

Abstract
Research into the underlying mechanisms of cognitive dysfunction in Alzheimer’s disease (AD) has relied traditionally on tasks such as the water maze which evaluate spatial learning and memory. Since non-spatial forms of memory are also disrupted by AD, it is critical to establish other paradigms capable of investigating these deficits. Utilizing a non-spatial learning task, acquisition of conditioned taste aversion (CTA) was evaluated in a mouse model of AD. This line of transgenic mice encode a mutated allele of the human amyloid precursor protein (APP) and presenilin 1 (PS1) genes and exhibit extensive amyloid plaque deposition in the brain by 6-7 mo of age. Compared to wildtype mice, 10-17 month old APP/PS1 mice failed to acquire CTA to saccharin. Mice that only possessed one of the two mutations were able to acquire CTA to the saccharin. In 2-5 month old APP/PS1 mice acquisition of CTA was disrupted despite the lack of extensive plaque deposition. However, further analysis indicated a potential gender difference in both the CTA deficit and onset of plaque deposition with females showing greater conditioned aversion.
Alzheimer’s disease (AD) is one of the most prominent neurodegenerative disorders associated with aging. The hallmark symptom of AD is impaired learning and memory function that progresses from mild to severe. AD is characterized pathologically by extra-cellular deposition of beta-amyloid (Aβ) plaques and intra-cellular neurofibrillar tangles consisting of aggregrates of hyperphosphorylated protein tau (Selkoe, 1989, Mandelkow and Mandelkow, 1998). The pathology is first observed in the hippocampal and cortical regions and has been shown to be correlated with deficits in learning and memory (Convit et al., 1997, De Leon et al., 1997), although this linkage remains controversial (Morgan, 2003).
Over the past decade the use of mice expressing human familial mutations of AD has provided insight into the relationship between the pathological mechanisms and memory impairments (Ashe, 2001). Mice expressing mutations in amyloid precursor protein (APP), presenilin-1 (PS1), presenilin-2 (PS2), tau or apolipoprotein (apoE) have been generated to model various behavioral physiological, pathological and biochemical aspects of AD (Corder et al., 1998, Ashe, 2000, Lewis et al., 2001, Gordon et al., 2002, Hwang et al., 2002, Jolas et al., 2002, Teter et al., 2002). The mice developed thus far to investigate the pathology and learning and memory associated deficits in AD do not include a mouse that possesses all of the pathophysiology of AD. However, even though they do not model all of the symptoms, these mice have proven useful tools for evaluating the contribution of specific genes to learning and memory deficits as well as potential therapeutics for alleviating these impairments.
The evaluation of learning and memory deficits in these mice to date has focused primarily on hippocampal-dependent spatial tasks, such as the water maze. However, the deficits associated with AD are not limited to spatial information (Pepin and Eslinger, 1989, Hom, 1992). A review of the human literature indicated AD patients display deficits in explicit memory as well as implicit memory deficits for verbal and visuoperceptual information (Carlesimo and Oscar-Berman, 1992). Therefore, it is important to evaluate if particular mouse models exhibit deficits on non-spatial learning and memory tasks involving brain regions other than the hippocampus.
Conditioned taste aversion (CTA) is a simple Pavlovian conditioning paradigm evaluating non-spatial learning and can be rapidly acquired in even a single trial. CTA involves pairing the taste of a novel conditioned stimulus, such as a saccharin solution, with an aversive unconditioned stimulus such as nausea. One advantage of CTA is that the pathways for processing of the gustatory conditioned stimulus and unconditioned stimulus information are relatively well-known (for a detailed review see Welzl et al., 2001). Parallel processing of both the conditioned and unconditioned stimulus occurs at the nucleus of the solitary tract, parabrachial nucleus of the pons, parvicellular thalamic ventral posteromedial nucleus, amygdala and agranaular insular cortex, indicating that an association between the conditioned stimulus and unconditioned stimulus could form at any of these locations. Other structures that have been implicated in the formation of a CTA include the hypothalamus (Roth et al., 1973), basal forebrain (Lopez-Garcia et al., 1993) and potentially the noradrenergic system (Dunn and Everitt, 1987). Lesions of the hippocampus impact CTA only mildly, or not at all (Best and Orr, 1973, Murphy and Brown, 1974, Yamamoto et al., 1995). Therefore, CTA appears to be an ideal paradigm for evaluating non-spatial forms of learning, not dependent on hippocampal function, in mouse models of AD.
A recent study investigating CTA in TgCRND8 mice with a double mutation in human APP genes (Janus et al., 2004) found that TgCRND8 mice exhibited a deficit in the acquisition of CTA. Another study reported no acquisition deficits in CTA in P301L transgenic mice exhibiting neurofibrillary tangles by 6 months of age; however, they did note accelerated extinction of the CTA (Pennanen et al., 2004). More recently a study has shown that mice with reduced levels of neprilysin, a major Aβ degrading enzyme, exhibit weaker CTA that extinguished faster than age-matched controls (Madani et al., 2006). Given these findings, CTA may be a useful model for assessing learning and memory deficits in mouse models of AD, but may depend on the AD-related genes involved.
To further validate CTA as a model to evaluate learning and memory deficits in mouse models of AD, we conducted a series of experiments to evaluate CTA in another mouse model of AD. The mice utilized were double transgenic mice expressing a human familial mutation in both APPswe & PS1Δe9. These mice exhibit Aβ deposition as well as learning and memory deficits by 6 months of age (Savonenko et al., 2005). The initial experiment was designed to replicate the findings reported by Janus et al. (2004) utilizing 10-17 month old mice after the reported onset of Aβ deposition. The second experiment examined the contribution of each transgene alone in 9-17 month old mice to assess if the deficit in CTA could be attributed to a single mutated gene, and the final experiment evaluated CTA in mice 2-5 months of age, prior to reported plaque deposition, in an attempt to assess if a deficit in CTA was correlated with Aβ deposition.
2. Materials and Methods
2.1.Animals
The mice used for this study were male and female APPswe/PS1ΔE9 dtg (+/+), heterozygous for APP (+/-) or PS1 (-/+), or wildtype (-/-) mice on a mixed strain background (primarily C57BL/6 and C3He/J) originally developed by D. Borchelt and colleagues at the Johns Hopkins School of Medicine (Borchelt et al., 1997). The mice were raised in the vivarium at the Gerontology Research Center under controlled experimental conditions (22±1 °C, 70±10% humidity) with a 12-hour light/dark cycle. All of the procedures were approved by the Animal Care and Use Committee of the Gerontology Research Center and followed the NIH guidelines for the Care and Use of Laboratory Animals.
2.2 Conditioning Procedures
At the initiation of the experiments, mice were moved to individual hanging wire cages with solid Plexiglas inserts placed on the bottom of the cage, and paper bedding was provided to cover the bottom of the cage. The mice had ad libitum access to food for the duration of the experiment. Water was provided through two 15 ml tubes attached to the outside of each cage. Access to water was limited to 7 hr each day from 08:00 to 15:00 hr. The water intake of mice for the first 30 min was measured, and on average the mice consumed around 1 ml of water during the first 30 min each day. The mice were also weighed each day prior to the bottles being attached to the cages. After 6 days of habituation where only water was provided, mice were given a conditioning day where a 0.5% saccharin solution serving as the conditioned stimulus (Saccharin sodium salt, Sigma Chemical Co., St. Louis, MO) was provided in one bottle during the first 30 min period. For the remainder of the 7-hr period, water was available in both bottles. The location of the saccharin bottle on the conditioning day was counter-balanced across experimental groups. One hour after the saccharin solution was removed, mice were given an i.p. injection of either lithium chloride (LiCl: 0.14 M, 2% of the body weight; Sigma Chemical Co., St. Louis, MO: L7026), or a corresponding amount of saline. The LiCl served as the unconditioned stimulus by producing a behavioral malaise. The following day consisted of a recovery period where mice had normal access to water in both bottles. Two days following conditioning, mice were given a two-bottle choice test in which the 0.5% saccharin solution was presented in one bottle with water in the other. The location of the saccharin on the cage was pseudo-randomizecd across experimental groups and the location of the saccharin solution on the conditioning day. Saccharin intake was expressed as a percentage of the total fluid intake (ml saccharin/(ml saccharin - ml water) X 100).
The group numbers for the three experiments were: Experiment 1 (10-17 month old APP/PS1 or wildtype mice; mean age = 14.1): APP/PS1 - saline (N=13; mean age = 13.6); APP/PS1 - LiCl (N=13: mean age = 14.2); wildtype - saline (N=6: mean age = 15.0); wildtype - LiCl (N=7: mean age = 14.0): Experiment 2 (9-17 month old APP alone, PS1 alone or wildtype mice; mean age = 13.4): APP - saline (N=8: mean age = 13.6); APP - LiCl (N=9: mean age = 13.5); PS1 - saline (N=8: mean age = 13.6); PS1 - LiCl (N=8: mean age = 13.0): Experiment 3 (2-5 month old APP/PS1 or wildtype mice; mean age = 3.5): APP/PS1 - saline (N=11: mean age = 3.5); APP/PS1 - LiCl (N=11: mean age = 3.6); wildtype - saline (N=11: mean age = 3.5); wildtype - LiCl (N=12: mean age = 3.4). The numbers of males and females were roughly equivalent within groups, and the age range of mice was counterbalanced across groups., e.g, the mean ages for the mice in experiment 1 were: APP/PS1 - saline = 13.6 months; APP/PS1 - LiCl = 14.2 months; wildtype - saline = 15.0 months; wildtype - LiCl = 14.0 months.
2.3 Histology
At the completion of the behavioral testing, mice were perfused with phosphate buffered saline followed by paraformaldehyde. The brains were removed and frozen for sectioning. The brain slices were stained with Congo red solution and counterstained with cresyl violet. The hippocampus, amygdala and cortex were visually examined for the presence of congofilic amyloid deposits, but the number of plaques was not quantified.
2.4 Data Analysis
Planned comparisons were conducted to evaluate saccharin preference after treatment with saline versus LiCl within each genotype. The critical α level was set at 0.05 for all statistical tests. The values in the figures represent the means ± S.E.M. The data was analyzed using the SPSS statistical program version 11.0.
3. Results
3.1 CTA in 10-17 month old APP/PS1 mice
Results of t-tests indicated 10-17 month old wildtype (-/-) mice administered LiCl exhibited significant avoidance of the saccharin solution during the choice test (t(11) = 4.82, p = 0.001) compared to mice given saline (see Figure 1A). However, for the 10-17 month old APP/PS1 (+/+) mice, there was no significant difference between the saline and LiCl injected mice for saccharin preference (t(25) = 1.53, p = 0.140), indicating a failure of the 10-17 month old APP/PS1 mice to acquire a CTA. An ANOVA indicated no significant interaction or main effects for saccharin consumption on the initial conditioning day suggesting no initial group biases for saccharin preference. Histological examination revealed extensive Aβ deposition in the hippocampus, amygdala and cortex of the 11-17 month old APP/PS1, but not wildtype mice of the same age (see Figure 1B).
Figure 1.
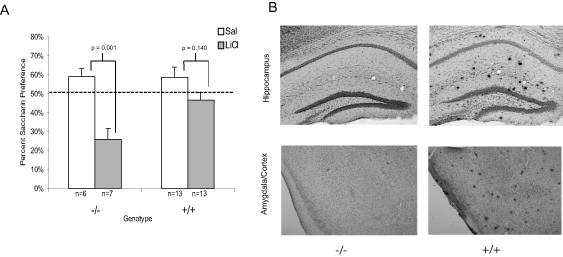
A) Mean saccharin preference (+/- 1 s.e.m.) after LiCl or saline administration in older APP/PS1 and wildtype mice. B) Images of the hippocampus, amygdala and cortex from representative brains of 10-17 month old APP/PS1 and wildtype mice, stained with Congo red and counterstained with cresyl violet. The images were taken at 5X and then cropped with image editing software. The brains were examined visually and no quantitative analysis was conducted.
3.2 CTA in 9-17 month old heterozygous mice
The results from this study indicated both the mice with the APP (+/-) (t(15) =2.89, p = 0.01) or the PS1 (-/+) (t(14) = 3.083, p = 0.008) mutation alone administered LiCl exhibited avoidance of the saccharin solution compared to saline treated mice (see Figure 2A). An ANOVA for saccharin consumption on conditioning day suggested no baseline differences in saccharin preference between the groups. Histology showed the lack of Aβ deposition in the hippocampus, amygdala and cortex of both groups of mice (see Figure 2B).
Figure 2.
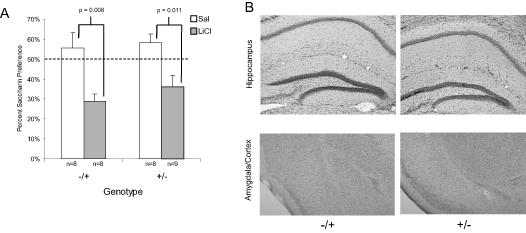
A) Mean saccharin preference (+/- 1 s.e.m.) after LiCl or saline administration in mice with either the mutation in APP (+/-) or PS1 (-/+). B) Images of the hippocampus, amygdala and cortex from representative brains of 9-17 month old APP (+/-) and PS1 (-/+), stained with Congo red and counterstained with cresyl violet. The images were taken at 5X and then cropped with image editing software.
3.3 CTA in 2-5 month old APP/PS1 mice
Similar to the first experiment with older mice, the 2-5 month old APP/PS1 (+/+) mice treated with LiCl exhibited saccharin preference similar to those treated with saline (t(20) = 1.21, p = 0.24) indicating a failure to acquire CTA to the saccharin solution (See Figure 3A). The wildtype mice of the same age treated with LiCl were able to acquire CTA to the saccharin solution (t(21) = 7.06, p < 0.001). Further analysis of the data from this experiment revealed a potential gender difference in the onset of memory deficits in the young APP/PS1 mice. While the 2-5 month old female +/+ mice administered LiCl (n=5) exhibited saccharin preference similar to 2-5 month old female +/+ mice administered saline (n=6) (t(10) = 0.315, p = 0.76), indicating a failure to acquire CTA, the 2-5 month old male +/+ mice administered LiCl (n=6), approached, but did not exhibit, significantly different preference from 2-5 month old male +/+ mice administered saline (n=6) (t(10) = 2.22, p = 0.051) suggesting a trend to acquire CTA (see Figure 4A). This raises the possibility the presence of a gender difference in the age of onset for memory deficits in the APP/PS1 mice. Based on this finding, the data from Experiment 1 was further analyzed to evaluate potential gender differences. As shown in Figure 4B, by 10 months of age (1st experiment) any gender differences appear to have disappeared, since both 10-17 month old male (t(13) = 1.88, p = 0.08) and female (t(10) = 0.27, p = 0.79) +/+ mice failed to acquire CTA to the saccharin solution. However, it must be noted that the group numbers, when broken down by gender, were rather small making it difficult to reach a substantial conclusion about any potential gender differences. This may be an area warranting future research.
Figure 3.
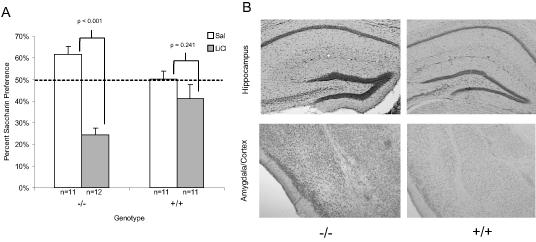
A) Mean saccharin preference (+/- 1 s.e.m.) after LiCl administration in 2-5 month old APP/PS1 and wildtype mice. B) Images of the hippocampus, amygdala and cortex from representative brains of 2-5 month old APP/PS1 and wildtype mice, stained with Congo red and counterstained with cresyl violet. The images were taken at 5X and then cropped with image editing software. The brains were examined visually and no quantitative analysis was conducted.
Figure 4.
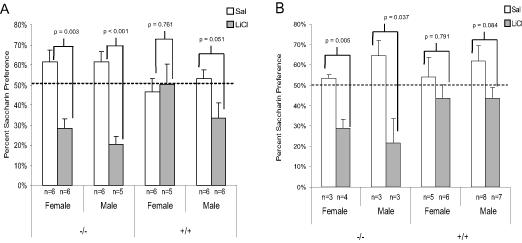
A) Mean saccharin preference (+/- 1 s.e.m.) in 2-5 month old APP/PS1 and wildtype mice from experiment 3 broken into groups by gender and genotype. B) Mean saccharin preference (+/- 1 s.e.m.) in 10-17 month old APP/PS1 and wildtype mice from experiment 1 broken down into groups by gender and genotype.
Examination of Aβ deposition in the brains of a subset of the young male and female +/+ mice suggested there might be some gender differences in plaque deposition. In general, the 2-5 month old female mice exhibited intermittent Aβ deposition in the hippocampus, amygdala and cortex, while the male 2-5 month old mice had little or no deposits in the same regions (see Figure 5). This observation might suggest that the failure to acquire CTA was correlated with Aβ deposition. However, examination of CTA in individual animals, and Aβ deposition in their brains, did not indicate such a correlation. Evaluation of individual brains from 2-5 month old female mice showed that despite the presence of sporadic plaque deposition mouse 42 did acquire CTA, while both mouse 32 and 38 failed to acquire CTA. Within the group of 2-5 month old male brains, both mice without detectable Aβ deposition (2 & 7) acquired CTA while mouse 34 with sporadic Aβ deposition failed to acquire CTA. So while among the 2-5 month old male mice, the presence of Aβ was consistent with the behavioral results, the results were not so clear in the 2-5 month old female mice.
Figure 5.
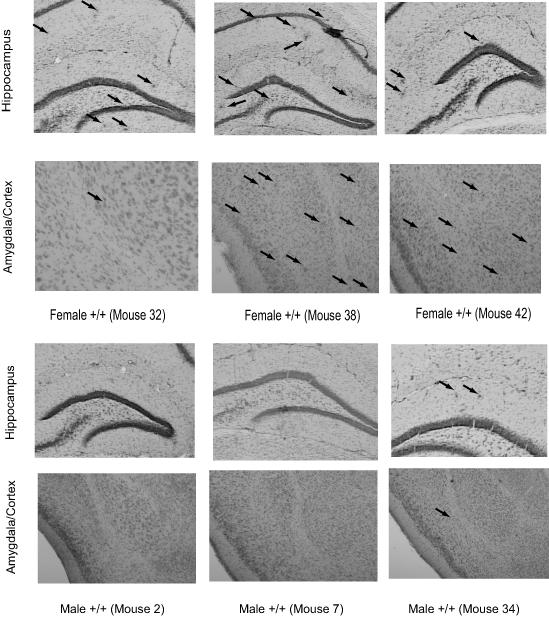
Images of the hippocampus, amygdala and cortex from representative brains of 2-5 month old male and female APP/PS1 mice, stained with Congo red and counterstained with cresyl violet, showing the sporadic Aβ deposition present in some of the brains. The images were taken at 5X and then cropped with image editing software. The brains were examined visually and no quantitative analysis was conducted. Because the plaques were more difficult to visualize in black & white, for the younger mice, arrows indicate the presence and location of plaques.
4. Discussion
Results from the current study indicate CTA is a suitable paradigm to evaluate learning and memory deficits in AD transgenic mice. Similar to results reported by Janus et al (2004) in TgCRND8 mice, both old (10-17 month) and young (2-5 month) APP/PS1 mice exhibited deficits in CTA. In the case of the old APP/PS1 mice, this deficit only appeared when both the APP and PS1 mutation were present, as mice carrying only the APP or PS1 mutation were capable of acquiring CTA. Importantly, these results, along with others (Janus et al., 2004, Madani et al., 2006), indicate that CTA acquisition deficits are not limited to one transgenic model of AD suggesting that the paradigm is generalizable to a variety of AD models. One interesting discovery was the potential gender difference observed in the 2-5 month old APP/PS1 mice. However, the group numbers for gender comparison were not large enough to reach any conclusions, therefore making it mostly speculative at this point in time.
Another important aspect of the results from the current study, in conjunction with those of Janus et al. (2004), is the ability to study forms of non-spatial learning and memory. CTA is a relatively non-invasive means to evaluate learning and memory using a task where the learning is rapidly acquired (in a single conditioning trial). CTA provides a rapid form of learning that is quickly assessed and does not require a significant training regimen. CTA is also not as sensitive to potential declines in motor function and visual perception, two potential confounds when evaluating behavior in aged mice with paradigms such as the water maze.
One surprising finding from this study was the gender difference that was observed in the young 2-5 month old APP/PS1 +/+ mice. Although based on the examination of the data, it is not possible to prescribe the differences to the presence of Aβ deposition, there is evidence for gender based differences in AD pathology in these mice. Wang et al. (2003) evaluated levels of Aβ40, Aβ42 and Aβ deposition in hippocampus of 2-, 12-and 17-month old male and female APP/PS1 mice. They found that at each age the female mice had higher levels for all measures. While this still does not provide an explanation of the underlying mechanism involved in the learning deficits, it does suggest that there may be an accelerated pathology in the female mice that may reflect the earlier onset of CTA deficits and might be one of the reasons that the 2-5 month old female mice, as a group, failed to acquire CTA. While there is an increased risk in women for developing AD, that may be linked to decreases in estrogen levels associated with menopause (Zheng et al., 2002, Yue et al., 2005), the age of the mice where the gender differences occurred suggests a decline in estrogen from menopause is unlikely in this case, making it difficult to easily link a decline in estrogen to the observed behavioral deficit. However, because this study was not designed to evaluate gender differences, and the group sizes when broken by gender were too small for adequate assessment, a more directed study would need to be conducted in order to verify the current results.
One debate in research utilizing AD transgenic mice has centered on the relationship between Aβ deposition and memory deficits. While AD is associated with various amounts of Aβ deposition, neurofibrillary tangles, changes in synaptic function and neuronal loss, the factors that lead to the onset of these pathological functions remain unclear. In more recent years evidence indicates that prior to Aβ deposition increased levels of soluble oligomer forms of Aβ may be toxic to neuronal function and highly correlated with the extent of cognitive dysfunction (Cherny et al., 1999, Lue et al., 1999, Naslund et al., 2000). Electrophysiological studies indicate synthetic or cell-produced forms of soluble Aβ can inhibit long-term potentiation in vitro and in vivo (Lambert et al., 1998, Kim et al., 2003), and in various mouse models of AD, cognitive deficits appear before plaque deposition is evident (Ashe, 2005). Although soluble forms of Aβ were not measured in the current studies, it may be an explanation for the CTA acquisition failure in the young 3-5 month old mice where there was lack of extensive plaque deposition.
Overall it appears that CTA is potentially a reliable paradigm for evaluating non-spatial learning and memory in various mouse models of AD. Along with the current study, deficits in the acquisition or extinction of CTA have been found to be present in more than one AD mouse model (Janus et al., 2004, Pennanen et al., 2004, Madani et al., 2006). Therefore, it may offer a rapidly acquired form of learning that can be used to evaluate potential therapeutic interventions. Based on the gender differences observed in the current study it may be worthwhile to further investigate the mechanisms underlying them. CTA appears to offer an additional viable avenue for investigating AD-induced learning and memory deficits in areas of the brain outside of the hippocampus, but further support would be provided by investigation of CTA in additional mouse models of AD.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute on Aging. Christopher Quigley provided assistance with the histological preparation. Collection of the brain slice images utilized the facilities of the Cell Biology and Bioimaging Core facilities at the Pennington Biomedical Research Center supported by NIH Grant 1P20 RR02/1945 and a CNRU center grant # 1P30 DK072476 sponsored by NIDDK.
List of Abbreviations
- CTA
conditioned taste aversion
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- PS1
presenilin 1
- Aβ
beta-amyloid
- LiCl
lithium chloride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashe KH. Synaptic structure and function in transgenic APP mice. Ann N Y Acad Sci. 2000;924:39–41. doi: 10.1111/j.1749-6632.2000.tb05558.x. [DOI] [PubMed] [Google Scholar]
- Ashe KH. Learning and memory in transgenic mice modeling Alzheimer’s disease. Learn Mem. 2001;8:301–308. doi: 10.1101/lm.43701. [DOI] [PubMed] [Google Scholar]
- Ashe KH. Mechanisms of memory loss in Abeta and tau mouse models. Biochem Soc Trans. 2005;33:591–594. doi: 10.1042/BST0330591. [DOI] [PubMed] [Google Scholar]
- Best P, Orr J. Effects of hippocampal lesions on passive avoidance and taste aversion learning. Physiol Behav. 1973;10:193–196. doi: 10.1016/0031-9384(73)90296-5. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Carlesimo GA, Oscar-Berman M. Memory deficits in Alzheimer’s patients: a comprehensive review. Neuropsychology Review. 1992;3:119–169. doi: 10.1007/BF01108841. [DOI] [PubMed] [Google Scholar]
- Cherny RA, Legg JT, McLean CA, Fairlie DP, Huang X, Atwood CS, Beyreuther K, Tanzi RE, Masters CL, Bush AI. Aqueous dissolution of Alzheimer’s disease Abeta amyloid deposits by biometal depletion. J Biol Chem. 1999;274:23223–23228. doi: 10.1074/jbc.274.33.23223. [DOI] [PubMed] [Google Scholar]
- Convit A, De Leon MJ, Tarshish C, De Santi S, Tsui W, Rusinek H, George A. Specific hippocampal volume reductions in individuals at risk for Alzheimer’s disease. Neurobiol Aging. 1997;18:131–138. doi: 10.1016/s0197-4580(97)00001-8. [DOI] [PubMed] [Google Scholar]
- Corder EH, Lannfelt L, Bogdanovic N, Fratiglioni L, Mori H. The role of APOE polymorphisms in late-onset dementias. Cell Mol Life Sc. 1998;54:928–934. doi: 10.1007/s000180050223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leon MJ, George AE, Golomb J, Tarshish C, Convit A, Kluger A, De Santi S, McRae T, Ferris SH, Reisberg B, Ince C, Rusinek H, Bobinski M, Quinn B, Miller DC, Wisniewski HM. Frequency of hippocampal formation atrophy in normal aging and Alzheimer’s disease. Neurobiol Aging. 1997;18:1–11. doi: 10.1016/s0197-4580(96)00213-8. [DOI] [PubMed] [Google Scholar]
- Dunn L, Everitt B. The effects of lesions to noradrenergic projections from the locus coeruleus and lateral tegmental cell groups on conditioned taste aversion in the rat. Behav Neurosci. 1987;101:409–422. doi: 10.1037//0735-7044.101.3.409. [DOI] [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O’Callaghan JP, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol. 2002;173:183–195. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- Hom J. General and specific cognitive dysfunctions in patients with Alzheimer’s disease. Arch Clin Neuropsychol. 1992;7:121–133. [PubMed] [Google Scholar]
- Hwang DY, Chae KR, Kang TS, Hwang JH, Lim CH, Kang HK, Goo JS, Lee MR, Lim HJ, Min SH, Cho JY, Hong JT, Song CW, Paik SG, Cho JS, Kim YK. Alterations in behavior, amyloid beta-42,caspase-3,and Cox-2 in mutant PS2 transgenic mouse model of Alzheimer’s disease. FASEB J. 2002;16:805–813. doi: 10.1096/fj.01-0732com. [DOI] [PubMed] [Google Scholar]
- Janus C, Welzl H, Hanna A, Lovasic L, Lane N, St George-Hyslop P, Westaway D. Impaired conditionedtaste aversion learning in APP transgenic mice. Neurobiol Aging. 2004;25:1213–1219. doi: 10.1016/j.neurobiolaging.2003.11.007. [DOI] [PubMed] [Google Scholar]
- Jolas T, Zhang X-S, Zhang Q, Wong G, Del Vecchio R, Gold L, Priestley T. Long-term potentiation is increased in the CA1 area of the hippocampus of APP(swe/ind) CRND8 mice. Neurobiol Dis. 2002;11:394–409. doi: 10.1006/nbdi.2002.0557. [DOI] [PubMed] [Google Scholar]
- Kim H-J, Chae S-C, Lee D-K, Chromy B, Lee SC, Park Y-C, Klein WL, Krafft GA, Hong S-T. Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein. FASEB J. 2003;17:118–120. doi: 10.1096/fj.01-0987fje. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Lopez-Garcia J, Fernandez-Ruiz J, Escobar M, Bermudez-Rattoni F, Tapia R. Effects of excitotoxic lesions of the nucleus basilis magnocellularis on conditioned taste aversion and inhibitory avoidance in the rat. Pharmacol Biochem Behav. 1993;23:439–444. doi: 10.1016/0091-3057(93)90098-e. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Path. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madani R, Poirier R, Wolfer DP, Welzl H, Groscurth P, Lipp H-P, Lu B, El Mouedden M, Mercken M, Nitsch RM, Mohajeri MH. Lack of neprilysin suffices to generate murine amyloid-like deposits in the brain and behavioral deficit in vivo. J Neurosci Res. 2006;84:1871–1878. doi: 10.1002/jnr.21074. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Mandelkow E. Tau in Alzheimer’s disease. Trends Cell Bio. 1998;8:425–427. doi: 10.1016/s0962-8924(98)01368-3. [DOI] [PubMed] [Google Scholar]
- Morgan D. Learning and memory deficits in APP transgenic mouse models of amyloid deposition. Neurochem Res. 2003;28:1029–1034. doi: 10.1023/a:1023255106106. [DOI] [PubMed] [Google Scholar]
- Murphy L, Brown T. Hippocampal lesions and learned taste aversion. J Comp Physiol Psychol. 1974;2:60–64. [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAM. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Pennanen L, Welzl H, D’Adamo P, Nitsch RM, Gotz J. Accelerated extinction of conditioned taste aversion in P301L tau transgenic mice. Neurobiol Dis. 2004;15:500–509. doi: 10.1016/j.nbd.2003.11.020. [DOI] [PubMed] [Google Scholar]
- Pepin EP, Eslinger PJ. Verbal memory decline in Alzheimer’s disease: a multiple- processes deficit. Neurology. 1989;39:1477–1482. doi: 10.1212/wnl.39.11.1477. [DOI] [PubMed] [Google Scholar]
- Roth S, Schwartz M, Teitelbaum P. Failure of recovered lateral hypothalamic rats to learn specific food aversions. J Comp Physiol Psychol. 1973;83:184–187. doi: 10.1037/h0034415. [DOI] [PubMed] [Google Scholar]
- Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzalez V, Wong MPF, Price DL, Tang F, Markowska AL, Borchelt DR. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: Relationships to β- amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis. 2005;18:602–617. doi: 10.1016/j.nbd.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Biochemistry of altered brain proteins in Alzheimer’s disease. Annu Rev Neurosci. 1989;12:463–490. doi: 10.1146/annurev.ne.12.030189.002335. [DOI] [PubMed] [Google Scholar]
- Teter B, Raber J, Nathan B, Crutcher KA. The presence of apoE4, not the absence of apoE3, contributes to AD pathology. J Alzheimers Dis. 2002;4:155–163. doi: 10.3233/jad-2002-4305. [DOI] [PubMed] [Google Scholar]
- Wang J, Tanila H, Puoliväli J, Kadish I, van Groen T. Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol Dis. 2003;14:318–327. doi: 10.1016/j.nbd.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Welzl H, D’Adamo P, Lipp H-P. Conditioned taste aversion as a learning and memory paradigm. Behav Brain Res. 2001;125:205–213. doi: 10.1016/s0166-4328(01)00302-3. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Fujimoto Y, Shimura T, Sakai N. Conditioned taste aversion in rats with excitotoxic brain lesions. Neurosci Res. 1995;22:31–49. doi: 10.1016/0168-0102(95)00875-t. [DOI] [PubMed] [Google Scholar]
- Yue X, Lu M, Lancaster T, Cao P, Honda S-I, Staufenbiel M, Harada N, Zhong Z, Shen Y, Li R. Brain estrogen deficiency accelerates Abeta plaque formation in an Alzheime’s disease animal model. Proc Natl Acad Sci U S A. 2005;102:19198–19203. doi: 10.1073/pnas.0505203102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Xu H, Uljon SN, Gross R, Hardy K, Gaynor J, Lafrancois J, Simpkins J, Refolo LM, Petanceska S, Wang R, Duff K. Modulation of A(beta) peptides by estrogen in mouse models. J Neurochem. 2002;80:191–196. doi: 10.1046/j.0022-3042.2001.00690.x. [DOI] [PubMed] [Google Scholar]