

Abstract
To unambiguously confirm the actual product in autoxidation of salvinorin A under basic conditions, deacetyl-1,10-didehydrosalvinorin G was synthesized from salvinorin C via intermediate salvinorin H. Furthermore, oxidation of salvinorin D with manganese dioxide gave salvinorin G in good yield.
Keywords: Salvinorin A, Oxidation, Manganese dioxide, Synthesis
Salvia divinorum, a Mexican medicinal plant, has been used traditionally for its psychoactive (hallucinogenic ) effects in divination rites.1 Previous phytochemical studies have resulted in the isolation of 34 compounds, including salvinorins A (1a), C (2a), D (2b), G (3) and H (2c).2–9 Of those compounds, 1a was identified as a potent and selective kappa opioid receptor (KOR) agonist.10,11 Because of the unique non-nitrogenous structure and potent binding activities to KOR, much effort has been directed toward a better understanding of structure-activity relationships (SAR) of 1a.12–26 Salvinorin derivatives readily underwent epimerization at C-8 under basic conditions.3,4,13–17 Surprisingly, treatment of 1a and its derivative with strong bases, such as Ba(OH)2,15 KOH18,26 and NaOH,25 yielded corresponding natural salvinorin analogs, and no epimerization at C-8 was observed. It was reported that treatment of 1a with KOH in methanol produced deacetyl-1,10-didehydrosalvinorin G (4a).18 Recently, we revised the structure of 4a to its 8-epimer (4b) based on comparison of 1H and 13C NMR data with those of 1a and 1b, NOESY data and chemical conversion.27 In order to unambiguously confirm the actual product in autoxidation of 1a under harsh basic conditions,18 it is necessary to synthesize the natural salvinorin derivative 4a and to complete its NMR data. In this Letter, we report the synthesis of 4a from 2a via intermediate 2c, and chemical conversion of 3 from 2b.
Following the published procedure,18 the diol 2c was prepared by deacetylation of 2a. Subsequent oxidation of 2c with manganese dioxide (Scheme 1) yielded 4a and deacetylsalvinorin G (5).28 Using 2D NMR techniques, including COSY, NOESY, HMQC and HMBC, permitted the full assignment of all 1H and 13C NMR chemical shifts of 4a (Tables 1 and 2), and the key 13C-1H correlations in the HMBC spectrum of 4a were shown in Figure 1. In the 1H NMR spectrum of 4a, the C-8-H shifted much upper field to δ 2.42, and the coupling constants (dd, J = 12.6 and 3.3 Hz) of H-8 are more suitable for axial orientation than those (δ 2.99, dd, J = 9.6 and 5.1 Hz) of 4b (Table 1). The C-12-H of 4a shifted low-field slightly compared with that of 4b (Table 1), and the H-12 of 4a showed the J values (10.5 and 6.6 Hz) for axial orientation. In addition, the H-11α (δ 3.76) of 4a shifted much lower field compared with that of 4b (δ 3.11), and the chemical shift changes are consistent with those of 2a (δ 2.49)4 and its 8-epimer (δ2.14).29 Comparison of the 13C resonances of C-6, C-8, C-12, C-13, C-17, C-19 and C-20 (Table 2) of 4a and 4b also confirms that H-8 in 4a is the β configuration. In the NOESY spectrum of 4a, H-12 (δ 5.62) showed cross peaks to H-11α (δ 3.76) and H-20 (δ 1.54), while H-19 (δ 1.76) related to H-6α (δ 2.51), H-7α (1.94) and H-20 (δ 1.54). It should be noted that there is no crossed peak between H-8 and H-12/H-19/H-20. Based on these data, the structure of 4a was confirmed as deacetyl-1,10-didehydrosalvinorin G.
Scheme 1.

Table 1.
1H NMR Data (300 MHz, CDCl3) for 4a and 4b [δ (ppm), m, J (Hz)]
| Proton | 4a | 4b |
|---|---|---|
| 3 | 6.88 s | 7.00 s |
| 6α | 2.51 dt (13.2, 3.3) | 2.54 dt (13.8, 7.5) |
| 6β | 1.46 td (13.5, 3.9) | 1.67–1.77 m |
| 7α | 1.94 ddt (14.4, 3.3, 13.5) | 2.24 ddd (14.4, 7.8, 5.1) |
| 7β | 2.27 dq (14.7, 3.6) | 1.98 ddd (14.1, 9.6, 6.6) |
| 8 | 2.42 dd (12.6, 3.3) | 2.99 dd (9.6, 5.1) |
| 11α | 3.76 dd (14.4, 6.3) | 3.11 dd (14.7, 3.0) |
| 11β | 2.09 dd (14.4, 10.5) | 2.02 dd (14.4, 12.3) |
| 12 | 5.62 dd (10.5, 6.6) | 5.44 dd (12.3, 2.4) |
| 14 | 6.42 dd (1.2, 0.9) | 6.42 dd (1.8, 0.9) |
| 15 | 7.41 t (1.8) | 7.41 t (1.8) |
| 16 | 7.44 d (0.9) | 7.49 d (0.9) |
| 19 | 1.76 s | 1.72 s |
| 20 | 1.54 s | 1.67 s |
| OH | 7.07 s | 6.92 s |
| CO2CH3 | 3.86 s | 3.85 s |
Table 2.
13C NMR Data (75 MHz, CDCl3) for 4a, 4b, 6a, 6b, 7a, 7b, 8a, 8b, 9a and 9b [δ (ppm)]
| Carbon | 4a | 4b | 6aa | 6bb | 7ab | 7bb | 8ab | 8bb | 9ac | 9bc |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 145.3 | 145.1 | 208.9 | 209.2 | 201.9 | 202.3 | 203.7 | 204.1 | 202.2 | 202.6 |
| 2 | 180.9 | 180.7 | 74.4 | 74.4 | 74.9 | 75.1 | 75.9 | 76.2 | 75.2 | 75.5 |
| 3 | 127.6 | 128.2 | 34.5 | 33.8 | 30.6 | 30.5 | 31.8 | 31.5 | 28.3 | 28.1 |
| 4 | 159.0 | 157.5 | 53.1 | 52.3 | 53.4 | 52.6 | 50.7 | 50.2 | 59.7 | 59.1 |
| 5 | 43.3 | 42.3 | 42.6 | 42.6 | 42.0 | 42.1 | 41.9 | 42.0 | 42.5 | 42.6 |
| 6 | 32.0 | 28.3 | 38.1 | 34.2 | 38.1 | 33.8 | 38.0 | 33.8 | 38.0 | 33.8 |
| 7 | 18.1 | 21.9 | 18.1 | 17.5 | 18.1 | 17.5 | 18.1 | 17.6 | 17.9 | 17.4 |
| 8 | 50.6 | 44.8 | 51.3 | 45.2 | 51.3 | 45.2 | 51.4 | 45.3 | 51.2 | 45.1 |
| 9 | 39.4 | 37.6 | 35.3 | 34.5 | 35.4 | 34.7 | 35.2 | 34.6 | 35.4 | 34.6 |
| 10 | 138.5 | 140.0 | 63.8 | 63.6 | 63.9 | 63.9 | 64.4 | 64.5 | 63.6 | 63.5 |
| 11 | 42.4 | 36.8 | 43.5 | 48.2 | 43.3 | 47.9 | 43.3 | 48.0 | 43.2 | 47.9 |
| 12 | 71.5 | 70.8 | 71.9 | 70.0 | 72.1 | 70.1 | 72.1 | 70.1 | 72.0 | 70.1 |
| 13 | 126.0 | 124.4 | 125.3 | 123.5 | 125.1 | 123.3 | 125.2 | 123.3 | 125.1 | 123.2 |
| 14 | 108.6 | 108.4 | 108.3 | 108.4 | 108.4 | 108.5 | 108.4 | 108.5 | 108.3 | 108.5 |
| 15 | 143.8 | 143.7 | 143.8 | 143.6 | 143.7 | 143.6 | 143.7 | 143.6 | 143.8 | 143.6 |
| 16 | 139.4 | 139.6 | 139.3 | 139.6 | 139.4 | 139.7 | 139.4 | 139.7 | 139.4 | 139.7 |
| 17 | 171.6 | 173.2 | 171.0 | 173.4 | 171.3 | 173.7 | 171.4 | 173.7 | 170.9 | 173.5 |
| 18 | 166.0 | 165.4 | 171.8 | 172.1 | 175.8 | 176.2 | 61.6 | 61.6 | 200.6 | 200.9 |
| 19 | 31.3 | 30.3 | 16.5 | 15.3 | 16.4 | 15.3 | 16.6 | 15.5 | 17.9 | 16.7 |
| 20 | 18.8 | 24.4 | 15.2 | 24.6 | 15.2 | 24.6 | 15.3 | 24.8 | 15.1 | 24.4 |
| CO2CH3 | 52.7 | 52.6 | 51.9 | 51.6 | - | - | - | - | - | - |
| -COCH3 | - | - | - | - | 170.0 | 169.9 | 170.1 | 169.9 | 170.0 | 169.8 |
| -COCH3 | - | - | - | - | 20.5 | 20.5 | 20.6 | 20.6 | 20.6 | 20.6 |
Figure 1.
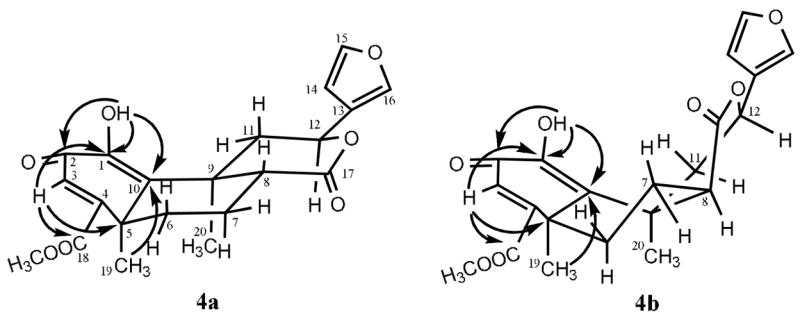
Key HMBC correlations of 4a and 4b
The coupling constants of H-6 and H-7 (Table 1) in 4a are significantly different from those of 4b. The J values of H-6β (td, 13.5, 3.9) and H-7α (dtd, 14.4, 3.3, 13.5) of 4a indicate that both protons are axial. It has been reported that the protons in anti-periplanar relationships show stronger correlations in the COSY spectrum.30 This was also evidenced by the protons (H-7α and H-6β, H-7α and H-8) of 4a in the COSY spectrum. On the other hand, only H-7β and H-8 of 4b exhibited stronger correlations in the COSY spectrum. These findings suggest that the B-ring in 4a should be an identical chair conformation (Figure 1), which is different from that of 4b. Obviously, the double bond between C-1 and C-10 in 4a and 4b distorts the B-ring conformation to a different extent.
Compounds 4a and 5 were screened for binding affinity at opioid receptors in vitro, as reported previously.7 Both compounds were inactive at mu, delta and kappa opioid receptors at 3 μM.
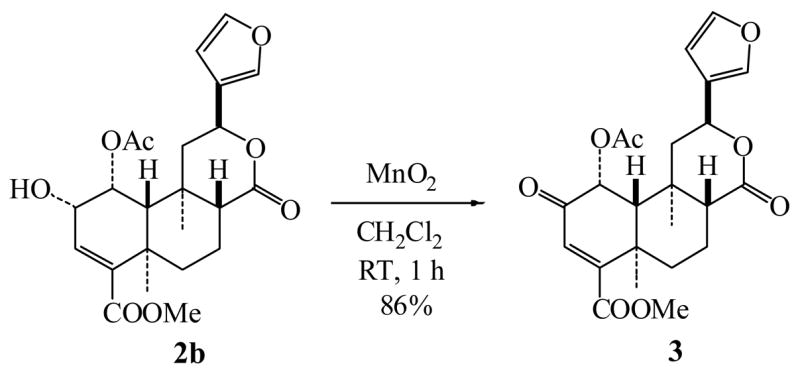
Salvinorin G (3) presents in S. divinorum in much lower level than 1a and 2a, and it showed a moderate binding affinity at KOR.7 3 can be prepared by oxidation of the natural occurring salvinorin D (2b)7 with manganese dioxide in an excellent yield (Scheme 2).31 This reaction not only confirms the structure of 3 but also demonstrates that no epimerization occurs under the mild oxidation conditions as shown in Scheme 1.
Scheme 2.
Numerous salvinorin derivatives have been prepared in recent years for SAR study and improvement of KOR binding affinity.12–26 Compounds 6a, 6b, 7a, 7b, 8a and 8b have served as key intermediates for C-2 and C-18 SAR studies. Among these compounds, only 6a and 8a were reported with full NMR assignments.30,13 However, 6a was measured in acetone-d6 at higher temperature (40 °C).30 Furthermore, 1b is the only compound with full 1H and 13C NMR assignments in numerous 8-epi-salvinorin derivatives.13 Therefore, we assigned all 13C NMR chemical shifts of 6a, 6b, 7a, 7b, 8a and 8b in comparison with those of 1a and 1b (Table 2).27 On the other hand, both aldehydes 9a and 9b were synthesized in our laboratory, and the incorrect 1H and 13C NMR data of 9a were presented in our previous paper.21 The 13C NMR data of 9a were revised and shown in Table 2.
In conclusion, deacetyl-1,10-didehydrosalvinorin G (4a) was readily synthesized from salvinorin H (2c). The product obtained by the treatment of 1a with hydroxides in MeOH18 has been unambiguously identified as 8-epi-deacetyl-1,10-didehydrosalvinorin G (4b). Finally, conversion of salvinorin G (3) from salvinorin D (2b) provides an authentic sample with intact stereochemistry at C-8 for further confirmation of 4a.
Supplementary Material
Supplementary data
Copies of 1H and 13C NMR spectra of 4a.


Acknowledgments
The authors wish to thank Ms. Jane F. McNeil for EI-MS determination and Drs. Wei Xu and Lee-Yuan Liu-Chen for the binding assay at opioid receptors. This work was supported by a research grant (R01-DA019688) from NIDA to D.Y.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Siebert DJ. J Ethnopharmacol. 1994;43:53–56. doi: 10.1016/0378-8741(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 2.Ortega A, Blount JF, Manchand PS. J Chem Soc, Perkin Trans 1. 1982:2505–2508. [Google Scholar]
- 3.Valdes LJ, III, Butler WM, Hatfield GM, Paul AG, Koreeda M. J Org Chem. 1984;49:4716–4720. [Google Scholar]
- 4.Valdes LJ, III, Chang H-M, Visger DC, Koreeda M. Org Lett. 2001;3:3935–3937. doi: 10.1021/ol016820d. [DOI] [PubMed] [Google Scholar]
- 5.Munro TA, Rizzacasa MA. J Nat Prod. 2003;66:703–705. doi: 10.1021/np0205699. [DOI] [PubMed] [Google Scholar]
- 6.Bigham AK, Munro TA, Rizzacasa MA, Robins-Browne RM. J Nat Prod. 2003;66:1242–1244. doi: 10.1021/np030313i. [DOI] [PubMed] [Google Scholar]
- 7.Lee DYW, Ma Z, Liu-Chen LY, Wang Y, Chen Y, Carlezon WA, Cohen B. Bioorg Med Chem. 2005;13:5635–5639. doi: 10.1016/j.bmc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- 8.Harding WW, Tidgewell K, Schmidt M, Shah K, Dersch CM, Snyder J, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. Org Lett. 2005;7:3017–3020. doi: 10.1021/ol0510522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shirota O, Nagamatsu K, Sekita S. J Nat Prod. 2006;69:1782–1786. doi: 10.1021/np060456f. [DOI] [PubMed] [Google Scholar]
- 10.Roth BL, Baner K, Westkaemper R, Siebert DJ, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc Natl Acad Sci. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sheffler DJ, Roth BL. Trends Pharmacol Sci. 2003;24:107–109. doi: 10.1016/S0165-6147(03)00027-0. [DOI] [PubMed] [Google Scholar]
- 12.Chavkin C, Sub S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen SJ, Roth BL. J Pharmacol Exp Ther. 2004;308:1197–1203. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- 13.Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan F. J Med Chem. 2005;48:345–348. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beguin C, Richards MR, Wang Y, Chen Y, Liu-Chen LY, Ma Z, Lee DYW, Carlezon WA, Cohen BM. Bioorg Med Chem Lett. 2005;15:2761–2765. doi: 10.1016/j.bmcl.2005.03.113. [DOI] [PubMed] [Google Scholar]
- 15.Lee DYW, Karnati VVR, He M, Liu-Chen LY, Kondaveti L, Ma Z, Wang Y, Chen Y, Beguin C, Carlezon WA, Cohen B. Bioorg Med Chem Lett. 2005;15:3744–3747. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 16.Lee DYW, He M, Kondaveti L, Liu-Chen LY, Ma Z, Wang Y, Chen Y, Li JG, Beguin C, Carlezon WA, Cohen B. Bioorg Med Chem Lett. 2005;15:4169–4173. doi: 10.1016/j.bmcl.2005.06.092. [DOI] [PubMed] [Google Scholar]
- 17.Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. J Med Chem. 2005;48:4765–4771. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- 18.Munro TA, Goetchius GW, Roth BL, Vortherms TA, Rizzacasa MA. J Org Chem. 2005;70:10057–10061. doi: 10.1021/jo051813e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harding WW, Schmidt M, Tidgewell K, Kannan P, Holden KG, Dersch CM, Rothman RB, Prisinzano TE. Bioorg Med Chem Lett. 2006;16:3170–3174. doi: 10.1016/j.bmcl.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 20.Beguin C, Richards MR, Li JG, Wang Y, Xu W, Liu-Chen LY, Carlezon WA, Cohen BM. Bioorg Med Chem Lett. 2006;16:4679–4685. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]
- 21.Lee DYW, He M, Liu-Chen LY, Wang Y, Li JG, Xu W, Ma Z, Carlezon WA, Cohen B. Bioorg Med Chem Lett. 2006;16:5498–5502. doi: 10.1016/j.bmcl.2006.08.051. [DOI] [PubMed] [Google Scholar]
- 22.Tidgewell K, Harding WW, Lozama A, Cobb H, Shah K, Kannan P, Dersch CM, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. J Nat Prod. 2006;69:914–918. doi: 10.1021/np060094b. [DOI] [PubMed] [Google Scholar]
- 23.Munro TA, Duncan KK, Staples RJ, Xu W, Liu-Chen L-Y, Beguin C, Carlezon WA, Cohen BM. Beilstein J Org Chem. 2007;3 doi: 10.1186/1860-5397-3-1. [ http://bjoc.beilstein-journals.org/content/pdf/1860-5397-3-1.pdf] [DOI] [PMC free article] [PubMed]
- 24.Bikbulatov RV, Yan F, Roth BL, Zjawiony JK. Bioorg Med Chem Lett. 2007;17:2229–2232. doi: 10.1016/j.bmcl.2007.01.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holden KG, Kidgewell K, Marquam A, Rothman RB, Navarro H, Prisinzano TE. Bioorg Med Chem Lett. 2007;17:6111–6115. doi: 10.1016/j.bmcl.2007.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bikbulatov RV, Stewart J, Jin W, Yan F, Roth BL, Ferreiraa D, Zjawiony JK. Tetrhedron Lett. 2008;49 doi: 10.1016/j.tetlet.2007.12.041. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma Z, Lee DYW. Tetrhedron Lett. 2007;48:5461–5464. [Google Scholar]
- 28.Synthesis of 4a. To a solution of 2c (12 mg, 31 μmol) in CH2Cl2 (3 ml) was added manganese dioxide (50 mg, 575 μmol), and the suspension was stirred at room temperature for 3 h. The solution was filtered and evaporated in vacuo. The residue was purified by silica gel column [CH2Cl2:AcOEt (10:1)] to give 4a (6.2 mg, yield 52%) and 5 (2.5 mg, yield 21%). Data for 4a: 1H and 13C NMR data, see Tables 1 and 2; EI-MS m/z 386 (M+). Data for 5: 1H NMR (CDCl3, 300 MHz)δ 7.46 (1H, br s, H-16), 7.43 (1H, br s, H-15), 6.41 (1H, br s, H-14), 6.38 (1H, s, H-3), 5.61 (1H, dd, J = 11.7 and 5.4 Hz, H-12), 4.33 (1H, d, J = 3.3 Hz, H-1), 3.84 (3H, s, COOCH3), 2.54 (1H, brs, OH), 2.52 (1H, dd, J = 12.6 and 5.1 Hz, H-11a), 2.12–2.32 (3H, m, H-6a, H-7a, H-8), 1.70–1.94 (3H, m, H-7b, H-10, H-11b), 1.73 (3H, s, H-19), 1.53 (3H, s, H-20), 1.34 (1H, m, H-6b); 13C NMR (CDCl3, 75 MHz)δ 198.9 (C-2), 171.4 (C-17), 166.4 (C-18), 162.3 (C-4), 143.9 (C-15), 139.4 (C-16), 127.1 (C-3), 125.4 (C-13), 108.3 (C-14), 71.8 (C-12), 70.1 (C-1), 54.0 (C-10), 52.5 (C-8), 52.0 (COOCH3), 43.2 (C-11), 38.0 (C-5), 37.3 (C-9), 35.1 (C-6), 23.3 (C-19), 18.2 (C-7), 16.8 (C-20); EI-MS m/z 388 (M+).
- 29.Munro TA. Ph.D. Thesis. University of Melbourne; Victoria, Australia: 2006. The Chemistry of Salvia divinorum; p. 212. [ http://eprints.infodiv.unimelb.edu.au/archive/00002327] [Google Scholar]
- 30.Giner JL, Kiemle DJ, Kutrzeba L, Zjawiony J. Magn Reson Chem. 2007;45:351–354. doi: 10.1002/mrc.1972. [DOI] [PubMed] [Google Scholar]
- 31.The quaternary carbon at C-17 and the carbonyl carbon of the acetyl group in 3 should be reassigned as δ 170.9 and δ 169.7, respectively.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Copies of 1H and 13C NMR spectra of 4a.