

Abstract
Cells released from primary tumors seed metastases to specific organs by a non-random process, implying the involvement of biologically selective mechanisms. Based on clinical, functional and molecular evidence, we show that the cytokine TGFβ in the breast tumor microenvironment primes cancer cells for metastasis to the lungs. Central to this process is the induction of angiopoietin-like 4 (ANGPTL4) by TGFβ via the Smad signaling pathway. TGFβ induction of ANGPTL4 in cancer cells that are about to enter the circulation enhances their subsequent retention in the lungs, but not in the bone. Tumor cell-derived ANGPTL4 disrupts vascular endothelial cell-cell junctions, increases the permeability of lung capillaries, and facilitates the trans-endothelial passage of tumor cells. These results suggest a mechanism for metastasis whereby a cytokine in the primary tumor microenvironment induces the expression of another cytokine in departing tumor cells, empowering these cells to disrupt lung capillary walls and seed pulmonary metastases.
INTRODUCTION
The identification of metastasis genes and mechanisms is essential for understanding the basic biology of this lethal condition and its implications for clinical practice (Fidler, 2003; Gupta, 2006). The predisposition of primary tumors to selectively invade different organs has been long recognized (Paget, 1889). Recent work has functionally identified and clinically validated sets of genes whose overexpression in breast cancer cells confers a selective advantage for the colonization of bones (Kang et al., 2003b; Lynch et al., 2005) or lungs (Minn et al., 2005). There is also the possibility that the microenvironment of a primary tumor may influence the fate of cancer cells that escape from this tumor. Among the factors in the tumor microenvironment that might play such a role, we chose to focus on the cytokine TGFβ. Accumulating evidence indicates that this cytokine can modulate tumor progression in various experimental systems (Bierie and Moses, 2006; Dumont and Arteaga, 2003; Siegel and Massagué, 2003).
TGFβ is a multifunctional cytokine with diverse effects on virtually all cell types and with key roles during embryo development and tissue homeostasis (Massagué et al., 2000). It regulates the production of microenvironment sensors and modulators, including cytokines, extracellular matrix components and cell surface receptors. Additionally, TGFβ has potent inhibitory effects on cell proliferation and, as such, it can deter tumor growth (Bierie and Moses, 2006; Dumont and Arteaga, 2003; Siegel and Massagué, 2003). Within the tumor microenvironment, TGFβ is produced by macrophages, mesenchymal cells and the cancer cells themselves, as a natural response to the hypoxic and inflammatory conditions that occur during tumor progression. The TGFβ receptors, which are membrane serine/threonine protein kinases, and their substrates, the Smad transcription factors, are tumor suppressors that frequently suffer inactivation in gastrointestinal, pancreatic, ovarian and hepatocellular cancinomas and subsets of gliomas and lung adenocarcinomas (Bierie and Moses, 2006; Levy and Hill, 2006). However, in breast carcinoma, glioblastoma, melanoma and other types of cancer, selective losses of growth inhibitory responses often accrue through alterations downstream of Smad, leaving the rest of the TGFβ pathway operational and open to co-option for tumor progression advantage (Massagué and Gomis, 2006). Low level expression of TGFβ receptors in the ER negative (ER−) breast tumors is associated with better overall outcome (Buck et al., 2004), whereas overexpression of TGFβ1 is associated with a high incidence of distant metastasis (Dalal et al., 1993). Studies in mouse models of breast cancer have implicated TGFβ in the suppression of tumor emergence (Bierie and Moses, 2006; Siegel and Massagué, 2003), but also in the induction of epithelial-mesenchymal transitions and tumor invasion (Thiery, 2002; Welch et al., 1990), the production of osteoclast-activating factors in the bone metastasis microenvironment (Kang et al., 2003b; Mundy, 2002), and the context-dependent induction of metastasis (Dumont and Arteaga, 2003; Siegel and Massagué, 2003). Thus, the effects of TGFβ on breast cancer progression in mouse models are as profound as they are disparate, making it difficult to discern from these models the role that TGFβ may be playing in human breast cancer.
To investigate the contextual role of the TGFβ pathway in human cancer and the mechanism by which TGFβ may instigate metastasis, we based our present work on the weight of clinical evidence and the use of a bioinformatics tool that classifies tumors based on the status of their TGFβ transcriptional readout. Applying this tool to a wealth of clinically annotated samples and gene expression data sets, we made the surprising observation that TGFβ activity in primary breast tumors is associated with an increased propensity of these patients to develop lung metastasis but not bone metastasis. This phenomenon implies a biologically selective TGFβ-dependent mechanism that favors tumor targeting of the lungs. We identify this mechanism based on ANGPTL4 as a critical TGFβ target gene, whose induction in cancer cells in the primary tumor primes these cells for disruption of lung capillary endothelial junctions to selectively seed lung metastasis.
RESULTS
Development of a TGFβ response bioinformatics classifier
In order to investigate the role of TGFβ in cancer progression, we set out to develop a bioinformatics classifier that would identify human tumors containing a high level of TGFβ activity. A gene expression signature typifying the TGFβ response in human epithelial cells was obtained from transcriptomic analysis of four human cell lines (Figure 1A, Supplementary Figure 1). These cell lines include HaCaT keratinocytes, HPL1 immortalized lung epithelial cells, MCF10A breast epithelial cells, and MDA-MB-231 breast carcinoma cells. The cells were treated with TGFβ1 for 3 h in order to capture direct TGFβ gene responses (Kang et al., 2003a). The resulting 153-gene TGFβ response signature (TBRS) (174 probe sets; Supplementary Table 1) was used to generate a classifier by means of “meta-gene” analysis with the cell lines as references (Bild et al., 2006). The meta-gene analysis resulted in a continuous variable ranging from 0 to 1 that designates the relative level of TGFβ pathway activity in tissue samples. Using 0.5 as a threshold, most tumors could be unambiguously assigned to a TBRS− class or a TBRS+ class. When applied to metastatic lesions extracted from bones, lungs and other sites representing the natural metastatic spectrum of human breast cancer, the TBRS classifier identified TGFβ activity in a 38/67 of these samples (Supplementary Table 2), which is in agreement with previous observations of activated Smad in a majority of human bone metastasis samples (Kang et al., 2005).
Figure 1. The TBRS associates with breast cancer metastasis in humans.
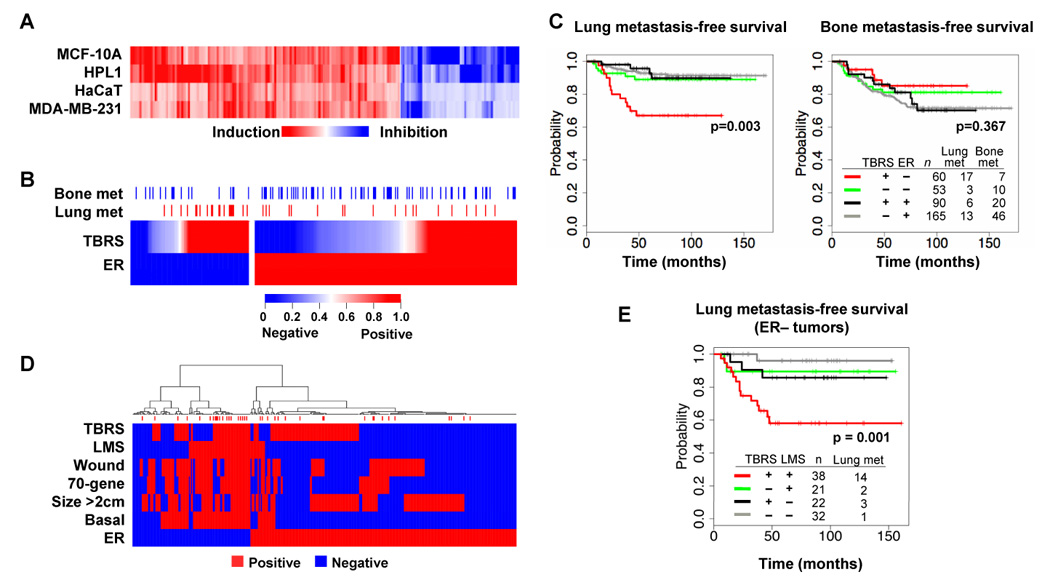
(A) The indicated epithelial cell lines were incubated for 3 h with TGFβ and then total RNA was subjected to microarray analysis. The heat map represents the change in expression levels of the 153 genes within the TBRS.
(B) TBRS status was assessed in a MSK/EMC cohort of 368 primary breast cancer tumors with known lung or bone metastatic outcomes. Red denotes a strong correlation between individual tumor gene expression profiles and the TBRS while blue indicates no correlation. Estrogen receptor α(ER) expression status is also indicated. Blue and red marks above the heat map indicate tumors that at any time developed bone or lung metastases, respectively.
(C) Kaplan-Meier curves representing the probability of cumulative lung (left panel) or bone (right panel) metastasis-free survival for this cohort. Tumors are categorized according to their TBRS and ER status. The P values for the ER-negative tumor comparisons are shown.
(D) Hierarchical clustering was performed on the MSK/EMC cohort with the indicated pathological and genomic markers including the TBRS, the lung metastasis signature (LMS), the wound response signature (Wound), the 70-gene prognosis signature (70-gene), size (Size >2cm), the basal molecular subtype (Basal), and the ER status. Red marks above the map indicate tumors that developed lung metastasis.
(E) Lung metastasis-free survival restricted to patients with ER-negative tumors. Patients were categorized according to their TBRS and LMS status. P value shown for the LMS+ tumor comparisons.
TGFβ activity in primary breast tumors is selectively linked to lung metastasis
We applied the TBRS classifier to a series of primary breast carcinomas that were analyzed on the same microarray platform (Minn et al., 2007; Minn et al., 2005; Wang et al., 2005). This series includes 82 tumors collected at Memorial Sloan-Kettering Cancer Center (MSK cohort) and 286 tumors from the Erasmus Medical Center (EMC cohort). Both cohorts comprised a mix of breast cancer subtypes, with tumors in the MSK cohort being more locally advanced than those in the EMC cohort (Minn et al., 2007). Out of a combined total of 368 patients, 39 patients developed lung metastases and 83 developed bone metastasis after a median follow-up of 10 years, with some patients developing metastases in both sites (Figure 1B). TBRS+ tumors were similarly distributed between estrogen receptor-positive (ER+) and ER− tumors (Figure 1B). Microarray analysis revealed that the TBRS+ tumors expressed significantly higher mRNA levels for TGFβ1, TGFβ2, and the latent TGFβ activating factor, LTBP1. TBRS− tumors had lower mRNA levels for type II TGFβ receptor, Smad3 and Smad4. The expression level of other TGFβ pathway components was the independent of TBRS status (Supplementary Figure 2).
TBRS status in ER+ tumors did not correlate with distant metastasis. However, in ER− tumors there was a striking association between TBRS+ status and relapse to the lungs (Figure 1C). This association was observed regardless of whether the tumor ER status was assigned using the clinical pathology reports, which are based on immunohistochemical analysis (Figure 1B,C), or using a microarray probe level designation (Supplementary Figure 3A,B). No link was observed between TBRS status and bone metastasis (Figure 1D, Supplementary Figure 3C) or liver metastasis, and a brain metastasis association did not attain statistical significance (Supplementary Figure 4). In univariate as well as multivariate analyses, the expression level of TGFβ pathway components was much inferior to the TBRS at linking these tumors with metastasis outcome (Supplemental Table 3). These results indicate that TGFβ activity in ER− breast tumors is selectively associated with lung metastasis.
Cooperation between TGFβ and the Lung Metastasis Signature
The association of TBRS with lung relapse prompted us to search for links between the TBRS and a previously described lung metastasis signature (LMS) (Minn et al., 2005). The LMS is a set of 18 genes whose expression in ER− tumors indicates a high risk of pulmonary relapse in patients (Minn et al., 2007). Several of these genes have been validated as mediators of lung metastasis (Gupta et al., 2007a; Gupta et al., 2007b; Gupta, 2007; Minn et al., 2005). The TBRS+ subset of ER− tumors partially overlapped the LMS+ subset (Figure 1D). Remarkably, tumors that were positive for both the TBRS and LMS were associated with a high risk of pulmonary relapse, whereas single-positive tumors were not (Figure 1E). Within poor-prognosis tumor subsets defined by other features, such as size >2cm, basal subtype gene-expression signature (Sorlie et al., 2003), 70-gene poor prognosis signature (van de Vijver et al., 2002), or wound signature (Chang et al., 2005), TBRS status was associated with risk of lung metastasis in nearly every case (Figure 1D). The TBRS performed independently of these other prognostic features (Supplementary Figure 5), as did the LMS (Supplementary Figure 6 (Minn et al., 2007).
TGFβ signaling in mammary tumors enhances lung metastatic dissemination
To functionally test whether TGFβ signaling in primary tumors contributes to lung metastasis, we used a xenograft model of ER− breast cancer (Minn et al., 2005). The MDA-MB-231 cell line was established from the pleural fluid of a patient with ER− metastatic breast cancer (Cailleau et al., 1978). MDA-MB-231 cells have a functional Smad pathway and evade TGFβ growth inhibitory responses through alterations downstream of Smads (Gomis et al., 2006). The lung metastatic subpopulation LM2-4175 (henceforth LM2) was isolated by in vivo selection of MDA-MB-231 cells (Minn et al., 2005). We perturbed the TGFβ pathway in LM2 cells by overexpressing a kinase-defective, dominant-negative mutant form of the TGFβ type I receptor (Weis-Garcia and Massagué, 1996), or by reducing the expression of Smad4, which is an essential partner of Smad2/3 in the formation of transcriptional complexes (Massagué et al., 2005). Using a validated SMAD4 short-hairpin RNA (shRNA) (Kang et al., 2005) we reduced Smad4 levels by 80–90% in LM2 cells (Figure 2B). As a control, we generated SMAD4 rescue cells by expressing a shRNA-resistant SMAD4 cDNA in SMAD4 knockdown cells (Figure 2B).
Figure 2. TGFβ signaling enhances mammary tumor dissemination to the lungs.
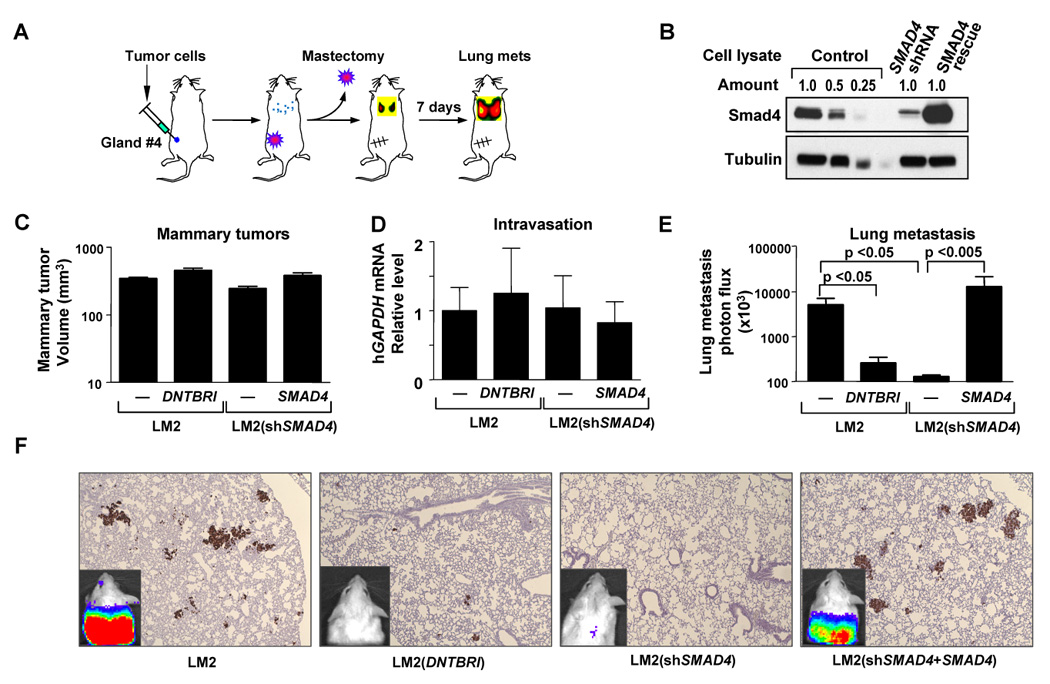
(A) Schematic of lung metastasis assay from an orthotopic breast cancer inoculation.
(B) Immunoblots using indicated antibodies were performed on whole-cell extracts from control, Smad4 knockdown, and Smad4-Rescue LM2 cells.
(C) Mice injected with 5×105 cells into the fourth mammary fat pads were measured for tumor size at day 28. n=14; error bars indicated s.e.m
(D) Blood from tumor-bearing mice was isolated and red blood cells lysed. RNA from the remaining cells was extracted for qRT-PCR. The presence of circulating tumor cells was assessed as a function of human-specific GAPDH expression relative to murine β2-microglobulin, in 3 mL of mouse blood perfusate. n=8; error bars indicate s.e.m.
(E) Bioluminescent quantification of lung seeding elicited from orthotopically implanted breast tumors. Orthotopic tumors were grown to approximately 300mm3, mastectomies were performed and lung seeding was quantified using bioluminescence imaging seven days later. n=7–15; error bars indicated s.e.m; p-values calculated using the one-tailed unpaired t-test.
(F) Representative bioluminescent images (inset) and lung histology of mice with the median value of bioluminescence in each experimental group in (D). Breast cancer cells were stained with human vimentin.
Neither the dominant negative TGFβ receptor nor the Smad4 knockdown decreased mammary tumor growth as determined by tumor volume measurements, or the extent of tumor cell passage into the circulation, as determined by qRT-PCR analysis of human GAPDH mRNA in blood cellular fractions (Figure 2C, 2D). Tumors inoculated into the mammary glands of immunocompromised mice and allowed to grow to 300 mm3, were surgically removed and the emergence of disseminated cells to the lungs after the mastectomy was determined (Figure 2A). Inactivation of TGFβ signaling markedly inhibited the lung metastatic seeding of the tumors as determined by quantitative luciferase bio-luminescence imaging (Figure 2E; Figure 2F insets) (Ponomarev et al., 2004) and histological examination (Figure 2F). These results suggest that the canonical TGFβ pathway enhances mammary tumor dissemination to the lungs.
TGFβ primes tumor cells to seed lung metastases
We wondered whether TGFβ within the breast tumor microenvironment could endow tumor cells with the ability to seed the lungs as these cells enter the circulation. To test this possibility, we mimicked the exposure of tumor cells to TGFβ by incubating LM2 cells with TGFβ for 6h prior to inoculation of these cells into the tail veins of mice. Interestingly, this pre-treatment with TGFβ significantly increased the lung colonizing activity of LM2 cells, as determined by a higher retention of these cells in the lungs 24 h after inoculation (Figure 3A). In this time frame LM2 cells extravasate into the lung parenchyma (Gupta et al., 2007a). A similar effect was observed when we carried out this experiment with malignant cells (CN34.2A) obtained from the pleural fluid of a breast cancer patient treated at MSKCC. The pre-treatment with TGFβ increased the lung seeding activity of LM2 and CN34.2A cells three- and five-fold, respectively (Figure 3B). The initial advantage provided by a transient exposure to TGFβ was sustained but not expanded during the ensuing outgrowth of metastatic colonies (Figure 3A, and data not shown).
Figure 3. TGFβ primes tumor cells for metastatic seeding of the lungs.
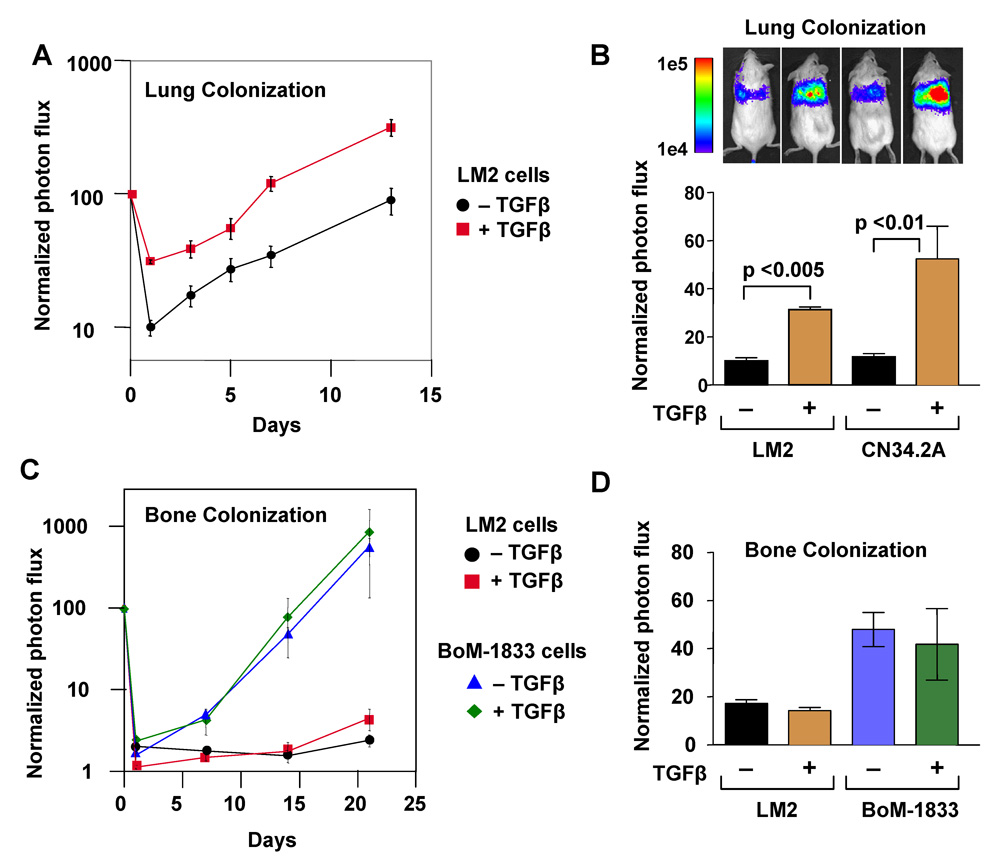
(A) LM2 cells and a clinically-derived pleural effusion sample (CN34.2A) were pretreated with TGFβ for 6 h. LM2 (2×105) and CN34.2A (4×105) cells were injected into the lateral tail vein and lung colonization was analyzed by in vivo bioluminescence imaging.
(B) Bar graph represents 24h time point measurements of the normalized photon flux from animals injected with either LM2 or CN34.2A cells. In vivo bioluminescent mouse images shown are from representative animals. n=7 for the LM2 experiment while n=6 for the CN34.2A experiment; error bars indicate s.e.m; p-values calculated using the one-tailed unpaired t-test.
(C) Bone colonization assays were performed by intracardiac injection of LM2 or BoM-1833 cells (3×104). Samples were pretreated with 100 pM of TGFβ for 6 h and compared to an untreated control. Plot represents normalized photon flux from mouse hindlimbs
(D) Bar graphs represent seven-day time point analysis of the normalized photon flux from the mouse hind limbs. n=8; error bars indicate s.e.m.
To investigate the selectivity of this lung metastasis-priming effect, we tested the effect of TGFβ pre-incubation on the establishment of bone metastases. LM2 cells have limited bone metastatic activity in addition to their high lung metastatic activity (Minn et al., 2005). The pre-treatment of LM2 cells with TGFβ prior to their inoculation into the arterial circulation did not increase the ability of these cells to colonize the bone (Figure 3C). We also tested the effect of TGFβ on the metastatic seeding of an MDA-MB-231 sub-population (BoM-1833) that is highly metastatic to bone (Kang et al., 2003b) and responsive to TGFβ (Kang et al., 2005). Pre-incubation of BoM-1833 cells with TGFβ did not increase their bone colonizing ability (Figure 3C), and had no discernible effect on the early seeding of the bones (Figure 3D). Thus, TGFβ stimulation primes tumor cells for an early step in lung metastasis but not bone metastasis, which is concordant with the selective association of TBRS+ status in primary tumors with risk of lung metastasis in clinical cohorts (refer to Figure 1C).
The TBRS/LMS gene ANGPTL4 is a TGFβ target in breast cancer
Given the convergence of the TBRS and the LMS in linking human primary tumors to risk of lung metastasis, we wondered whether TGFβ may act by augmenting the activity of a LMS gene(s). The LMS includes 15 candidate mediators of lung metastasis and three suppressors (Minn et al., 2005) (see Figure 4C). Interestingly, the LMS genes ANGPTL4, which encodes the multifunctional factor angiopoietin-like 4 (Oike et al., 2004), and NEDD9, which encodes an adaptor protein implicated in focal contact formation and cell motility (Kim et al., 2006), were present in the TBRS (Supplementary Table 1). An induction of ANGPTL4 by TGFβ was observed in four different epithelial cell types tested (Figure 4A). Moreover, among ER− tumors ANGPTL4 expression was significantly higher in the TBRS+ tumors (median-centered intensity value=1.07) than in TBRS− tumors (median value=0.30). NEDD9 expression was not different between these two groups (Figure 4B). TBRS+ and TBRS− tumors in the ER+ group showed a smaller difference in ANGPTL4 expression (Supplementary Figure 7).
Figure 4. The TBRS/LMS gene ANGPTL4 is a Smad-dependent TGFβ target.
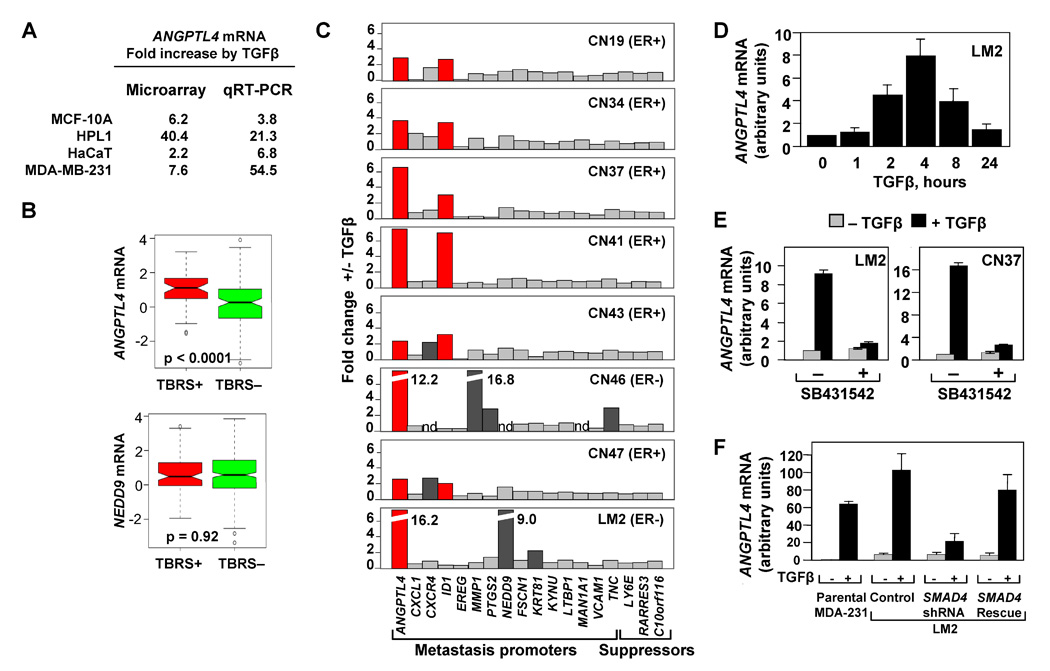
(A) Microarray and qRT-PCR analysis for the four epithelial cell lines treated with TGFβ. Fold change values for the TGFβ induction of ANGPTL4 are indicated.
(B) Box-and-whisker plot comparing ANGPTL4 and NEDD9 TBRS-negative and -positive ER-negative tumors from the MSK/EMC cohorts. P value was calculated using the Wilcoxon rank sum test.
(C) TGFβ-induced changes in the mRNA expression of LMS genes in a panel of clinically derived pleural effusion samples and LM2 cells. Cells were treated with 100 pM of TGFβ for 3 h and analyzed by qRT-PCR using primers for the indicated genes. ER status for each breast cancer patient is designated.
(D) LM2 breast cancer cells were treated with 100pM of TGFβ for the indicated lengths of time and ANGPTL4 mRNA levels were analyzed using qRT-PCR.
(E) Treatment of LM2 (left panel) and pleural effusion-derived CN37 sample (right panel) with TGFβ and the TGFβ-receptor kinase inhibitor, SB431542. qRT-PCR expression levels are shown relative to the untreated control sample.
(F) MDA-231, LM2 control, LM2-Smad4-depleted, and LM2-Smad4-Rescue cell lines were treated with 100 pM TGFβ for 3 h. TGFβ-induced fold changes of ANGPTL4 were analyzed by qRT-PCR analysis, n=3; error bars indicate standard deviation.
To determine the effect of TGFβ on individual LMS genes, we used tumor cells isolated from pathological pleural fluids from patients with ER− and ER+ metastatic breast cancer. Lung metastasis was diagnosed in 6/7 of these cases. All samples were obtained from routine therapeutic procedures, and were used under institutionally approved protocols and informed consent (Gomis et al., 2006). Carcinoma cells were isolated from these samples using the epithelial cell surface marker EpCAM (Kielhorn et al., 2002). TGFβ addition increased ANGPTL4 expression between 2- and 12-fold in all metastatic samples, and 16-fold in the LM2 cells, as determined by quantitative (q)RT-PCR (Figure 4C). These results confirm that the LMS gene ANGPTL4 is a TGFβ target gene in breast cancer cells.
None of the other LMS genes, NEDD9 included, was consistently regulated by TGFβ in this set of samples, with one exception: the transcriptional inhibitor of cell differentiation ID1 was induces approximately two-fold by TGFβ in most samples (Figure 4C). As a component of the LMS, ID1 mediates tumor re-initiation after ER− cells enter the lung parenchyma (Gupta et al., 2007b). This induction of ID1 by TGFβ is interesting less for its restricted magnitude than for the fact that TGFβ represses ID1 in untransformed breast epithelial cells (Kang et al., 2003a). This switched responsiveness of ID1 is consistent with the pattern of loss of TGFβ growth inhibitory responses in metastatic breast cancer cells (Gomis et al., 2006).
The induction of ANGPTL4 expression by TGFβ was observed in all 13 malignant pleural cell samples tested, regardless of the ER, progesterone receptor or ERBB2 receptor status and type of the original primary tumor (Table 1). The induction of ANGPTL4 by TGFβ was rapid and lasted for 8h (Figure 4D). Addition of SB431542, an ATP analogue inhibitor of the TGFβ type I receptor kinase (Laping et al., 2002), abolished the ANGPTL4 response in LM2 and CN37 cells (Figure 4E). Smad4 knockdown markedly inhibited the ANGPTL4 response to TGFβ, whereas a shRNA-resistant SMAD4 cDNA containing two silent mutations in the shRNA-targeted sequence rescued this response (Figure 4F). Additionally, we tested ANGPTL4 induction by a variety of cytokines that are typical of the tumor microenvironment. In this group, TGFβ was the strongest inducer of ANGPTL4 in the MDA-MB-231 cells (Supplementary Figure 8). Thus, ANGPTL4 induction in metastatic breast cancer cells is mediated by the canonical TGFβ-receptor-Smad pathway.
Table 1.
Clinical and histological staging of malignant pleural effusion samples, and theirANGPTL4 response to TGFβ
| ER Statusa | PR Statusa | ERBB2 Statusa | Tumor Typea | Metastasis Sites | ANGPTL4 mRNA +/− TGFβ | |
|---|---|---|---|---|---|---|
| CN19 | + | + | + | ductal | Pl, Lu, LN | 2.9 |
| CN34 | + | − | ++ | ductal | Pl, Lu, Bo, LN | 3.8 |
| CN37 | + | + | ++ | ductal | Pl, Lu, LN, Br, Li | 6.7 |
| CN41 | + | + | ++ | ductal | Pl, Lu?, Bo, Br, CW | 8.7 |
| CN43 | + | + | + | ductal | Pl, Lu, Bo, Li | 2.3 |
| CN46 | − | − | +++ | ductal | Pl, Bo, ST, LN | 12.8 |
| CN47 | + | + | + | lobular | Pl, Lu, Li, LN, Ca, Co | 2.8 |
| CN90 | − | − | − | ductal | Pl, Lu, Bo, Li | 24.7 |
| BCN5 | + | + | + | ductal | Pl, Lu, Bo, LN, Sc | 9.5 |
| BCN6 | + | + | +++ | ductal | Pl, Lu, Bo, Li | 10.9 |
| BCN7 | + | + | + | ductal | Pl, Lu, Bo, Li, LN, Ad | 10.2 |
| BCN8 | + | − | − | lobular | Pl, Bo, As, ST | 3.0 |
| BCN9 | − | − | − | ductal | Pl, LN | 13.7 |
Status in primary tumor
Pl, pleura; Lu, lung; Bo, bone; Br, brain; Sc, subcutaneous; LN, lymph nodes; Li, liver; Ad, adrenal; As, ascites; ST, soft tissue; Ca perinoteal carcinomatosis; Co, colon; CW, chest wall
ANGPTL4 participates in TGFβ priming for lung metastasis
To investigate whether ANGPTL4 participates in the pro-metastatic effects of TGFβ, we knocked down its expression in LM2 cells by means of a shRNA. LM2 cells expressing a rescue ANGPTL4 cDNA together with this shRNA serves as a control (Figure 5A). This knockdown did not decrease the ability of LM2 cells to grow as mammary tumors (Figure 5B) and to pass into the circulation (Figure 5C). The incidence of lymph node metastases in LM2 tumor-bearing mice was also not affected by ANGPTL4 knockdown, as determined by ex-vivo analysis of luciferase activity the excised lymph nodes (Figure 5D). However, the dissemination to the lungs from orthotopically implanted LM2 cells was decreased more than 10-fold by the ANGPTL4 knockdown, and this decrease could be prevented with the ANGPTL4-rescue construct (Figure 5E). ANGPTL4 knockdown did not decrease the residual bone metastatic activity of LM2 cells (data not shown). These results provided functional evidence that ANGPTL4 is involved in metastatic dissemination to the lungs by orthotopically implanted LM2 tumors.
Figure 5. ANGPTL4 mediates TGFβ priming for mammary tumor dissemination to the lungs.
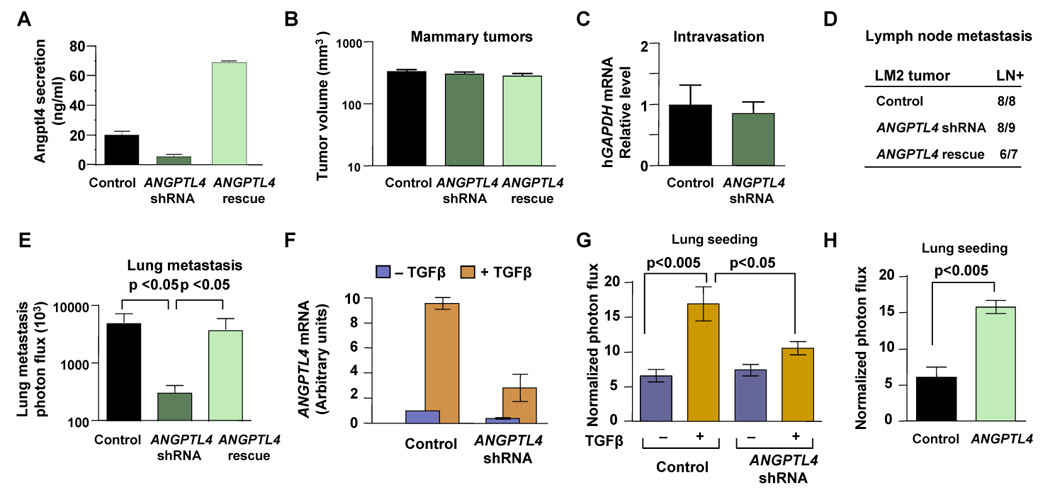
(A) Secreted Angptl4 protein levels in the control, ANGPTL4 knockdown, or ANGPTL4-rescue LM2 cells were analyzed by ELISA. n=3; error bars indicate standard deviation
(B) Tumors size measurements were taken from mice inoculated into the fourth mammary fat pads with 5×105 control, ANGPTL4 knockdown, or ANGPTL4-rescue LM2 cells. Tumor size was measured at day 28. n=14; error bars indicated s.e.m
(C) The presence of circulating tumor cells was assessed by qRT-PCR as a function of human-specific GAPDH expression relative to murine β2-microglobulin, in 3 mL of mouse blood perfusate. n=10; error bars indicate s.e.m.
(D) Peri-aortic lymph node metastasis from indicated tumors was analyzed by ex-vivo detection of luciferase activity. Positive bioluminescent signal in extracted lymph nodes indicated presence of metastasized tumor cells.
(E) Photon flux measurements of breast cancer cells seeding the lung from orthotopically injected tumors as indicated. n=13–15 error bars indicated s.e.m; p-values calculated using the one-tailed unpaired t-test.
(F) ANGPTL4 mRNA levels were determined by qRT-PCR analysis in cells that were incubated for 6 h with or without TGFβ. n=3; error bars indicate standard deviation.
(G) Lung colonization analysis was performed by injecting 2×105 cells into the lateral tail vein. Prior to injection, cells were treated as indicated with 100 pM TGFβ for 6 h. Bar graphs represent 24 h time point measurements of the normalized photon flux. n=14–21; error bars indicate s.e.m; p-values calculated using the one-tailed unpaired t-test.
(H) Normalized photon flux measurements from tail vein injected animals. Lung colonization measurements were taken from animals injected with control or Angptl4 overexpressing LM2 cells. n=6; error bars indicate s.e.m; p-values calculated using the one-tailed unpaired t-test.
When orthotopically implanted, LM2 tumors accrue TGFβ activity that primes lung metastasis seeding (refer to Figure 2D). We subjected the ANGPTL4 knockdown LM2 cells to the ex-vivo TGFβ priming assay. Of note, the induction of ANGPTL4 expression by TGFβ was blunted but not completely eliminated in the knockdown cells (Figure 5F). This notwithstanding, the knockdown of ANGPTL4 significantly blunted the priming effect of TGFβ on lung seeding by LM2 cells (Figure 5G). The constitutive overexpression of exogenous ANGPTL4 in LM2 cells increased lung colonization by these cells (Figure 5H). These results provide evidence that ANGPTL4 expression is necessary for the ability of TGFβ to prime LMS+ breast cancer cells and sufficient for increasing seeding of the lungs.
ANGPTL4 mediates endothelial disruption and trans-endothelial tumor cell passage
The ability of TGFβ to promote lung seeding through an induction of ANGPTL4 suggested that this process may target an early pulmonary seeding step. Extravasation, or the passage of circulating tumor cells through the tight lung capillary endothelial junctions, is an important initial step in lung colonization. We, therefore, investigated whether Angptl4 might affect endothelial cell layers in a manner that would facilitate the passage of tumor cells across endothelia. HUVEC human vascular endothelial cells were allowed to grow to form tight monolayers on tissue culture dishes, and at this point the monolayers were exposed to media containing human recombinant Angptl4 or no addition (Figure 6A), or media conditioned by control LM2 cells or by cells overexpressing Angptl4 (Figure 6B). In both cases Angptl4 caused an acute disruption of endothelial cell-cell junctions. Staining with antibodies against the tight junction component zonula occludens 1 (ZO-1), against the adherens junction component β-catenin, or staining of the actin cytoskeleton with phalloidin (Dejana, 2004), revealed that the monolayer integrity was dramatically perturbed by Angptl4 (Figure 6A and B).
Figure 6. ANGPTL4 mediates endothelial monolayer disruption, lung capillary permeability, and trans-endothelial tumor cell migration.
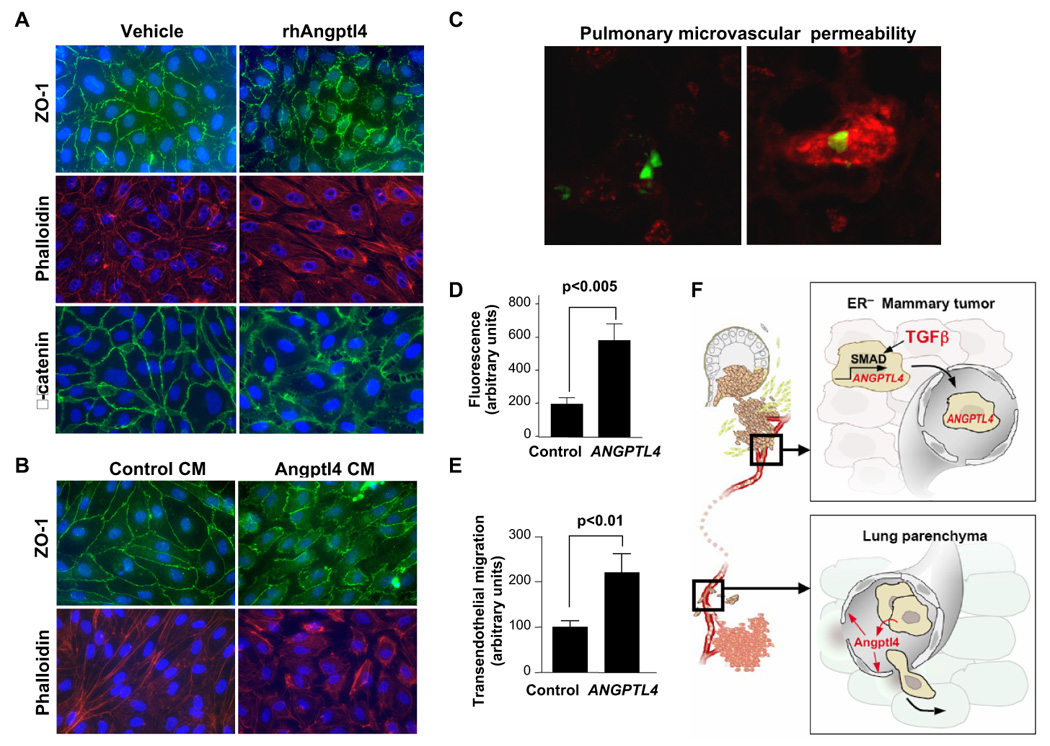
(A) HUVEC monolayers were grown to confluence on fibronectin coated slides and then treated for 24 h with rhAngptl4. Slides were subsequently fluorescently stained with anti-ZO-1 antibody, phalloidin, and anti-β-catenin antibody.
(B) HUVEC monolayers were treated for 24 h with media conditioned by control LM2 cells or LM2 cells that overexpress Angptl4. Samples were stained for ZO-1 and phalloidin.
(C) GFP-labeled MDA-231 cells were injected via the tail vein and allowed to lodge in the lungs. One day post injection, a rhodamine-dextran dye was injected into circulation. Three hours after dye injection, lungs were extracted and frozen sections were obtained. Representative confocal images are shown here of cells with and without accumulation of dye in the lung parenchyma.
(D) Images were obtained as described in (C) with control or Angptl4 overexpressing MDA-MB-231 cells. A region of interest was drawn around the GFP-labeled cells and the amount of dextran dye was quantified based on rhodamine emissions. n=40 cells; error bars indicate s.e.m; p-values calculated using the one-tailed unpaired t-test.
(E) Indicated cell lines were seeded into trans-well inserts that were previously covered with a HUVEC monolayer. Cells that migrated cross the endothelial layer into the bottom side of the transwell membrane were quantified with Volocity software. n=15, error bars indicate s.e.m; p-values calculated using the one-tailed unpaired t-test.
(F) Schematic model of the cytokine relay set up by TGFβ activity in the primary tumor. ER− primary tumor cells that are exposed to TGFβ respond with ANGPTL4 induction via the Smad pathway. As they enter the circulation and reach the lung capillaries, these cells secrete Angptl4 which disrupts endothelial cell junctions thereby enabling the cancer cells to more efficiently enter the lung parenchyma.
To determine if tumor cell-derived Angptl4 can disrupt the integrity of endothelia in pulmonary capillaries, we performed in vivo lung capillary permeability assays. We used parental MDA-MB-231 cells or these cells stably expressing an ANGPTL4 vector, rather than using LM2 cells, in order to avoid potential confounding effects of the other LMS genes that are expressed in LM2 cells (Gupta et al., 2007a). GFP-labeled MDA-MB-231 cells either expressing a control vector or expressing ANGPTL4 were inoculated into NOD/SCID mice. One day post inoculation, the animals were injected with a rhodamine-conjugated dextran, in order to measure vessel permeability. The lungs were then extracted and analyzed for retained rhodamine using fluorescent microscopy. No rhodamine signal was present in the lungs of mice that were not inoculated with cancer cells (data not shown). In inoculated animals, however, diffuse areas of rhodamine signal surrounded the cancer cells that lodged in the lungs (Figure 6C). Cells overexpressing Angptl4 showed a 3-fold increase in surrounding rhodamine signal, as determined by quantitative analysis of the fluorescent area (Figure 6D; Supplemental Figure 9).
To test the effect of Angptl4 on cell migration across an endothelial layer, endothelial monolayers were set on trans-well tissue culture inserts. LM2 cells overexpressing Angptl4 passed twice as efficiently through these layers into the lower chamber of the trans-well compared to control LM2 cells (Figure 6E). Collectively, these data demonstrate that Angptl4 disrupts the integrity of vascular endothelial cell layers both in vitro and in the lungs, facilitating the passage of breast cancer cells.
DISCUSSION
Primary tumor microenvironments may promote metastasis by selecting for highly invasive and resistant cancer cell phenotypes (Bernards and Weinberg, 2002) and systemically fostering the mobilization of marrow-derived progenitor cells (Kaplan et al., 2005). The ability to subsequently colonize distant organs depends on the organ colonizing faculties of disseminated tumor cells as well as on certain permissive conditions that may be present in the otherwise restrictive microenvironment of target organs (Gupta, 2006). The present results suggest a distinct mechanism for the colonization of a distant organ, one that relies on a stimulus in the primary tumor microenvironment to enhance the ability of departing tumor cells to seed the lungs (Figure 6F).
Angptl4 as an inhibitor on endothelial integrity that mediates lung metastasis seeding
Angptl4 is expressed in the liver, adipose tissue, and placenta, as well as in ischemic tissues (Oike et al., 2004). It was identified in a search for new members of the angiopoietin family of vascular regulators, and independently in a search for targets of the PPAR family of metabolic response transcription factors (Oike et al., 2004). While Angptl4’s role in lipid metabolism has been well-characterized, little is known about its role in vascular biology. Indeed, the effects of angiopoietin-like proteins in experimental systems are complex, at times acting as general endothelial cell survival factors (Kim et al., 2000), modulating endothelial cell adhesion (Cazes et al., 2006), or paradoxically stimulating (Hermann et al., 2005; Le Jan et al., 2003) as well as inhibiting angiogenesis (Ito et al., 2003). Chronic systemic secretion of Angptl4 from a transgene expressed in muscle tissue in mice inhibited metastasis by xenografted melanoma cells (Galaup et al., 2006). These diverse and at times opposing responses are suggestive of a context, tissue specific activity of this multifaceted molecule.
ANGPTL4 is one of the top performing genes in the LMS with a highly significantly association with lung relapse (p< 0.000001; (Minn et al., 2005). In the present work, we show that TGFβ stimulation sharply increased the expression of ANGPTL4 in both cell populations, and we have functionally validated ANGPTL4 as a mediator of breast cancer lung metastasis. ANGPTL4 knockdown in LMS+ cells inhibits their ability to seed the lungs, and it does so without affecting the growth of these cells as mammary tumors, their passage into the circulation, or their invasion of lymph nodes. Angptl4 antagonizes vascular endothelial tight junctions and adherens junctions, and disrupts the integrity of capillary walls when secreted from metastatic breast cancer cells that have lodged in the lungs. These results strongly suggest that Angptl4 acts as an enhancer of breast cancer cell extravasation by transiently suppressing the integrity of capillaries These observations fit with the role of Angptl4 as a vascular regulator in ischemia and tumor hypoxia conditions (Le Jan et al., 2003), and are in line with the role of the angiopoietin and angiopoietin-like factors in vascular remodeling (Camenisch et al., 2002; Gale et al., 2002; Parikh et al., 2006). Together with the presence of ANGPTL4 in two distinct gene expression signatures –the LMS and the TBRS− that are associated with lung metastasis in breast cancer patients, this evidence suggests that Angptl4 is a clinically relevant mediator of lung metastasis in breast cancer.
TGFβ activity in primary breast tumors is linked to lung metastasis
Studies in breast cancer patients have shown correlations between the expression of TGFβ pathway components and disease outcome (Levy and Hill, 2006). However, the role of TGFβ in breast cancer progression has remained baffling given the disparate results from various animal models. In transgenic mouse models, TGFβ action can enhance extravascular lung metastasis formation (Bierie and Moses, 2006), whereas a conditional knockout of TGFβ receptor in the mammary epithelium showed that TGFβ can suppress both primary tumor growth and lung metastases (Forrester et al., 2005). Therefore, the causal relationship between TGFβ and breast cancer progression in human, and the identity of downstream TGFβ targets that may be involved in this action, has remained unknown.
To address this problem, we have developed a bioinformatics classifier, the TBRS, based on the TGFβ gene response signature of human epithelial cells. The TBRS can not only classify tumor tissue samples that have a gene expression profile corresponding to TGFβ signaling but can also help identify key downstream TGFβ mediators, as shown in this work. Using this tool to interrogate a wealth of existing clinical breast cancer datasets, we have found that the presence of TGFβ activity in primary tumors is selectively associated with risk of lung metastases. Surprisingly, this association is restricted to ER− tumors. Both ER+ and ER− cancer cells exhibit ANGPTL4 induction by TGFβ, although the ANGPTL4 expression level is higher in TBRS+/ER− than in TBRS+/ER+ tumors. An explanation for the selective association with lung metastasis in the ER− group may lie with the fact that the contributions of TGFβ and ANGPTL4 to lung metastasis occur in the context of the LMS+ phenotype. The TBRS+ status is not associated with metastasis in the ER−/LMS− tumor subset or in ER+ tumors, which are generally LMS− (refer to Figure 1D). ER− tumors that score positive for both TBRS and LMS are the ones with a high risk of lung metastasis (refer to Figure 1E).
We observed a high expression level of TGFβ1, TGFβ2 and LTBP1 in TBRS+ tumors, which is consistent with the TGFβ activity typified by the TBRS, and is in line with a reported association of high TGFβ1 levels with lung metastasis (Dalal et al., 1993). Other reports have shown that among ER− tumors, a low expression of the TGFβ type II receptor is associated with favorable outcome (Buck et al., 2004). Our data are also in line with these findings, in that the TBRS− tumors display a significantly lower expression level of the type II TGFβ receptor. Additionally, we find that the Smad levels are differentially expressed with TBRS+ tumors expressing higher levels of Smad3 and Smad4 while expressing lower levels of Smad2. Indeed, Smad3, more than Smad2, is critical for the induction of TGFβ gene responses (Chen et al., 2001; Chen et al., 2002; Gomis et al., 2006; Seoane et al., 2004). Despite these interesting links, the TGFβ pathway components tested individually or as a group did not perform as strongly as did the TBRS at linking ER− primary tumors with lung metastasis.
A TGFβ-Angptl4 relay system primes mammary tumors for seeding of lung metastases
Several activities have been ascribed to TGFβ that would favor tumor progression in general, including the maintenance of a mesenchymal phenotype (Shipitsin et al., 2007) or the dampening of immune functions (Gorelik and Flavell, 2002). However, it is not obvious how these effects of TGFβ would favor metastasis to one particular organ over another. Yet, our clinical and functional evidence selectively links TGFβ in the primary breast tumor microenvironment to lung metastasis and not bone metastasis. This observation implies a biologically selective mechanism, and our results point at Angptl4 induction by TGFβ as a centerpiece of this mechanism. We provide evidence that TGFβ stimulation of mammary carcinoma cells before they enter the circulation primes these cells for seeding of the lungs through a transient induction of Angptl4. This effect is mediated by the canonical TGFβ receptor and Smad signaling pathway, which in normal breast epithelial cells would suppress cell proliferation, but in metastatic breast cancer cells fails to efficiently trigger cytostatic gene responses (Gomis et al., 2006). Given the disruptive effect of Angptl4 on endothelial cell junctions, we suggest that TGFβ-mediated induction of this factor increases the extravasation capabilities of breast cancer cells as they arrive in the lungs. Thus, a cytokine in the microenvironment of mammary tumors can endow departing cancer cells with increased expression of another cytokine to more efficiently seed a distant organ.
A vasculature disruptive mechanism may provide a selective invasive advantage in lung but not bone because of the inherent differences in the microvasculature of these two tissues. Lung vascular endothelial junctions act as a barrier that restricts the passage of cells. In contrast, the bone marrow vasculature consists of capillary vascular channels, called sinusoids, which have a discontinuous endothelium to facilitate the passage of hematopoietic and other cells (Oghiso and Matsuoka, 1979). Therefore, lung metastasis may require robust extravasation functions such as those provided by Angptl4 and other factors (Gupta et al., 2007a), and additional lung colonizing functions (Gupta et al., 2007b). In contrast, osteolytic metastasis by breast cancer cells may principally require their adaptation to the bone microenvironment and the recruitment and activation of osteoclasts (Mundy, 2002).
The ability of TGFβ to prime disseminating breast cancer cells for lung metastasis is clinically and mechanistically distinct from the advantage that metastatic colonies may later extract from locally produced TGFβ. TGFβ released in the bone microenvironment can foster the expansion of osteolytic colonies through an osteoclast activation cycle (Kang et al., 2003b; Mundy, 2002; Yin et al., 1999). Indeed, of 67 samples of human breast cancer metastasis to bone, lung, brain liver and other sites, that we analyzed, more than half scored as TBRS+. This result is also consistent with our previous observation of activated Smad in a majority of bone metastases from breast cancer patients (Kang et al., 2005) and the involvement of several TGFβ target genes in the bone osteolytic process (Kang et al., 2003b; Mundy, 2002). TGFβ metastatic lesions might support subsequent rounds of metastatic dissemination by the mechanism outlined here.
The TGFβ–Angptl4 cytokine relay system described here provides an example of how stimuli in the primary tumor can affect distant metastases. We envision that TGFβ and other factors in different tumor microenvironments may act in this manner to influence metastases from other tumor types, or to other organ sites. Further validation of this concept may provide impetus for specific therapeutic approaches designed to prevent the presentation of metastatic dissemination during disease progression.
Experimental Procedures
Additional methods can be found in the Supplementary Section.
Cell culture and reagents
MDA-MB-231 and its metastatic derivatives LM2-4175 and BoM-1833 have been described previously (Kang et al., 2003b; Minn et al., 2005). Breast carcinoma cells were isolated from the pleural effusion of patients with metastatic breast cancer treated at our institution upon written consent obtained following IRB regulations as previously described (Gomis et al., 2006). BCN samples were obtained and treated as per Hospital clinic de Barcelona guidelines (CEIC-approved).
TGFβ and TGFβ-receptor inhibition used 100pM TGFβ1 (R&D Systems) for 3 or 6 h as indicated and 10 µM SB431542 (Tocris) with 24 h pretreatment. Epithelial cell lines were treated for 3h with BMP2 (25 ng/mL, R&D), Wnt3a (50 ng/mL, R&D), FGF (5 ng/mL, Sigma), EGF (100 ng/mL, Invitrogen), IL6 (20 ng/mL, R&D), VEGF-165 (100 ng/mL, R&D), and IL1β (100 ng/mL, R&D). Conditioned media experiments were performed by growing cells in serum-deprived media for 48 hours. Recombinant human Angptl4 (Biovendor) was used at 2.5 µg/mL for 24 h.
RNA isolation, labeling, and microarray hybridization
Methods for RNA extraction, labeling and hybridization for DNA microarray analysis of the cell lines have been described previously (Kang et al., 2003b; Minn et al., 2005). The EMC and MSK tumor cohorts and their gene expression data have been previously described (Minn et al., 2007; Minn et al., 2005; Wang et al., 2005). Bone or lung recurrence at any time is indicated.
TGFβ response gene-expression signature and TBRS classifier
Cell lines with and without TGFβ1 treatment (3 h, 100 pM) were subject to expression profiling using Affymetrix U133A or U133 plus2 microchips. Microarray results were pre-processed using RMA algorithm (carried with affy package of R statistical program). The first comparison was conducted between all TGFβ treated samples versus all untreated samples. Three hundred and fifty genes that yielded a p value of 0.05 or less (after Benjamini and Hochberg correction for multiple tests) were kept. Among these genes, we chose to focus on the genes that are significantly changed in at least two different cell lines when the cell lines are considered separately. This step resulted in 174 probe sets corresponding to 153 distinct human genes, which were collectively designated as the TGFβ gene response signatures.
To generate a TBRS classifier, we carried out a “meta-gene” analysis based on this gene set and using the cell lines as references (Bild et al., 2006) and references therein. In short, expression values of the 153 TGFβ responsive genes in cell lines were linearly transformed and encapsulated into one or two “Meta genes”. A Bayesian Probit model was then trained based the cell line data and applied to the Meta genes of the tumor samples. For each tumor, a number between 0 and 1 was derived, indicating the likelihood that the TGFβ signaling is active in that tumor.
Generation of retrovirus and knockdown cells
Knockdown of SMAD4 and ANGPTL4 was achieved using pRetroSuper technology (Brummelkamp et al., 2002) targeting the following 19-nucleotide sequences: 5′-GGTGTGCAGTTGGAATGTA -3′ (SMAD4) and 5′-GAGGCAGAGTGGACTATTT-3′ (ANGPTL4). To produce retrovirus for knockdown, the hairpin vector was transfected into the GPG29 amphotropic packaging cell line (Ory et al., 1996).
Immunofluorescence
HUVECs were grown to confluence on fibronectin coated chamber slides (BD Biosciences). The cells were fixed for 10 min in 4% paraformaldehyde in PBS, and incubated for 5 min on ice in 0.5% Triton X-100 in PBS. After blocking with 2% BSA, the monolayers were processed for staining with anti-ZO1 (Zymed), anti-beta-catenin (Santa Cruz), rhodamine phalloidin (Molecular Probes) for F-actin staining and DAPI (Vector Labs) for nuclear staining. Fluorescence images were obtained using an AxioImager Z1 microscopy system (Zeiss).
Animal studies
All animal work was done in accordance with a protocol approved by the MSKCC Institutional Animal Care and Use Committee. NOD/SCID female mice (NCI) age-matched between 5–7 weeks were used for xenografting studies. For experimental metastasis assays from bilateral orthotopic inoculations, the tumors were extracted from both mammary glands when they each reached 300 mm3, approximately 30 days. Seven days after mastectomies, lung metastases were monitored and quantified using non-invasive bioluminescence as previously described (Minn et al., 2005).
In vivo lung permeability assays
To observe in vivo permeability of lung blood vessels, tumor cells were labeled by incubating with 5µM cell tracker green (Invitrogen) for 30 min and inoculated into the lateral tail vein. One day post inoculation, mice were injected intravenously with rhodamine-conjugated dextran (70 kDa, Invitrogen) at 2 mg per 20 g body weight. After 3 h, mice were sacrificed; lungs were extracted and fixed by intra-tracheal injection of 5 mL of 4% PFA. Lungs were fixed-frozen and 10µm sections were taken to be examined by fluorescence microscopy for vascular leakage. Images were acquired on an AxioImager Z1 microscopy system (Zeiss). To analyze, a uniform ROI of approximately 3 nuclei in diameter was drawn around the tumor cells and applied to each image. A second larger ROI was also applied with similar results. Signal from the ROI was quantified using Volocity (Improvision).
Statistical analysis
Results are reported as mean ± standard error of the mean unless otherwise noted. Comparisons between continuous variables were performed using an unpaired one-sided t-test. Statistics for the orthotopic lung metastasis assays were performed using log-transformation of raw photon flux.
Supplementary Material
Acknowledgements
We would like to thank P. Bos, A. Chiang, G. Gupta, M.-Y. Kim, D. Nguyen, T. Oskarsson, C. Palermo, and S. Tavazoie for helpful discussions and technical suggestions, and J. Foekens for facilitating access to data set clinical annotations. We would also like to acknowledge E. Montalvo, A. Shaw, W. Shu and the members of the Molecular Cytology Core Facility and the Genomic Core Facility for expert technical assistance. This work was funded by grants from the National Institutes of Health, the Kleberg Foundation, the Hearst Foundation, and the BBVA Foundation. D.P. is supported by an NIH Medical Scientist Training Program grant GM07739. J.M. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bernards R, Weinberg RA. A progression puzzle. Nature. 2002;418:823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- Bierie B, Moses HL. Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer. Nat Rev Cancer. 2006;6:506–520. doi: 10.1038/nrc1926. [DOI] [PubMed] [Google Scholar]
- Bild AH, Potti A, Nevins JR. Linking oncogenic pathways with therapeutic opportunities. Nat Rev Cancer. 2006;6:735–741. doi: 10.1038/nrc1976. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell. 2002;2:243–247. doi: 10.1016/s1535-6108(02)00122-8. [DOI] [PubMed] [Google Scholar]
- Buck MB, Fritz P, Dippon J, Zugmaier G, Knabbe C. Prognostic significance of transforming growth factor beta receptor II in estrogen receptor-negative breast cancer patients. Clin Cancer Res. 2004;10:491–498. doi: 10.1158/1078-0432.ccr-0320-03. [DOI] [PubMed] [Google Scholar]
- Cailleau R, Olive M, Cruciger QV. Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro. 1978;14:911–915. doi: 10.1007/BF02616120. [DOI] [PubMed] [Google Scholar]
- Calonge MJ, Massagué J. Smad4/DPC4 silencing and hyperactive Ras jointly disrupt transforming growth factor-beta antiproliferative responses in colon cancer cells. J Biol Chem. 1999;274:33637–33643. doi: 10.1074/jbc.274.47.33637. [DOI] [PubMed] [Google Scholar]
- Camenisch G, Pisabarro MT, Sherman D, Kowalski J, Nagel M, Hass P, Xie MH, Gurney A, Bodary S, Liang XH, et al. ANGPTL3 stimulates endothelial cell adhesion and migration via integrin alpha vbeta 3 and induces blood vessel formation in vivo. J Biol Chem. 2002;277:17281–17290. doi: 10.1074/jbc.M109768200. [DOI] [PubMed] [Google Scholar]
- Cazes A, Galaup A, Chomel C, Bignon M, Brechot N, Le Jan S, Weber H, Corvol P, Muller L, Germain S, et al. Extracellular matrix-bound angiopoietin-like 4 inhibits endothelial cell adhesion, migration, and sprouting and alters actin cytoskeleton. Circ Res. 2006;99:1207–1215. doi: 10.1161/01.RES.0000250758.63358.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, Sorlie T, Dai H, He YD, van't Veer LJ, Bartelink H, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci U S A. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CR, Kang Y, Massagué J. Defective repression of c-myc in breast cancer cells: A loss at the core of the transforming growth factor beta growth arrest program. Proc Natl Acad Sci U S A. 2001;98:992–999. doi: 10.1073/pnas.98.3.992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CR, Kang Y, Siegel PM, Massagué J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell. 2002;110:19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- Dalal BI, Keown PA, Greenberg AH. Immunocytochemical localization of secreted transforming growth factor-beta 1 to the advancing edges of primary tumors and to lymph node metastases of human mammary carcinoma. Am J Pathol. 1993;143:381–389. [PMC free article] [PubMed] [Google Scholar]
- Dejana E. Endothelial cell-cell junctions: happy together. Nat Rev Mol Cell Biol. 2004;5:261–270. doi: 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- Dumont N, Arteaga CL. Targeting the TGF beta signaling network in human neoplasia. Cancer Cell. 2003;3:531–536. doi: 10.1016/s1535-6108(03)00135-1. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- Forrester E, Chytil A, Bierie B, Aakre M, Gorska AE, Sharif-Afshar AR, Muller WJ, Moses HL. Effect of conditional knockout of the type II TGF-beta receptor gene in mammary epithelia on mammary gland development and polyomavirus middle T antigen induced tumor formation and metastasis. Cancer Res. 2005;65:2296–2302. doi: 10.1158/0008-5472.CAN-04-3272. [DOI] [PubMed] [Google Scholar]
- Galaup A, Cazes A, Le Jan S, Philippe J, Connault E, Le Coz E, Mekid H, Mir LM, Opolon P, Corvol P, et al. Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumor cell motility and invasiveness. Proc Natl Acad Sci U S A. 2006;103:18721–18726. doi: 10.1073/pnas.0609025103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale NW, Thurston G, Hackett SF, Renard R, Wang Q, McClain J, Martin C, Witte C, Witte MH, Jackson D, et al. Angiopoietin-2 is required for postnatal angiogenesis and lymphatic patterning, and only the latter role is rescued by Angiopoietin-1. Dev Cell. 2002;3:411–423. doi: 10.1016/s1534-5807(02)00217-4. [DOI] [PubMed] [Google Scholar]
- Gomis RR, Alarcon C, Nadal C, Van Poznak C, Massagué J. C/EBPbeta at the core of the TGFbeta cytostatic response and its evasion in metastatic breast cancer cells. Cancer Cell. 2006;10:203–214. doi: 10.1016/j.ccr.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Gorelik L, Flavell RA. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol. 2002;2:46–53. doi: 10.1038/nri704. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K, Massagué J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007a;446:765–770. doi: 10.1038/nature05760. [DOI] [PubMed] [Google Scholar]
- Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorova-Manova K, Gerald WL, Brogi E, Benezra R, Massagué J. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc Natl Acad Sci U S A. 2007b doi: 10.1073/pnas.0709185104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Perk J, Acharyya S, de Candia P, Mittal V, Todorava-Manova K, Gerald WL, Brogi E, Benezra R, Massagué J. ID genes mediate tumor re-initiation during breast cancer lung metastasis. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0709185104. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, aM J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Hermann LM, Pinkerton M, Jennings K, Yang L, Grom A, Sowders D, Kersten S, Witte DP, Hirsch R, Thornton S. Angiopoietin-like-4 is a potential angiogenic mediator in arthritis. Clin Immunol. 2005;115:93–101. doi: 10.1016/j.clim.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Ito Y, Oike Y, Yasunaga K, Hamada K, Miyata K, Matsumoto S, Sugano S, Tanihara H, Masuho Y, Suda T. Inhibition of angiogenesis and vascular leakiness by angiopoietin-related protein 4. Cancer Res. 2003;63:6651–6657. [PubMed] [Google Scholar]
- Kang Y, Chen CR, Massagué J. A self-enabling TGFbeta response coupled to stress signaling: Smad engages stress response factor ATF3 for Id1 repression in epithelial cells. Mol Cell. 2003a;11:915–926. doi: 10.1016/s1097-2765(03)00109-6. [DOI] [PubMed] [Google Scholar]
- Kang Y, He W, Tulley S, Gupta GP, Serganova I, Chen CR, Manova-Todorova K, Blasberg R, Gerald WL, Massagué J. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc Natl Acad Sci U S A. 2005;102:13909–13914. doi: 10.1073/pnas.0506517102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massagué J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003b;3:537–549. doi: 10.1016/s1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielhorn E, Schofield K, Rimm DL. Use of magnetic enrichment for detection of carcinoma cells in fluid specimens. Cancer. 2002;94:205–211. doi: 10.1002/cncr.10193. [DOI] [PubMed] [Google Scholar]
- Kim I, Kim HG, Kim H, Kim HH, Park SK, Uhm CS, Lee ZH, Koh GY. Hepatic expression, synthesis and secretion of a novel fibrinogen/angiopoietin-related protein that prevents endothelial-cell apoptosis. Biochem J. 2000;346(Pt 3):603–610. [PMC free article] [PubMed] [Google Scholar]
- Kim M, Gans JD, Nogueira C, Wang A, Paik JH, Feng B, Brennan C, Hahn WC, Cordon-Cardo C, Wagner SN, et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell. 2006;125:1269–1281. doi: 10.1016/j.cell.2006.06.008. [DOI] [PubMed] [Google Scholar]
- Laping NJ, Grygielko E, Mathur A, Butter S, Bomberger J, Tweed C, Martin W, Fornwald J, Lehr R, Harling J, et al. Inhibition of transforming growth factor (TGF)- beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol Pharmacol. 2002;62:58–64. doi: 10.1124/mol.62.1.58. [DOI] [PubMed] [Google Scholar]
- Le Jan S, Amy C, Cazes A, Monnot C, Lamande N, Favier J, Philippe J, Sibony M, Gasc JM, Corvol P, et al. Angiopoietin-like 4 is a proangiogenic factor produced during ischemia and in conventional renal cell carcinoma. Am J Pathol. 2003;162:1521–1528. doi: 10.1016/S0002-9440(10)64285-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy L, Hill CS. Alterations in components of the TGF-beta superfamily signaling pathways in human cancer. Cytokine Growth Factor Rev. 2006;17:41–58. doi: 10.1016/j.cytogfr.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Lynch CC, Hikosaka A, Acuff HB, Martin MD, Kawai N, Singh RK, Vargo-Gogola TC, Begtrup JL, Peterson TE, Fingleton B, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7:485–496. doi: 10.1016/j.ccr.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Massagué J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- Massagué J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Padua D, Bos P, Nguyen DX, Nuyten D, Kreike B, Zhang Y, Wang Y, Ishwaran H, et al. Lung metastasis genes couple breast tumor size and metastatic spread. Proc Natl Acad Sci U S A. 2007;104:6740–6745. doi: 10.1073/pnas.0701138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagué J. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–524. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- Oghiso Y, Matsuoka O. Distribution of colloidal carbon in lymph nodes of mice injected by different routes. Jpn J Exp Med. 1979;49:223–234. [PubMed] [Google Scholar]
- Oike Y, Yasunaga K, Suda T. Angiopoietin-related/angiopoietin-like proteins regulate angiogenesis. Int J Hematol. 2004;80:21–28. doi: 10.1532/ijh97.04034. [DOI] [PubMed] [Google Scholar]
- Ory DS, Neugeboren BA, Mulligan RC. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc Natl Acad Sci U S A. 1996;93:11400–11406. doi: 10.1073/pnas.93.21.11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paget S. The distribution of seondary growths in cancer of the breast. Lancet. 1889;1:571–573. [PubMed] [Google Scholar]
- Parikh SM, Mammoto T, Schultz A, Yuan HT, Christiani D, Karumanchi SA, Sukhatme VP. Excess circulating angiopoietin-2 may contribute to pulmonary vascular leak in sepsis in humans. PLoS Med. 2006;3:e46. doi: 10.1371/journal.pmed.0030046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev V, Doubrovin M, Serganova I, Vider J, Shavrin A, Beresten T, Ivanova A, Ageyeva L, Tourkova V, Balatoni J, et al. A novel triple-modality reporter gene for whole-body fluorescent, bioluminescent, and nuclear noninvasive imaging. Eur J Nucl Med Mol Imaging. 2004;31:740–751. doi: 10.1007/s00259-003-1441-5. [DOI] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massagué J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–223. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- Shipitsin M, Campbell LL, Argani P, Weremowicz S, Bloushtain-Qimron N, Yao J, Nikolskaya T, Serebryiskaya T, Beroukhim R, Hu M, et al. Molecular definition of breast tumor heterogeneity. Cancer Cell. 2007;11:259–273. doi: 10.1016/j.ccr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Siegel PM, Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- Weis-Garcia F, Massagué J. Complementation between kinase-defective and activation-defective TGF-beta receptors reveals a novel form of receptor cooperativity essential for signaling. Embo J. 1996;15:276–289. [PMC free article] [PubMed] [Google Scholar]
- Welch DR, Fabra A, Nakajima M. Transforming growth factor beta stimulates mammary adenocarcinoma cell invasion and metastatic potential. Proc Natl Acad Sci U S A. 1990;87:7678–7682. doi: 10.1073/pnas.87.19.7678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, Massagué J, Mundy GR, Guise TA. TGF-beta signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103:197–206. doi: 10.1172/JCI3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.