

Abstract
We report the proton second moment obtained directly from the Free Induction Decay (FID) of the NMR signal of variously hydrated bovine serum albumin (BSA) and hen egg white lysozyme (HEWL) and from the width of the NMR Z-spectrum of the cross-linked protein gels of different concentrations. The second moment of the proteins decreases in a continuous stepwise way as a function of increasing water content, which suggests that the structural and dynamical changes occur in small incremental steps. Although the second moment is dominated by the short range distances of nearest neighbors, the changes in the second moment show that the protein structure becomes more open with increasing hydration level. A difference between the apparent liquid content of the sample as found from decomposition of the FID and the analytically determined water content demonstrates that water absorbed in the early stages of hydration is motionally immobilized and magnetically indistinguishable from rigid protein protons while at high hydration levels some protein side-chain protons move rapidly contributing to liquid-like component of the NMR signal.
Keywords: protein hydration, wideline NMR, proton second moment, structural rearrangements, water
Introduction
It is widely known that structural and dynamical changes accompany dehydration of proteins [1–4]; however, it is difficult to monitor these changes quantitatively or even qualitatively. In solid systems, i.e., ones where rapid motional averaging does not provide extreme spectral narrowing, the NMR linewidths are large and obscure the spectral features usually used for structure assignment. Nevertheless, NMR measurements may provide interesting structural information because the linewidths or the second moments of the resonance lines are a strong function of proton-proton intermoment distances and local dynamical averaging of the dipolar couplings. The second moment of the solid lineshape may, therefore, provide a direct report of the average protein structure.
Historically, the second moment was difficult to measure accurately because of spectral broadening by saturation effects in continuous wave experiments. In pulse experiments, the receiver dead time following an excitation pulse requires extrapolation of the detectable signal to zero time which, in turn, requires knowledge of the lineshape function for the extrapolation [5]. However, high field NMR spectrometers have short receiver ring-down times, so that such extrapolations are not long, and the receiver bandwidths now are sufficient to capture accurate NMR signals in the time domain. The signal following a single short excitation pulse, or FID, is a Gaussian decay for a solid protein-proton-spin system:
| [1] |
where Mxy(t) is the transverse magnetization decay at time t and M2 is the second moment. The second moment is related to the intermoment distance by the equation [6],
| [2] |
where γ is the magnetogyric ratio, ℏ is Planck's constant, N is the number of nuclei per unit volume, is the intermoment distance between nuclei j and k, and θjk is the angle between the applied magnetic field and . For a rigid system, where all orientations of the vector rjk are equally probable, the second moment must be averaged over all θjk which lead to the expression
| [3] |
Although there is no structural detail in the second moment for the complex arrangement of protein spins, this simple measurement provides a global structural assessment for disordered systems. The second moment also reflects the changes in the local dynamics of the observed spins. The effect of local motion is observed as a reduction of the second moment. For a protein, rapid motions such as, for example, methyl group Markovian jumps may partially average the dipole-dipole couplings and reduce the second moment due to decrease in the average value of (1 – 3cos2 θjk) and terms in Eq. [2]. The second moment then reports the combined impact of the high frequency averaging on the effective structure. We describe here an interesting example of the progressive hydration of proteins from the lyophilized to the fully hydrated state. Changes in the protein structure and dynamics with hydration result in a decrease in the 1H second moment, but the response is not linear.
Experimental
Sample Preparation
Bovine Serum Albumin (BSA Fraction V, Sigma-Aldrich, Inc, #A3059) and lysozyme from chicken egg white (Sigma Chemical Co., #L-6876) were each dialyzed against four changes of deionized water and lyophilized to constant weight using a mechanical vacuum line at room temperature. Approximately 150 mg of each purified protein was dissolved in 2 mL water and the measured pH was 6.5 for BSA and 5.2 for lysozyme. In order to avoid possible denaturation or disulfide bond reorganization the temperature was not increased in the drying process to achieve completely dry protein, as some authors reported [7]. The residual water content in the lyophilized proteins was determined by a coulometric Karl-Fischer titration using a Photovolt Aquatest 8 titrator and was found to be 3.5 ± 0.04 % (% water weight/total weight) for lysozyme and 5.4 ± 1.08 % for BSA. This residual water content corresponds to approximately 29 water molecules per lysozyme molecule and 212 water molecules per BSA molecule. Thus, a NMR measurement of completely dry protein was not made.
Hydrated protein powders
Both BSA and lysozyme were hydrated isopiestically by standing over water-glycerol (Fisher Scientific, Fair Lawn, NJ 07410) solutions of different composition [8]. The dried protein sample (150–200 mg) contained in a plastic boat was weighed accurately (Mettler AT 261 Delta Range, ±0.00001 g), placed on a support inside a sealed glass container over a water-glycerol mixture, and allowed to equilibrate for over 3 weeks. Plastic boats with protein were then removed and immediately weighed; the mass increase was attributed to water absorbed by the protein. The total water content in the hydrated protein powders was computed as the sum of the initial water content for the dried stock sample plus the gain from the isopiestic hydration procedure. Hydration results are summarized in Fig. 1 and are consistent with earlier reports [7, 9]. We note that BSA adsorbs water more strongly than lysozyme throughout the accessible range of relative humidity.
Figure 1.

Hydration level of BSA (◆) and hen egg white lysozyme (○) as a function of the relative humidity for the proteins hydrated isopiestically over aqueous glycerol solutions.
Protein gels
BSA and lysozyme gels were prepared by dissolving 50, 150, 250 and 400 mg of dried BSA and 250 and 340 mg of dried lysozyme each in 0.9 ml of water. These protein solutions were transferred into individual 5 mm NMR tubes. 0.1 ml of 25% aqueous glutaraldehyde (Sigma Chemical Co, St. Louis, MO) was added to each tube using a long glass pipette, and vigorously mixed. Gels formed within 10 minutes. The corresponding water content of BSA gels was then 73.0, 81.1, 87.7, 95.5 mass % and lysozyme gels 75.5 and 80.7 mass %.
The effect of cross-linking proteins on their structure has been reported to be negligible as studied by X-ray diffraction method [10]. The differences between root-mean-square displacements of native and glutaraldehyde cross-linked proteins lied between 0.12 and 0.2 Å.
NMR Measurements and Data Analysis
Nuclear magnetic resonance data were collected using a Varian Unity Plus NMR spectrometer operating at a 1H resonance frequency of 500 MHz.
Hydrated protein powders
Proton free induction decays for hydrated protein samples were recorded at 298 K following a single 8 µs excitation pulse using a 5 mm triple-resonance z-gradient probe. The acquisition delay time following the transmitter pulse was 3.27 µs. Figure 2 shows the initial portion of representative free induction decays for variously hydrated BSA powders. In the case of a hydrated protein, the FID of the transverse signal is a superposition of a solid component and a liquid component, which are easily separable since the time constants for the decay are significantly different as seen in Fig. 2. Because the samples are spatially and dynamically heterogeneous, the liquid component line is well characterized by a Gaussian shape (Eq.[1]). The fit of the total decay to a sum of Gaussian functions was robust with the correlation coefficients always larger than 0.999. The correlation coefficients indicate how well the calculated curve fits the original data and assume values from zero to one. For the driest samples, a single Gaussian accounts for the data. At the highest water content a small contribution from a very long component was noted and the first 330 µs of the 512 µs decay was fit reliably to a sum of Gaussian components plus a constant.
Figure 2.

The early portion of the normalized transverse magnetization decay for BSA powders hydrated isopiestically to different levels: (○) 8.5% water, (□) 14.3% water, (◇) 23.9% water, (△) 33.8% water by mass. The total acquisition time for all measurements of hydrated powders was 512 µs.
Protein gels
For cross-linked protein gel samples, the water-proton signal far exceeds the protein-proton signal, making accurate measurement of the protein-proton second moment difficult. The dynamic range problem was circumvented by measuring the Z-spectra of the gel samples. In this experiment, the water resonance is observed as a function of the frequency offset of a long weak saturating rf pulse. The pulse sequence consisted of a soft preparation pulse of duration 1 or 3 s with a nutation frequency not exceeding 120 Hz. The preparation pulse was offset by a variable frequency from the water resonance and, after an 80 µs delay, was followed by a 12.5 µs 90° observation pulse and acquisition. The saturation is transferred to the water resonance and the dependence of the water signal intensity on the frequency offset of the preparation pulse reports the line shape of the solid component spectrum [11]. Representative Z-spectra are summarized in Fig. 3. The most central portion of the spectra, where a narrow peak due to water signal saturation is usually observed [11], was deleted for clarity. The solid lines in Fig. 3 are Gaussian fits to the experimental line shape. The second moment is related to the full width at half height of the absorption line shape (FWHH, Δν) by the relation,
| [4] |
To eliminate the effects of saturation on the linewidth, (FWHH)2 versus the square of the preparation pulse power was extrapolated to zero as shown in Fig. 4. These extrapolated values were used in determining the second moment of protein gels. The extrapolated FWHH values differ by a relative standard deviation of less than 2%.
Figure 3.

The normalized Z-spectra for a 40% BSA gel in water as a function of the amplitude of the preparation pulse at 298 K: (●) 56 Hz, (■) 63 Hz, (◆) 71 Hz, (▲) 89 Hz, (▼) 112 Hz. The solid lines are Gaussian fits to the experimental line shapes simulated with the non-linear Levenburg-Marquardt least-squares procedure.
Figure 4.

The square of the full width at half height of the z-spectrum as a function of the preparation pulse amplitude for BSA gels at different mass per cent protein at 298 K: (○) 5%, (▲) 15%, (◇) 25%, (■) 40%. The solid lines are linear fits to the experimental data points.
Results and Discussion
The 1H second moments are shown in Figure 5A as a function of mass percent water for BSA and lysozyme hydrated powders and gels, and the portion of the data limited to hydrated powders is plotted as a function of the number of water molecules per protein molecule in Fig. 5B. The putatively dry protein remains partly hydrated as determined by Karl-Fischer titrations, although these samples show no liquid or slowly decaying transverse magnetization component. Therefore, the residual water in the vacuum-dried protein at levels below 5% is not rotationally mobile and the water-proton NMR signal is indistinguishable from the FID of the solid protein protons.
Figure 5.

The 1H second moment for the rapidly decaying portion of the FID for BSA (◆) and hen egg white lysozyme (○) as a function of (A) the mass per cent water (samples above 40% were cross-linked protein gels); (B) water molecules per protein molecule for hydrated powders.
The proton second moment for both proteins decreases by approximately 60% with increasing hydration to the level of the fully hydrated protein in a gel. The second moment change may result from two causes: structural changes in the protein that increase the intermoment distances contributing to the sum in Eq. 2 and motional averaging of the inter-proton dipolar couplings. The addition of water, which has a significant electric dipole moment, may relax the electrostatic constraints caused by the charged groups or dipoles in the protein. The resulting structural reorganization may facilitate local motion of some groups because of changes in the effective free volume in the folded structure. Of course, it is well known that such motions as N-H small librations [12] or methyl rotations [13, 14] are present at 298 K whether the protein is dry or hydrated; however, it is clear from earlier work that solvation of side chains permits side chain motions that lengthen the transverse decay significantly [15]. The present analysis of the transverse decay isolates the fast component, which then corresponds to the majority of the protein protons that do not experience significant dynamical averaging. The most rapidly moving side-chain proton contributions are present in the slower part of the decay that is not included in the second moment plotted. In this sense, the second moment shown in Fig. 5 represents a report on the structure and dynamics of the protein core.
For BSA, the beginning of the hydration process, up to approximately 8% water by mass, results in 42% of the second moment decrease. At this point there are approximately 320 water molecules per BSA molecule. These changes parallel significant decreases in the enthalpy of water adsorption to the protein, which have been attributed to hydration of protein charged groups [9, 16]. The second part of the M2 response for BSA hydration extends to approximately 30% water by mass, which corresponds to approximately 1600 water molecules per protein molecule, and represents a nearly fully hydrated system. We note that the second moment at this point is still somewhat larger than that of the protein gel, although 88% of the total change in the second moment has occurred. Similar numbers of water molecules (1200–1600) per protein have been reported for fully hydrated albumin [17]. Some discrepancy in these numbers may result from differences in the pH of the solutions from which the samples were prepared because the hydration response should be a function of the effective charge distribution on the protein. This distribution may be a reason why the second moment profile for lysozyme is qualitatively different at low water contents from the albumin. Even at 100% relative humidity, lysozyme does not reach full hydration and the proton second moment decreases an additional factor of 1/3 from the hydrated powder with 35% water content to the cross-linked gel. This observation is consistent with low (10%) lysozyme activity in the isopiestically hydrated powder [16]. The partial molar heat capacity reported for lysozyme as a function of hydration shows that at 5–7 and 21 mass percent there are peaks on the otherwise rather smooth curve that are identified with the proton transfer from the carboxylic acid to basic protein groups at the lowest level of hydration, and with specific interactions of water with carbonyl oxygen atoms and other polar sites at 21% [9]. At approximately 20% water by mass, we observe a change in the slope of the second moment for lysozyme versus water content that correlates with this earlier report.
Generally, the protein second moments shown in Fig. 5 respond to increasing hydration similarly to the changes in the enthalpy of adsorption for water, or for the partial molar heat capacity reported by others [9]. This parallel behavior is expected because the energetic changes undoubtedly result in small structural and dynamical rearrangements of the protein that are reflected in the second moment.
It is instructive at this point to compare measurements with calculations of the second moment. To calculate the second moment, the coordinates of protein protons within three-dimensional structures from the protein data bank were created with the aid of Insight II software (Biosym Technologies, Inc.). Two different PDB files were used to calculate (and compare) the average second moment of HEWL. The structure was determined by solution NMR in 1E8L while in 1F10 file the X-Ray diffraction (XRD) measurements were conducted on protein hydrated at 88% relative humidity. The structure of bovine serum albumin is not reported yet but the amino sequence of the protein is 76% homologous to the amino acid sequence of human serum albumin (HSA). Moreover, computational and mass spectroscopy studies showed high degree of similarity in 3D structures between these two proteins [18–20]. Therefore, PDB file 1E78, where the structure of fully hydrated HSA was measured by XRD, was used for calculation of the second moment.
In general, the calculation of the protein second moment is made by taking the time average of (1 – 3 cos2 θjk) and in Eq. [2] since protein side-chain and back-bone protons can participate in fast motions, and after that averaging over all possible powder orientations. The accuracy of this calculation depends on the accuracy of determination that are deduced from solution NMR structures or XRD spectroscopy with the certain assumptions that are made about bond length and angles relating hydrogens to heavier atoms that can be seen by XRD. A few M2 values greater than 3×1010 s−2 are not shown in Fig.5 and were dismissed in the calculation since they corresponded to unrealistically close proton-proton separation.
The computation of the second moment yields an average value of 1.39×1010 s−2 (1E8L) and 1.33×1010 s−2 (1F10) for HEWL and 1.32×1010 s−2 for HSA. These values are essentially equal to the measured values for the dry proteins. However, the calculation is based on the structure of the protein in a fully hydrated state, not the lyophilized state. Furthermore, the calculation presumes that the methyl protons are rigid at 298 K, but it is well established that methyl groups rotate rapidly at this temperature [13, 14]. Correction of the calculated average by the order parameter associated with methyl rotation with appropriate weight (183 methyl protons in HEWL and 939 in HSA) reduces the calculated average second moment to 〈M2〉rigid = 1.06×1010 s−2 (1E8L) for lysozyme and 0.97×1010 s−2 for HSA. The assumption is that all remaining protein protons are not moving fast enough to average the dipolar coupling strength.
The distribution of HSA second moments is shown in Fig. 6 where the three distinct populations derive largely from CH, CH2, and CH3 contributions. The inset, which illustrates dependence of the second moment on the intermoment distance, shows that the second moment is dominated by nearest neighbor interactions such as interactions between the two protons in a methylene group and is not, therefore, very sensitive to longer range secondary or tertiary structure of the protein. Applying the correction for methyl group rotational jumps makes the distribution of the second moment bimodal, with the first peak corresponding to mostly CH and CH3 protons and the second one to CH2 protons. Consequently, the relative amplitude of the first peak in this distribution is larger than the weight of the second peak. A similar calculation was reported [21] and referred to the dipolar energy that enters the intramolecular rotational spin-lattice relaxation equation [22] and differs from the present values of the second moment [23] by the numerical factor of 10/3.
Figure 6.

Calculated distribution of the second moment based on human serum albumin PDB structure 1E78. It was assumed in this calculation that all protein protons are rigid and the few values greater than 3×1010 s−2 are not shown. The inset illustrates the dependence of the second moment on the intermoment distance.
If one neglects probable dynamical averaging, comparison of this computed second moment with the measured values for the lyophilized state provide a crude estimate for the magnitude of the structural change on hydration. The changes will not derive from the distances of protons bonded to the same atom, i.e, CH2 or CH3 functions, but from the next nearest proton distances. Figure 6 (inset) suggests that the average change is small, of order 0.1 Å or so, which is an upper bound because this comparison neglects likely motional averaging effects. This structural change constitutes approximately 24% of the decrease in the second moment observed on hydration. The observation of the structural changes on hydration is consistent with previous X-Ray and nuclear magnetic relaxation dispersion reports [24–27]. The remaining 76% decrease in the second moment is ascribed to the increase in motional freedom of protein protons.
Based on the measured value of the second moment for a fully hydrated protein (gel) and the calculated rigid average value of the second moment, one can compute an average “collective” order parameter [21, 28]. For lysozyme (1E8L) this parameter, , is equal to 0.54 and it is equal to 0.58 for BSA. It is not clear how to interpret this global value of the order parameter even though it may be accounted for by small displacements of order 0.1 Å. One expects that surface residues will suffer greater displacements than interior core residues or backnone N – H protons, but this simple solid state measurement does not provide resolution to make this distinction.
The free induction decay for the variously hydrated protein may be used to extract the water content from the ratio of the slowly decaying transverse component to the total, extrapolated to zero time. Indeed, NMR has been successfully used to estimate liquid fractions in dynamically heterogeneous materials such as fats for many years [29–31]. This approach applied to the present protein samples is summarized in Fig. 7. If the spectroscopic and gravimetric/analytical measurements are exactly equivalent, a linear dependence with a slope of unity should result as shown by the solid line in Fig. 7. A difference between the analytical and the spectroscopic measurement is expected if water is not mobile at low hydration levels and is essentially as dynamically rigid as the protein. At lowest water contents, the BSA and lysozyme samples show a negative deviation indicating that at least some of the adsorbed water is sufficiently rotationally immobilized on the protein to have the same transverse decay as the protein protons. For albumin, the difference is negative until nearly 15% water by mass when it becomes positive and gradually increases until reaching a plateau at approximately 30% water by mass. For lysozyme, there is almost no difference between the spectroscopic and the gravimetric/analytical measurements until water content reaches 35% by mass.
Figure 7.
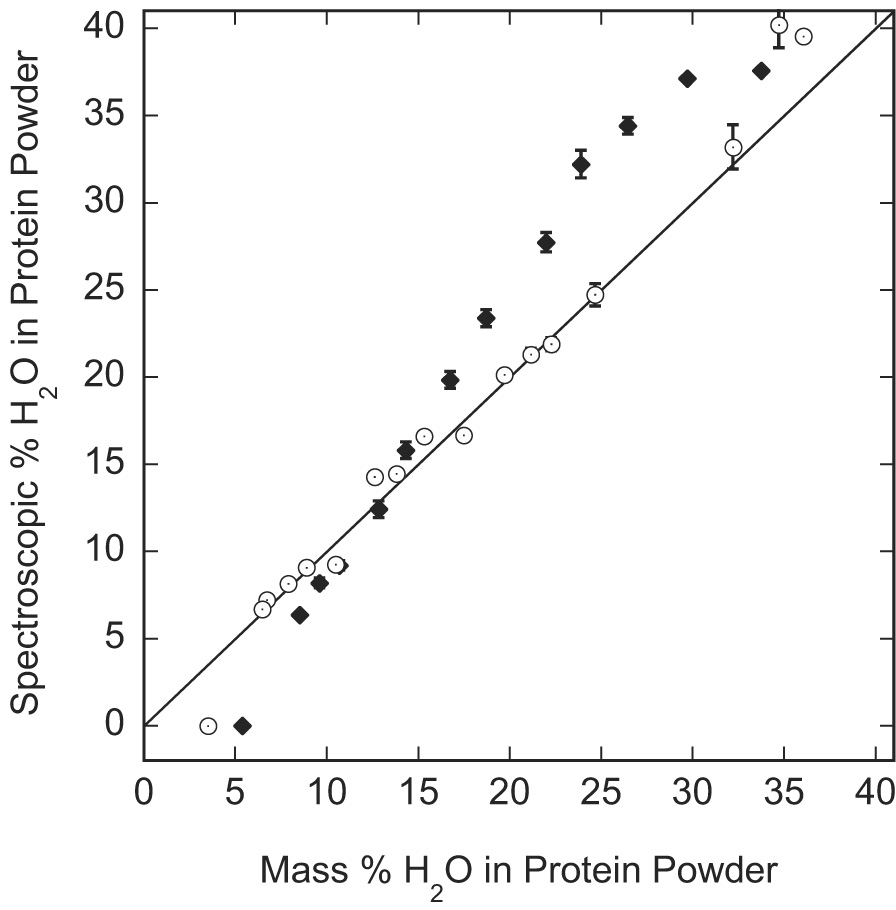
The water content determined from the ratio of the slowly decaying portion of the 1H FID to the total extrapolated initial amplitude as a function of the analytically determined mass per cent water in variously hydrated BSA (◆) and hen egg white lysozyme (○) powders. The solid line with the slope of 1 is drawn for reference.
A positive deviation of the spectroscopic measurement is expected at high hydration levels when there is enough water to mobilize some protein side chains so that the side-chain protons contribute to the slowly relaxing transverse component [15]. Both proteins show this positive deviation at high water contents, but it is clear that lysozyme requires much more water than BSA to mobilize the protein side chains. Once the side chains are mobilized, additional water will not increase this contribution. The side-chain contribution will remain constant, but the water proton contribution will increase linearly with increasing water content. Therefore, the side-chain contribution to the slowly relaxing component will decrease as can be seen in Fig. 7 for the BSA data.
Conclusion
Studies of the proton second moment provide a simple and efficient means for monitoring structural and dynamical changes in proteins as a function of hydration level. For both bovine serum albumin and hen egg white lysozyme, the second moment of the rapidly decaying component of the FID decreases with the increase of the hydration level that indicates “swelling” of the protein structure and increase in motional freedom of the protein protons. Nevertheless, the two studied globular proteins exhibit qualitatively different changes in the second moment. This difference was attributed to the different effective charge distribution on the proteins. In the cross-linked protein gels, which have the same hydration level as protein solutions of the same composition, the second moment was found to be somewhat less than in the protein powder hydrated at 100% relative humidity.
It was found that at the lowest hydration levels, the water protons are dynamically indistinguishable from those of the protein, which causes a negative difference between the apparent liquid content of the sample as determined from decomposition of the FID and the gravimetric water contents. At higher hydration levels, there may be a positive deviation when protein side-chains that move rapidly contribute to the slowly decaying component of the transverse magnetization decay.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bagchi B. Water Dynamics in the Hydration Layer around Proteins and Micelles, Chemical Reviews. Vol. 105. Washington, DC, United States: 2005. pp. 3197–3219. [DOI] [PubMed] [Google Scholar]
- 2.Paulaitis ME, Pratt LR. Hydration theory for molecular biophysics. Advances in Protein Chemistry. 2002;62:283–310. doi: 10.1016/s0065-3233(02)62011-x. 283 plates. [DOI] [PubMed] [Google Scholar]
- 3.Prabhu N, Sharp K. Protein-Solvent Interactions, Chemical Reviews. Vol. 106. Washington, DC, United States: 2006. pp. 1616–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smith JC, Merzel F, Bondar A-N, Tournier A, Fischer S. Structure, dynamics and reactions of protein hydration water, Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences. 2004;359:1181–1190. doi: 10.1098/rstb.2004.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeener J, Broekaert P. Nuclear magnetic resonance in solids. Thermodynamic effects of a pair of radio-frequency pulses. Physical Review. 1967;157:232–240. [Google Scholar]
- 6.Slichter CP. Principles of magnetic resonance. 3rd ed. Springer, Berlin, Heidelberg, New York: 1980. [Google Scholar]
- 7.Kuntz IDJ, Kauzmann W. Hydration of proteins and polypeptides. Adv. Protein Chem. 1974;28:239–345. doi: 10.1016/s0065-3233(08)60232-6. [DOI] [PubMed] [Google Scholar]
- 8.Washburn EW. International critical tables of numerical data, physics, chemistry, and technology. First Electronic Edition. Vol. 1928 Knovel, Norwich, NY, USA: 2003. [Google Scholar]
- 9.Rupley JA, Yang P-H, Tollin G. Thermodynamic and related studies of water interacting with proteins. In: Rowland SP, editor. Water in Polymers. Washington, DC USA: American Chemical Society; 1980. pp. 111–132. [Google Scholar]
- 10.Salem M, Mauguen Y, Prange T. On the edge of the denaturation process: Application of X-ray diffraction to barnase and lysozyme cross-linked crystals with denaturants in molar concentrations. Biochimica et Biophysica Acta. 2006;1764:903–912. doi: 10.1016/j.bbapap.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 11.Grad J, Bryant RG. Nuclear magnetic cross-relaxation spectroscopy. Journal of Magnetic Resonance. 1990;90:1–8. doi: 10.1002/mrm.1910170216. [DOI] [PubMed] [Google Scholar]
- 12.Usha MG, Peticolas WL, Witterbort RJ. Deuterium Quadrupole Coupling in N-Acetylglycine and Librational Dynamics in Solid Poly(γ-benzyl-L-glutamate) Biochemistry. 1991;30:3955–3962. doi: 10.1021/bi00230a022. [DOI] [PubMed] [Google Scholar]
- 13.Andrew ER. Protein dynamics in the solid state by proton relaxation. Bulletin of Magnetic Resonance. 1983;5:104–106. [Google Scholar]
- 14.Andrew ER. N.M.R. of solid biopolymers. Polymer. 1985;26:190–192. [Google Scholar]
- 15.Goddard Y, Korb JP, Bryant RG. Structural and dynamical changes in serum albumin at low temperature: examination of the glass transition. Biophys J. 2006;91:3841–3847. doi: 10.1529/biophysj.106.090126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenhaber F, Argos P. Hydrophobic regions on protein surfaces: definition based on hydration shell structure and a quick method for their computation. Protein Engineering. 1996;9:1121–1133. doi: 10.1093/protein/9.12.1121. [DOI] [PubMed] [Google Scholar]
- 17.Franks F. Solvation interactions of proteins in solution. Phil. Trans. R. Soc. London B. 1977;278:89–96. doi: 10.1098/rstb.1977.0032. [DOI] [PubMed] [Google Scholar]
- 18.Maruyama T, Katoh S, Nakajima M, Nabetani H. Mechanism of Bovine Serum Albumin Aggregation During Ultrafiltration. Biotechnology and Bioengineering. 2001;75:233–238. doi: 10.1002/bit.10001. [DOI] [PubMed] [Google Scholar]
- 19.Huang BX, Kim H-Y, Dass C. Probing 3D structure of bovine serum albumin by chemical cros-linking and mass spectrometry. Journal od the American Soc of Mass Spectrometry. 2004;15:1237–1247. doi: 10.1016/j.jasms.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Grange AH, Zumwalt MC, Sovocol GW. Determination of ion and neutral loss compositions and deconvolution of product ion mass spectra using an orthogonal acceleration time-of-fligh mass spectrometer and an ion correlation program. Rapid Commun Mass Spectrom. 2006;20:89–102. doi: 10.1002/rcm.2277. [DOI] [PubMed] [Google Scholar]
- 21.Luchinat C, Parigi G. Collective Relaxation off Proteins at Very Low Magnetic Field: A New Window on Protein Dynamics and Aggregation. JACS. 2007;129:1055–1064. doi: 10.1021/ja0633417. [DOI] [PubMed] [Google Scholar]
- 22.Abragam A. The Priniciples of Nuclear Magnetism. Oxford University Press, Oxford, The Clarendon Press; 1961. Chapt. VIII; p. 300. [Google Scholar]
- 23.Abragam A. The Priniciples of Nuclear Magnetism. Oxford University Press, Oxford, The Clarendon Press; 1961. Chapt. IV; p. 112. [Google Scholar]
- 24.Krupyanskii YF, Eshchenko GV, Esin SV, Mikhailyuk MG, Vetrov OD, Korotina OA, Zakharova NI, Knox PP, Rubin AB. A study of protein structure changes during hydration by diffuse X-ray scattering. I. The intensity and the shape of 10-angstrom maximum. Biofizika. 2005;50:1002–1012. [PubMed] [Google Scholar]
- 25.Krupyanskii YF, Mikhailyuk MG, Esin SV, Eshchenko GV, Moroz AP, Okisheva EA, Seifullina NK, Knox PP, Rubin AB. A study of protein structure changes during hydration using diffuse X -ray scattering. II. Fourier transform analysis of X-ray scattering data. Biofizika. 2006;51:13–23. [PubMed] [Google Scholar]
- 26.Bell JA. The X-ray crystal structure of a highly desiccated protein, Biomacromolecules: From 3-D to Applications, Hanford Symposium on Health and the Environment, 34th; Oct. 23–26, 1995; Pasco, Wash.. 1997. pp. 231–238. [Google Scholar]
- 27.Korb JP, Bryant RG. The physical basis for the magnetic field dependence of proton spin-lattice relaxation rates in proteins. Journal of Chemical Physics. 2001;115:10964–10974. [Google Scholar]
- 28.Bertini I, Gupta Y, Luchinat C, Parigi G, Schlorb C, Schwalbe H. NMR Spectroscopic Detection of Protein Protons and Longitudinal Relaxation Rates between 0.01 and 50 MHz. Edition 44. Angewandte Chemie International; 2005. pp. 2223–2225. [DOI] [PubMed] [Google Scholar]
- 29.Bosin WA, Marmor RA. The determination of the solid content of fats and oils by nuclear magnetic resonance. Journal of the American Oil Chemists' Society. 1968;45:335–337. doi: 10.1007/BF02667105. [DOI] [PubMed] [Google Scholar]
- 30.Swindells CE, Ferguson PA. Measurement of the solid content of fats by nuclear magnetic resonance. Canadian Institute of Food Science and Technology Journal. 1972;5:82–86. [Google Scholar]
- 31.Wettstrom R. Wide-line NMR for product and process control in fat industries. Journal of the American Oil Chemists' Society. 1971;48:15–17. [Google Scholar]