol acetyl transferase (CAT) mRNA. J Virol Methods. 1994;48:325–338. doi: 10.1016/0166-0934(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 26.Kaur A, Hale CL, Noren B, Kassis N, Simon MA, Johnson RP. Decreased Frequency of Cytomegalovirus (CMV)-Specific CD4+ T Lymphocytes in Simian Immunodeficiency Virus-Infected Rhesus Macaques: Inverse Relationship with CMV Viremia. J Virol. 2002;76:3646–3658. doi: 10.1128/JVI.76.8.3646-3658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grissmer S, Dethlefs B, Wasmuth JJ, Goldin AL, Gutman GA, Cahalan MD, Chandy KG. Expression and chromosomal localization of a lymphocyte K+ channel gene. Proc Natl Acad Sci USA. 1990;87:9411–9415. doi: 10.1073/pnas.87.23.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 29.Bennett JS, Gossett KA, McCarthy MP, Simpson ED. Effects of ketamine hydrochloride on serum biological and hematological variables in rhesus monkeys (Macaca mulatta) Vet Clin Pathol. 1992;21:15–18. doi: 10.1111/j.1939-165x.1992.tb00576.x. [DOI] [PubMed] [Google Scholar]
- 30.Kim C-Y, Lee H-S, Han S-C, Heo J-D, Kwon M-S, Ha C-S, Han S-S. Hematological and serum biochemical values in cynomologus monkeys anesthesized with ketamine hydrochloride. J Med Primatol. 2005;34:96–100. doi: 10.1111/j.1600-0684.2005.00097.x. [DOI] [PubMed] [Google Scholar]
- 31.Kaur A, Daniel MD, Hempel D, Martin D, Hirsch S, Johnson RP. Cytotoxic T-Lymphocyte Responses to Cytomegalovirus Normal and Simian Immunodeficiency Virus-Rhesus Macaques. J Virol. 1996;70:7725–7733. doi: 10.1128/jvi.70.11.7725-7733.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villinger F, Brice GT, Mayne AE, Bostik P, Mori K, June CH, Ansari AA. Adoptive transfer of simian immunodeficiency virus (SIV) naive autologous CD4+ cells to macaques chronically infected with SIV is sufficient to induce long-term nonprogressor status. Blood. 2002;99:590–599. doi: 10.1182/blood.v99.2.590. [DOI] [PubMed] [Google Scholar]
- 33.Azam P, Sankaranarayanan A, Homerick D, Griffey S, Wulff H. Targeting effector memory T cells with the small molecule Kv1.3 blocker PAP-1 suppresses allergic contact dermatitis. J Invest Dermatol. 2007 doi: 10.1038/sj.jid.5700717. Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Freedman BD, Fleischmann BK, Punt J, Hashimoto Y, Gaulton G, Kotlikoff MI. Identification of Kv1.1 expression by murine CD4 − CD8 − thymocytes: A role for voltage-dependent K + channels in murine thymocyte development. J Biol Chem. 1995;270:22406–22411. doi: 10.1074/jbc.270.38.22406. [DOI] [PubMed] [Google Scholar]
- 35.Lewis RS, Cahalan MD. Subset-specific expression of potassium channels in developing murine T lymphocytes. Science. 1988;239:771–775. doi: 10.1126/science.2448877. [DOI] [PubMed] [Google Scholar]
- 36.Beeton C, Chandy KG. Potassium channels, memory T cells, and multiple sclerosis. Neuroscientist. 2005;11:550–562. doi: 10.1177/1073858405278016. [DOI] [PubMed] [Google Scholar]
- 37.Koo GC, Blake JT, Talento A, Nguyen M, Lin S, Sirotina A, Shah K, Mulvany KDH, Jr, Cunningham P, Wunderler DL, McManus OB, Slaughter R, Bugianesi R, Felix J, Garcia M, Williamson J, Kaczorowski G, Sigal NH, Springer MS, Feeney W. Blockade of the voltage-gated potassium channel Kv1.3 inhibits immune responses in vivo. J Immunol. 1997;158 [PubMed] [Google Scholar]
- 38.Asher DMCJG, Jr, Lang DJ, Gajdusek DC. Persistent shedding of cytomegalovirus in the urine of healthy monkeys (37897) Proc Soc Exp Biol Med. 1974;145:794–801. doi: 10.3181/00379727-145-37897. [DOI] [PubMed] [Google Scholar]
- 39.Baskin GB. Disseminated cytomegalovirus infection in immunodeficient rhesus monkeys. Am J Pathol. 1987;129:345–352. [PMC free article] [PubMed] [Google Scholar]
- 40.Osborn KG, Prahalada S, Lowenstine LJ, Gardner MB, Maul DH, Henrickson RV. The pathology of an epizootic of acquired immunodeficiency in in rhesus macaques. Am J Pathol. 1984;114:94–103. [PMC free article] [PubMed] [Google Scholar]
- 41.Vogel P, Weigler BJ, Kerr H, Hendrickx AG, Barry PA. Seroepidemiologic studies of cytomegalovirus infection in a breeding population of rhesus macaques. Lab Anim Sci. 1994;44:25–30. [PubMed] [Google Scholar]
- 42.Kaur A, Kassis N, Hale CL, Simon M, Elliott M, Gomez-Yafal A, Lifson JD, Desrosiers RC, Wang F, Barry P, Mach M, Johnson RP. Direct relationship between suppression of virus-specific immunity and emergence of cytomelgalovirus disease in simian. AIDS J Virol. 2003;77:5749–5758. doi: 10.1128/JVI.77.10.5749-5758.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kean LS, Adams AB, Strobert E, Hendrix R, Gangappa S, Jones TR, Shirasugi N, Rigby MR, Hamby K, Jiang J, Bello H, Anderson D, Cardona K, Durham MM, Pearson TC, Larsen CP. Induction of chimerism in rhesus macaques through stem cell transplant and costimulation blockade-based suppression. Am J Transplant. 2007;7:320–335. doi: 10.1111/j.1600-6143.2006.01622.x. [DOI] [PubMed] [Google Scholar]
- 44.Shah K, Blake JT, Huang C, Fischer P, Goo GC. Immunosuppressive effects of a Kv1.3 inhibitor. Cell Immunol. 2003;221:100–106. doi: 10.1016/s0008-8749(03)00063-7. [DOI] [PubMed] [Google Scholar]
- 45.Silvestri G, Fedanov A, Germon S, Kozyr N, Kaiser WJ, Garber DA, McClure H, Feinberg MB, Staprans SI. Divergent host responses during primary simian immunodeficiency virus SIVsm infection of natural sooty mangabey and nonnatural rhesus macaque hosts. J Virol. 2005;79:4043–4054. doi: 10.1128/JVI.79.7.4043-4054.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Silvestri G, Sodora DL, Koup RA, Paiardini M, O’Neil SP, McClure HM, Staprans SI, Feinberg MB. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity. 2003;18:441–452. doi: 10.1016/s1074-7613(03)00060-8. [DOI] [PubMed] [Google Scholar]