

Abstract
Aggregation of amyloid-β (Aβ) peptide into soluble and insoluble forms within the brain extracellular space is central to the pathogenesis of Alzheimer’s disease. Full length amyloid precursor protein (APP) is endocytosed from the cell surface into endosomes where it is cleaved to produce Aβ. Aβ is subsequently released into the brain interstitial fluid (ISF). We hypothesized that synaptic transmission results in more APP endocytosis, thereby increasing Aβ generation and release into the ISF. We found that inhibition of clathrin-mediated endocytosis immediately lowers ISF Aβ levels in vivo. Two distinct methods which increased synaptic transmission resulted in an elevation of ISF Aβ levels. Inhibition of endocytosis, however, prevented the activity-dependent increase in Aβ. We estimate that ~70% of ISF Aβ arises from endocytosis-associated mechanisms with the vast majority of this pool also dependent on synaptic activity. These findings have implications for AD pathogenesis and may provide insights into therapeutic intervention.
Introduction
Extracellular aggregation and accumulation of the amyloid-β (Aβ) peptide in different forms including plaques within the hippocampus and neocortex is one hallmark pathology of Alzheimer’s disease (AD) (Selkoe, 2001). Within the brain, Aβ is primarily produced within neurons by cleavage of the amyloid precursor protein (APP) followed by secretion into the brain interstitial fluid (ISF). Within the ISF, Aβ is found in its normal, soluble form throughout life. During the pathogenesis of AD, Aβ aggregates into forms that include insoluble species and probably soluble Aβ oligomers, both of which have been demonstrated under particular circumstances to be toxic to cells or to disrupt synaptic plasticity. While the initial polymerization seed for Aβ aggregates may form intracellularly or extracellularly (Gouras et al., 2000; Meyer-Luehmann et al., 2003), it is likely that much of the peptide necessary for Aβ aggregation as well as plaque formation and growth is derived from the soluble ISF pool of Aβ. Conversion of soluble Aβ into either plaques or oligomers is concentration-dependent, meaning that elevated levels of ISF Aβ are likely to hasten the formation of these toxic species. Consequently, knowing the factors that regulate ISF Aβ levels has implications for understanding disease pathogenesis as well as for developing therapeutics.
Although molecular events surrounding secretase cleavage of APP are well understood, the cellular processes regulating Aβ production and the subsequent release of Aβ by neurons are less characterized. Neuronal activity modulates Aβ levels. Activation of muscarinic M1 acetylcholine receptors increases β-secretase cleavage of APP and consequently reduces Aβ levels in animal models as well as in humans (Caccamo et al., 2006; Nitsch et al., 2000). Activation of NMDA receptors decreases α-secretase cleavage, thereby increasing Aβ levels (Lesne et al., 2005). Lesions of the cortical axon fibers that project to the dentate gyrus substantially reduce Aβ accumulation in APP transgenic mice (Lazarov et al., 2002; Sheng et al., 2002). Additionally, modulating synaptic transmission alters extracellular soluble Aβ levels in organotypic brain slice (Kamenetz et al., 2003). Our group demonstrated using APP transgenic and wild-type mice that synaptic activity dynamically modulates ISF Aβ level in vivo (Cirrito et al., 2005). Hippocampal seizures induced by electrical stimulation increased ISF Aβ levels by 30% within thirty minutes. Conversely, decreasing synaptic transmission using tetrodotoxin (TTX) or tetanus toxin rapidly reduced ISF Aβ levels by 30% and 80% respectively.
In acute brain slices, synaptic vesicle cycling alone, in the absence of neuronal depolarization, was sufficient to drive release of Aβ from neurons (Cirrito et al., 2005). Because Aβ is not located within synaptic vesicles (Ikin et al., 1996; Marquez-Sterling et al., 1997), we hypothesized that an event closely associated with vesicle exocytosis is actually the underlying mechanism responsible for Aβ production and release from neurons. Cell culture experiments have shown that full length APP is retrieved from the cell surface via clathrin-mediated endocytosis (Nordstedt et al., 1993), then APP is cleaved by β-secretase (BACE) and β-secretase within late and early endosomes to produce Aβ (Lah and Levey, 2000; Vassar et al., 1999). Aβ produced in the endocytic pathway is then brought to the cell surface where it is released into the extracellular fluid (Koo et al., 1996). Inhibition of endocytosis reduces APP internalization and reduces Aβ production and release in cell lines (Carey et al., 2005; Koo and Squazzo, 1994). During synaptic transmission, synaptic vesicles fuse with the plasma membrane followed by the synaptic vesicle membrane being retrieved from the cell surface, at least in part via clathrin-mediated endocytosis (Maycox et al., 1992). Within synaptosomes, clathrin-coated vesicles contain APP as well as several synaptic vesicle proteins including synaptophysin, synaptotagmin I, and SV2 (Marquez-Sterling et al., 1997). This suggests that synaptic proteins and APP can be internalized from the plasma membrane together via a common pathway. Because synaptic activity leads to elevated endocytosis of synaptic vesicle-related proteins, we hypothesized a synaptic activity-induced increase in endocytosis also drives more APP into the endocytic compartment, ultimately resulting in increased Aβ production and release. Herein, we provide direct evidence for this hypothesis in the brain in vivo.
Results
Aβ is primarily produced within neurons and secreted into the brain ISF. In order to assess ISF Aβ levels in living mice, we utilized an in vivo microdialysis technique that enables serial sampling of molecules within the brain ISF (Cirrito et al., 2003). Microdialysis probes were implanted unilaterally into the hippocampus of three month old Tg2576 APP transgenic mice (Hsiao et al., 1996). Following probe implantation under volatile anesthesia, mice remained awake with freedom of movement throughout the experiment. ISF Aβ levels were assessed every 30–60 minutes for up to 24 hours, enabling us to determine basal ISF Aβ levels and then intervene pharmacologically to assess the effect on ISF Aβ levels over time within the same mouse. In addition to measuring Aβ levels, microdialysis also permits the direct administration of small molecular weight compounds into the hippocampus via reverse microdialysis. Using this procedure, we administered compounds to the exact region of tissue from which Aβ was sampled. In a subset of mice, dual recording electrodes were attached to the microdialysis probe assembly to record electrical field potentials near sites of Aβ measurement (Cirrito et al., 2005).
Inhibition of endocytosis reduces ISF Aβ levels in vivo
We hypothesized that Aβ released into the brain ISF following synaptic transmission requires an intermediate event involving APP endocytosis. We first assessed the influence of endocytosis on ISF Aβ levels under normal conditions. A dynamin dominant negative inhibitory peptide (dynamin-DN) was infused directly into the hippocampus by reverse microdialysis. Dynamin-DN blocks dynamin binding to amphiphysin, thus inhibiting scission of clathrin-coated vesicles from the plasma membrane. This myristylated, cell-permeable peptide has been shown in cell culture to inhibit clathrin-mediated endocytosis (Marks and McMahon, 1998). In vivo, dynamin-DN reduced biotinylated transferrin uptake into cells within the dentate gyrus confirming the in vivo effectiveness of this peptide at reducing endocytosis (Supplementary Figure 1).
Basal ISF Aβ levels were established in three month old Tg2576 APP transgenic mice over eight hours, followed by four hours of continuous administration of dynamin-DN (Fig 1A). At this age, Tg2576 mice do not contain Aβ deposits and Aβ metabolism can be studied without the complicating factor of Aβ aggregates (Hsiao et al., 1996). Within thirty minutes of dynamin-DN treatment, ISF Aβ levels were significantly decreased with a maximal decrease of 70% by hour four of treatment (Fig. 1A). This is similar to studies in cell culture demonstrating that inhibition of endocytosis reduces Aβ levels in media (Koo and Squazzo, 1994). Following treatment, dynamin-DN was removed from the microdialysis perfusion buffer and ISF Aβ levels were measured for an additional 8 hours. Within two hours of washout, Aβ levels increased transiently by 250% over baseline levels and returned to basal levels within eight hours. ISF Aβ levels did not change significantly in mice treated with vehicle or in mice treated with 200μM scrambled peptide (Fig. 1A). Similarly, ISF levels of the neuropeptide orexin (also called hypocretin) were also unchanged in response to dynamin-DN infusion (Supplementary Fig, 2).
Figure 1. Inhibition of endocytosis reduces ISF Aβ levels.

(A) In vivo measurement of ISF Aβ levels during inhibition of endocytosis in 3 month old Tg2576 mice. Infusion of a dynamin-DN inhibitory peptide (200μM) into the hippocampus via reverse microdialysis reduced ISF Aβ1-x levels to 36.1 ± 4.2% of baseline levels over fours hours (p<0.0001, n=9 per group). When dynamin-DN was removed from the microdialysis perfusion buffer, ISF Aβ levels increased transiently by 246.7 ± 20.7% compared to baseline (p<0.0001) and returned to basal levels by eight hours of washout. ISF Aβ levels did not change significantly over the course of the study in either untreated mice or in mice treated with 200μM scrambled peptide. (B) A dose escalation of dynamin-DN demonstrated a maximum reduction of ISF Aβ levels with a dose of 200μM of inhibitory peptide (p<0.0001, n=3). (C) shows final Aβ levels achieved during each epoch depicted in B. (D–F) EEG recordings during basal and dynamin-DN administration. (D) Representative EEG traces during basal and dynamin-DN treatment. EEG amplitude standard deviation (E) and frequency (F) were not significantly different when endocytosis was inhibited (n=3). (G–I) Evoked fEPSPs from dentate gyrus during basal and dynamin-DN treatment. (G) Representative fEPSP traces elicited by medial perforant pathway stimulation. Arrow denotes initial fEPSP slope. (H) The initial slope of the fEPSP did not change during 4 hours of dynamin-DN treatment; however when the perfusion buffer was changed to contain 10μM TTX the fEPSP was significantly reduced by 89.8 ± 3.8% within 15 minutes of treatment (p<0.0001, n = 3). (I) Likewise, fEPSP slope was similar under basal and dynamin-DN treatment during 1 Hz trains of stimuli for 60 seconds (n=3). Data presented as mean ± SEM. ** represent p<0.001, *** represent p<0.0001.
To determine the dose of dynamin-DN that maximally reduces ISF Aβ levels in vivo, Tg2576 mice were administered a dose escalation of inhibitory peptide (Fig. 1B). Dynamin-DN had an IC50 of 100.4 ± 5.9 μM in vivo with a dose of 200μM maximally reducing ISF Aβ levels (Fig. 1C). These data demonstrate that inhibition of clathrin-mediated endocytosis can dramatically reduce ISF Aβ levels in vivo.
To assess the effect of dynamin-DN on cell surface APP levels, we treated a Neuro2A (N2A) neuroblastoma cell line that expresses APP695 (Thinakaran et al., 1996) with dynamin-DN or a myristylated, scrambled control peptide for four hours. Inhibition of endocytosis increased cell surface APP fluorescence by 50% compared to the control peptide treatment (Supplementary Fig. 3A). Dynamin-DN treatment did not alter total APP expression levels in this cell line; however it significantly reduced APP β-CTF levels (Supplementary Fig. 3B). Dynamin-DN also significantly decreased Aβ levels within the cells by 30% (Supplemental Fig. 3C). Consistent with the effects of dynamin-DN, another less specific inhibitor of clathrin-mediated endocytosis, chlorpromazine (Wang et al., 1993), reduced ISF Aβ levels by 20% (Supplementary Figure 4). Together, the data demonstrate that dynamin-DN reduces APP endocytosis, cleavage by β-secretase, and thus Aβ generation in N2A cells.
Inhibition of endocytosis does not depress synaptic transmission
Clathrin-mediated endocytosis is required for a variety of cellular functions, including in part, the recycling of synaptic vesicle membrane in order to replenish the readily releasable pool of synaptic vesicles (Heuser and Reese, 1973). Inhibition of dynamin can deplete synaptic vesicles (Newton et al., 2006). Interestingly, genetic deletion of dynamin I in mice does not affect spontaneous synaptic transmission; however it can result in depressed evoked postsynaptic potentials, especially following trains of strong exogenous stimuli (Ferguson et al., 2007). Because depressed synaptic activity reduces Aβ release (Cirrito et al., 2005; Kamenetz et al., 2003), it is conceivable that dynamin-DN may reduce ISF Aβ levels by inhibiting synaptic activity and not necessarily by having a direct effect on APP endocytosis or Aβ generation. In order to test this possibility during inhibition of endocytosis we assessed neuronal activity using spontaneous field potential recordings as well as evoked potential recordings.
Dual recording electrodes assessed extracellular field potentials, or depth EEG, within the hippocampus at the same location that Aβ was measured and that dynamin-DN was administered (Fig 1D). Inhibition of endocytosis did not alter the amplitude (Fig. 1E) or frequency (Fig. 1F) of EEG activity within the hippocampus at any point during administration. Mice remained awake with freedom of movement throughout these recordings. Therefore, these results suggest that global neurotransmission was not dramatically altered by dynamin-DN at the doses utilized.
EEG records spontaneous, physiological neuronal activity within a large volume of brain tissue and may not be sensitive to subtle, local changes in activity. Consequently, we assessed evoked field EPSPs (fEPSPs), which provides a more direct assessment of synaptic transmission within the hippocampus in an awake, behaving animal. Bipolar stimulating electrodes were implanted into the medial perforant pathway. In the same animal, a microdialysis probe with dual recording electrodes was implanted into the dentate gyrus, enabling evoked fEPSP measurement and local dynamin-DN administration (Fig. 1G). Dynamin-DN did not change the initial slope of the fEPSP measured at either 15 minutes, 2 hours, or 4 hours of administration compared to basal recordings from the same mouse (Fig. 1H). Given that dynamin-DN produced a negative result, we sought to confirm that a change in fEPSP could be detected if an appropriate compound was infused into the brain. Following dynamin-DN administration, the perfusion buffer was changed to contain tetrodotoxin (TTX), a sodium channel blocker that should potently inhibit evoked fEPSPs. Within 15 minutes of TTX treatment, the fEPSP slope was reduced by 95% and was undetectable by 1 hour of treatment. Single fEPSPs were not inhibited by dynamin-DN, so we also determined if trains of stimuli could elicit a difference in fEPSP slope. Stimuli at 1 Hz for 60 seconds also did not result in a difference in the evoked EPSP slope between basal and dynamin-DN conditions (Fig. 1I). The reduction in fEPSP slope in both groups over the course of the train is typical for this type of recording using medial perforant pathway stimulation (McNaughton, 1980). Given that neither spontaneous EEG activity nor evoked fEPSPs were altered in on the presence of dynamin-DN, it does not appear that inhibition of endocytosis substantially affects synaptic transmission in vivo at normal, spontaneous levels or at low-frequencies of stimulation. These data suggest that the small, remaining portion of clathrin-mediated endocytosis is capable of sustaining normal synaptic transmission or that some dynamin-1 independent mechanisms of vesicle retrieval are active (Ferguson et al., 2007). It appears from our data that administration of dynamin-DN does not adversely affect in vivo synaptic transmission over the timescale that we assessed.
Activity-dependent elevation of ISF Aβ levels requires endocytosis
Synaptic transmission dynamically modulates ISF Aβ levels in vivo. Depression of synaptic activity reduces ISF Aβ levels whereas seizures rapidly increase Aβ levels in Tg2576 mice (Cirrito et al., 2005). We sought to determine if subtle increases in synaptic transmission could also elevate ISF Aβ levels and if endocytosis was required for the activity-dependent Aβ production and release. Tg2576 mice were treated with 25μM picrotoxin, a non-competitive GABAA receptor antagonist, via reverse microdialysis (Fig. 2A). Picrotoxin caused a significant 50% increase in ISF Aβ levels by 4 hours of administration (Fig. 2B). In contrast, dynamin-DN reduced ISF Aβ levels and prevented the increase in Aβ levels when co-administered with picrotoxin. While high doses of picrotoxin can generate seizures, the low dose used here only produced occasional spikes in synchronous EEG activity (Fig. 2C). Similar spikes in EEG activity also occurred during dynamin-DN and picrotoxin co-administration. As a consequence of picrotoxin treatment, the amplitude of EEG activity increased similarly in both dynamin-DN and vehicle-treated mice (Fig. 2D).
Figure 2. Activity-dependent release of Aβ requires endocytosis.

(A) ISF A levels in Tg2576 mice treated with vehicle or 200μM dynamin-DN followed by 25μM picrotoxin via reverse microdialysis to increase neuronal activity (n = 4 per group). (B) Picrotoxin alone increased ISF A levels by 145.5 ± 6.8% averaged over 4 hours of administration (p<0.01). Pretreatment with 200μM dynamin-DN prevented the increase in ISF A levels caused by picrotoxin. (C) Representative EEG traces during basal conditions and during picrotoxin treatment with and without dynamin-DN administration. This low dose of picrotoxin did not generate seizures in any of the mice tested, but consistently caused synchronous spikes in EEG activity (arrows) in the presence and absence of dynamin-DN. (D) EEG amplitude increased similarly in picrotoxin-treated mice administered vehicle or dynamin-DN. Data presented as mean ± SEM. ** represents p<0.001, *** represents p<0.0001.
Metabotropic glutamate receptors 2/3 (mGluR2/3) are expressed presynaptically to modulate glutamate release. When these receptors are inhibited, glutamate release from the presynaptic terminal is enhanced (Losonczy et al., 2003). LY341495, a mGluR2/3 antagonist, was administered directly into the hippocampus to increase synaptic activity during microdialysis to determine the effect on ISF Aβ levels (Fig 3A). LY341495 caused a gradual increase in ISF Aβ levels to 20% of baseline over 8 hours (Fig 3B). In the presence of LY341495 and dynamin-DN, Aβ levels were not increased compared to dynamin-DN alone. The effects of both picrotoxin and LY341495 on ISF Aβ levels were prevented by inhibiting endocytosis. This suggests that endocytosis was a key event linking elevated synaptic activity and release of Aβ into the brain ISF in Tg2576 mice.
Figure 3. Increased glutamate release elevates ISF Aβ levels which requires endocytosis.

(A) ISF A levels in Tg2576 mice treated with dynamin-DN or vehicle followed by LY341495, an mGluR2/3 antagonist that increases pre-synaptic glutamate release. (B) LY341495 increased ISF A levels by 122.2 ± 8.4% during hours 6-8 of treatment (p<0.05, n = 4). The increase in A levels was prevented by pretreatment with dynamin-DN (n = 4). Data presented as mean ± SEM. * represents p<0.05.
One concern regarding any transgenically expressed protein is that it could be regulated and processed differently than the endogenous protein. Consequently, we assessed the link between synaptic activity, endocytosis, and murine ISF Aβ40 and Aβ42 levels in 3 month old C57Bl6 wild-type mice. Because murine Aβ levels are substantially lower and more difficult to detect than human Aβ in Tg2576 mice, we sampled ISF Aβ levels over 12 hour periods instead of every 30 minutes (Fig. 4). Similar to Tg2576 mice, 25μM picrotoxin increased murine ISF Aβ40 and Aβ42 levels by 130% over baseline. Dynamin-DN reduced ISF Aβ40 and Aβ42 levels by 60% and 75% respectively. Interestingly, the greater decrease in Aβ42 than Aβ40 was statistically significant. A subset of mice was pre-treated with dynamin-DN for 1 hour, then co-administered picrotoxin for 12 hours. Aβ levels in these mice were not statistically different than those treated with dynamin-DN alone. That endocytosis is a key event linking synaptic activity and Aβ generation in wild-type mice underscores that this phenomenon is part of a normal pathway for Aβ production.
Figure 4. Endocytosis is required for activity-dependent release of endogenous murine.

Aβ.
In C57Bl6 mice, 25μM picrotoxin increased ISF murine A 40 and Aβ42 levels to 133.4 ± 2.3 and 131.5 ± 6.1% respectively (p<0.0001) of baseline over 12 hours. 200μM dynamin-DN reduced murine A 40 levels to 38.3 ± 2.8% (p<0.0001) and Aβ42 levels to 23.8 ± 0.8% of baseline (p<0.0001). Aβ42 levels were significantly more reduced than Aβ40 levels (p<0.0001; noted by ###). Pretreatment with dynamin-DN for 1 hour then co-administration of dynamin-DN and picrotoxin caused ISF A 40 and A 42 levels to decrease to 43.9 ± 3.7% and 18.9 ± 1.5% of baseline which was not statistically different from dynamin-DN treatment alone. n = 6 per group. Data presented as mean ± SEM. *** represents p<0.0001.
Inhibition of endocytosis does not affect ISF Aβ elimination
It is possible that inhibition of endocytosis could affect both ISF Aβ production and ISF Aβ elimination. In the case of Aβ production, endocytosis is required for internalization of full length APP prior to Aβ generation (Koo and Squazzo, 1994). In the case of elimination, molecules such as LRP1 have been shown to internalize Aβ directly or bound to apoE (Deane et al., 2004). In order to determine if inhibition of endocytosis modulates ISF Aβ elimination, we measured the half-life of ISF Aβ in Tg2576 mice treated with vehicle or dynamin-DN. Basal ISF Aβ levels were determined followed by administration of dynamin-DN or vehicle for four hours, after which mice were injected with a potent β-secretase inhibitor (LY411575) subcutaneously to rapidly inhibit β-secretase activity and Aβ production (Fig. 5A) (Cirrito et al., 2003). ISF Aβ elimination followed first-order kinetics in both vehicle and dynamin-DN treated mice (Fig. 5B). The half-life of ISF Aβ was approximately 1 hour in both groups (Fig. 5C) which is similar to previously published values for this mouse model (Cirrito et al., 2005; Yin et al., 2006). This demonstrates that dynamin-DN does not dramatically affect ISF Aβ elimination. Though this assay may be insensitive to subtle changes in Aβ elimination, it suggests that inhibition of endocytosis alters ISF Aβ levels via changes in Aβ production and release following APP internalization and not via changes in Aβ clearance.
Figure 5. Dynamin-DN does not affect ISF Aβ elimination.
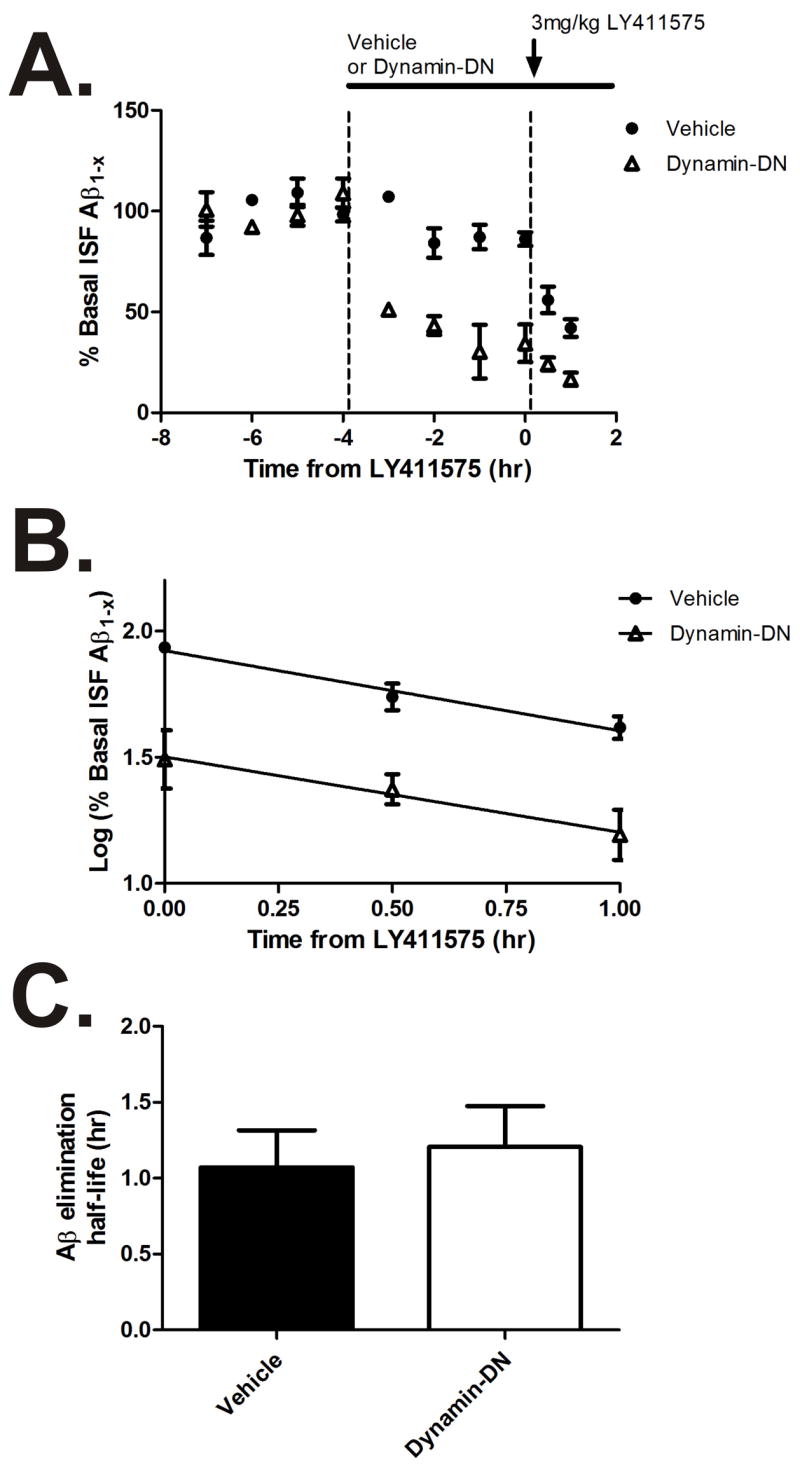
(A) Hippocampal ISF A levels were measured in Tg2576 mice that were treated with vehicle or 200μM dynamin-DN, followed by 3mg/kg LY411575 s.c. to inhibit -secretase activity (n = 4-6 per group). (B) Plots of log % basal ISF A levels were linear in both vehicle and dynamin-DN treated mice, demonstrating first-order kinetics of ISF A elimination in both groups. (C) The elimination half-life of ISF A (calculated from the slope in panel B) was the same in vehicle and dynamin-DN treated mice (1.1 ± 0.24 hr and 1.2 ± 0.27 hr, respectively). Data presented as mean ± SEM.
Distinct pathways for ISF Aβ generation
Both inhibition of synaptic activity (Cirrito et al., 2005) and inhibition of endocytosis reduce ISF Aβ levels. In order to determine the extent that activity and endocytosis each contribute to ISF Aβ levels, we co-administered TTX and dynamin-DN. If dynamin-DN was administered first, ISF Aβ levels declined by a maximum of 70% (Fig. 6A,B). If microdialysis perfusion buffer was then switched to contain dynamin-DN and TTX, then there was no additional change in ISF Aβ levels. This suggests that all of the ISF Aβ that is produced due to synaptic activity requires endocytosis. Conversely, if animals were pretreated with TTX, followed by dynamin-DN co-administration, then ISF Aβ levels were further reduced when endocytosis was inhibited (Fig. 6C,D). This demonstrates that there is a pool of ISF Aβ that is generated via endocytosis that is not affected by synaptic activity, but remains sensitive to dynamin-DN.
Figure 6. Distinct pathways for Aβ generation.

(A) Tg2576 mice were either treated with 200μM dynamin-DN for 12 hours or pre-treated with 200μM dynamin-DN for 4 hours followed by dynamin-DN and 10μM TTX together for an additional 8 hours. Hippocampal ISF A levels were measured by microdialysis. (B) Dynamin-DN reduced ISF A levels to 33.0 ± 3.2% of baseline over 12 hours (p<0.0001) and co-administration with TTX did not additionally change A levels compared to dynamin-DN alone (n=4 per group). (C) Mice were either treated with 10μM TTX alone for 12 hours or with TTX for 8 hours followed by TTX and 200μM dynamin-DN together for an additional 4 hours. 10μM TTX alone for 12 hours reduced ISF Aβ levels to 41.8 ± 3.2% of baseline (p<0.0001, n=8). When TTX was co-administered with dynamin-DN, ISF Aβ levels decreased to 30.0 ± 1.4% of baseline (p<0.0001, n=5). The 7% difference between TTX and TTX with dynamin-DN was significant (p<0.05). Data presented as mean ± SEM. * represents p<0.05.
Discussion
Many studies have postulated that the aggregation of Aβ in both soluble and insoluble forms in the brain is likely a key initiating factor in AD pathogenesis. Thus, influences on the aggregation process are potentially major treatment targets. Since the process of Aβ aggregation is concentration-dependent, understanding factors that regulate Aβ levels in the location that it aggregates (e.g. in the brain extracellular space) is likely to provide critical insights into the biology of AD. While it has been demonstrated that APP endocytosis is a key step in Aβ generation and release in vitro, we provide direct evidence that inhibition of clathrin-mediated endocytosis reduces ISF Aβ levels in awake mice, both transgenic and wild-type, under normal conditions. Importantly, in addition to endocytosis, our work shows that synaptic activity, and specifically synaptic vesicle exocytosis, is linked with neuronal Aβ release. Utilizing two distinct pharmacological interventions, we found that increasing synaptic activity increases ISF Aβ levels in a manner dependent on endocytosis. Clathrin-mediated endocytosis of APP is necessary for activity-dependent regulation of ISF Aβ levels in APP transgenic mice as well as in wild-type mice. This emphasizes that these results elucidate a normal pathway for regulating ISF Aβ levels. Blockade of endocytosis primarily affected Aβ generation with no detectable change in ISF Aβ elimination. We estimate that the majority of ISF Aβ in vivo is generated through endocytosis of APP and that in the brain, synaptic activity plays a primary role in that process. Overall, these findings define a pathway by which synaptic activity influences Aβ release into brain ISF that is likely relevant to AD pathogenesis.
One major pathway for Aβ generation is endocytosis of full length APP from the plasma membrane into the endocytic compartment, where BACE and γ-secretase act to produce Aβ. Aβ is then subsequently secreted from the neuron into the extracellular space. Our hypothesis is that synaptic activity drives more APP into the endocytic compartment, thus increasing Aβ generation and release. Action potential invasion of the synaptic terminal causes calcium influx (Fig. 7A). Calcium causes synaptic vesicles to fuse with the plasma membrane and to release neurotransmitter into the synaptic cleft (Fig. 7B). This is the basis of synaptic transmission. As more synaptic vesicles fuse with the plasma membrane, there is an increase in the amount and overall rate of endocytosis that recycles the vesicular membrane from the cell surface. When the synaptic vesicle membrane is recycled from the plasma membrane via clathrin-mediated endocytosis, more APP is internalized as well (Fig. 7C). It is possible that calcium entry may also cause endocytosis through other mechanisms, such as synthesis of PI(4,5)P2, which recruits the clathrin assembly and mediates clathrin-mediated endocytosis (Wenk et al., 2001). Increased endocytosis increases APP internalization into the endocytic compartment where Aβ is generated (Fig. 7D). Once generated, Aβ can be secreted from the neuron into the brain extracellular space (Fig. 7E).
Figure 7. Model of synaptic-dependent release of Aβ.

(A) Depolarization of the synaptic terminal causes calcium influx leading to synaptic vesicle release. (B) Synaptic vesicle membrane recycling from the cell surface via clathrin-mediated endocytosis causes more APP to internalized (C). Within endosomes, BACE and β-secretase cleave APP to produce Aβ (D) which is secreted from the neuron into the brain ISF (E).
Our previous studies provide direct evidence that Aβ is generated due to presynaptic mechanisms; causing synaptic vesicle exocytosis with α-latrotoxin in the absence of postsynaptic activation is still capable of generating Aβ (Cirrito et al., 2005). We cannot rule out a postsynaptic mechanism as well however. α-Latrotoxin in the presence of postsynaptic activation has a greater effect on extracellular Aβ than α-latrotoxin in the absence of the postsynaptic activation. The difference between these two conditions (~20% of Aβ generation) is likely due to postsynaptic mechanisms that also influence Aβ generation (Cirrito et al., 2005).
Two mechanistically distinct agents increased synaptic activity and increased ISF Aβ levels. Picrotoxin inhibits ionotropic GABAA receptors, thus increasing synaptic transmission by reducing inhibition of excitatory neurons. In contrast, LY341495 blocks perisynaptic G-protein coupled mGluR2/3s, thus increasing excitatory neurotransmission by directly enhancing glutamate release at the synapse. Both of these pathways increased Aβ levels by modestly increasing synaptic activity. In both cases, inhibition of endocytosis completely blocked the elevation the ISF Aβ levels. This phenomenon was also observed with wild-type murine Aβ40 and Aβ42 levels. Interestingly, inhibition of endocytosis had a larger affect on Aβ42 levels than Aβ40, perhaps suggesting that a greater percent of extracellular Aβ42 comes from endosomes than does Aβ40.
Dynamin-DN reduced ISF Aβ levels by over 60%. However, washout of dynamin-DN caused a transient surge in ISF Aβ levels. This suggests that APP may have been retained on the cell surface during inhibition of endocytosis and comprised a large pool of substrate capable of generating Aβ when endocytosis resumed. In the in vivo paradigm, for Aβ production to be elevated when endocytosis resumed, much of the APP retained on the plasma membrane must have remained as the full length, uncleaved protein.
Several studies have demonstrated that Aβ, particularly oligomeric species, can modulate various aspects of synaptic transmission (Hsieh et al., 2006; Shankar et al., 2007; Ting et al., 2007; Townsend et al., 2006). Our studies demonstrate that inhibition of endocytosis has no effect on spontaneous EEG activity or evoked potentials in vivo. Interestingly, blocking endocytosis reduced ISF Aβ levels by 70% but did not appear to modulate transmission. Given that Aβ levels were not reduced to zero, it is possible that the remaining 30% of ISF Aβ was sufficient to retain Aβ’s effect, if any, on synaptic transmission. It is also likely that the majority of ISF Aβ present in 3 month old Tg2576 mice is monomeric and not oligomeric Aβ. Further studies are required to address what role Aβ has on synaptic activity in vivo.
The data suggest there are at least three cellular mechanisms that contribute to ISF Aβ production. First, a pathway that is synaptic activity-dependent (TTX sensitive) and endocytosis-dependent which contributes to approximately 60% of ISF Aβ levels in Tg2576 mice. Second, a pool of ISF Aβ that requires endocytosis, but is independent of synaptic activity. This pathway is responsible for another 10% of ISF Aβ levels. Interestingly, inhibition of endocytosis does not reduce ISF Aβ levels to zero. The remaining pool of Aβ, comprising 30% of total ISF Aβ levels, may be the product of several mechanisms, including Aβ produced within the secretory pathway (Busciglio et al., 1993) or Aβ diffusing from brain areas that are not affected by dynamin-DN or TTX. Alternatively, this last pool may be a factor of incomplete inhibition of endocytosis or some small contribution of altered Aβ elimination. These values provide rough estimates for each of these pools and provide an interesting framework to consider the various pathways that contribute to the overall pool of ISF Aβ. A source of 70% of ISF Aβ in vivo is a tantalizing target for therapeutic development. While inhibiting all clathrin-mediated endocytosis is unlikely to be a feasible strategy for lowering extracellular Aβ levels, it may be possible to modulate individual components of the endocytic machinery or synaptic transmission to selectively affect Aβ generation.
Several molecules influence the rate and amount of APP endocytosis. LRP1 expression increases the rate of APP endocytosis from the plasma membrane whereas LRP1b expression reduces the rate of internalization (Cam et al., 2004; Cam et al., 2005). Recent evidence also implicates another LDL-R family member, LR11 or SorLa, in modulating APP trafficking through the endosomal compartment (Andersen et al., 2005; Dodson et al., 2006; Offe et al., 2006; Rogaeva et al., 2007). LR11 overexpression increases APP co-localization within the Golgi and reduces Aβ generation in culture whereas deletion of the LR11 gene in mice increases brain Aβ levels (Andersen et al., 2005). Given that several LDL-R family members modulate Aβ endocytosis, it will be important to elucidate if and how these molecules affect synaptic activity-dependent Aβ generation.
While our studies do not directly assess where Aβ is produced or released, it is likely that Aβ generation that is caused by events within the synaptic terminal would occur at or near the synapse. Kamenetz and colleagues demonstrated that neurons virally expressing APP can depress synaptic transmission in nearby neurons (Kamenetz et al., 2003). This also suggests that Aβ released from a neuron might even be able to feed back to inhibit activity within the same neuron. We have identified a mechanism of Aβ production and release near the synapse that is intimately associated with synaptic activity. This phenomenon produces a situation whereby synaptically-released Aβ is positioned to modulate synaptic activity. The conformation of Aβ that is released following synaptic activity will likely be a critical factor for modulation of neurotransmission. For instance, Aβ oligomers appear to be much more potent at depressing synaptic transmission than Aβ monomers (Townsend et al., 2006). The Aβ conformations that are released at synapses and whether those Aβ species change with age or disease stage is unknown.
Synaptic activity appears to affect Aβ levels in humans as well. First, a fraction of patients with temporal lobe epilepsy develop diffuse Aβ deposits in seizure-prone brain regions as early as 30 years of age (Gouras et al., 1997; Mackenzie and Miller, 1994). Second, there are several links between stress, neuronal activity, and AD. Individuals that are prone to psychological distress are more likely to develop AD (Wilson et al., 2005; Wilson et al., 2003). Also, plasma levels of the stress hormone, cortisol, are correlated with the rate of dementia progression in patients with AD (Csernansky et al., 2006). In APP transgenic mice subjected to restraint stress, ISF Aβ levels are significantly higher than control animals, however the acute increase in Aβ levels is blocked in the absence of neuronal activity (Kang et al., 2007). Final support for the idea of a casual link between synaptic activity and Aβ levels in humans comes from recent brain imaging studies. Brain regions that contain the most metabolic activity throughout life, and presumably have the highest levels of neuronal activity, are also the regions most vulnerable to Aβ accumulation and aggregation in AD patients (Buckner et al., 2005). In each of these cases, synaptic activity appears to play a role in regulating Aβ levels under physiologic conditions. We now identify a cellular pathway, endocytosis, that likely links synaptic activity and Aβ production.
Experimental procedures
Animals
All experimental procedures involving animals were performed in accordance with guidelines established by the Animal Studies Committee at Washington University. We bred Tg2576+/− hemizygous mice (a generous gift from Dr. K. Ashe, University of Minnesota) to wild-type C57Bl6/SJL mice (Taconic Farms, Germantown, NY), then aged the Tg2576+/− offspring to 3–5 months for use in experiments. Animals were screened for the Tg2576 transgene by PCR from tail DNA. Three-month old C57Bl6 mice were ordered from Jackson Laboratory (Bar Harbor, Maine).
Compounds
All pharmacological agents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted. Dynamin dominant negative (Dynamin-DN) inhibitory peptide (myristylated) and control scrambled peptide were purchased from Tocris Biosciences (Ellisville, MO). The mGluR2/3 antagonist, LY341495, and γ-secretase inhibitor, LY411575, were kind gifts from Darryle Schoepp and Patrick May (Eli Lilly and Company, Indianapolis, IN). All compounds were diluted in microdialysis perfusion buffer and administered via reverse microdialysis with the exception of LY411575 which was diluted in Mazola corn oil and administered subcutaneously.
Aβ ELISA
ISF Aβ levels were assessed using sandwich ELISAs as described (Cirrito et al., 2003). Briefly, to assess human Aβ1-x, a central domain, mouse monoclonal antibody (m266) was used to capture and a biotinylated N-terminal, human Aβ-specific antibody (m3D6) was used to detect, followed by streptavidin-poly-HRP-20 (Fitzgerald Industries, Concord, MA). Human Aβ1-40 and human Aβ1-42 was assessed similar to Aβ1-x, however mouse monoclonal antibodies m2G3 and m21F12, respectively, were used as coating antibodies. Murine Aβx-40 was assessed using an Aβ40-specific mouse monoclonal antibody, mHJ2, as a coating antibody and a biotinylated central domain antibody, mHJ5.1, as the detecting antibody, followed by streptavidin-poly-HRP-40 (Fitzgerald Industries). All ELISA assays were developed using Super Slow ELISA TMB (Sigma, St. Louis, MO) and absorbance read on a Bio-Tek FL-600 plate reader (Winooski, Vermont) at 650nm.
In vivo Aβ Microdialysis
In vivo microdialysis to assess brain ISF Aβ1-x in the hippocampus of awake, freely moving Tg2576 mice was performed similar to previously described (Cirrito et al., 2003). This technique samples soluble molecules within the extracellular fluid that are smaller than 30-kilodaltons, the molecular-weight cut off of the probe membrane. Aβ capable of entering the probe has been dubbed “exchangeable Aβ or eAβ” (Cirrito et al., 2003).
Under isoflurane volatile anesthetic, guide cannula (BR-style, Bioanalytical Systems, Indianapolis, IN) were cemented into the left hippocampus (bregma –3.1mm, 2.5 mm lateral to midline, and 1.2 mm below dura at a 12° angle). Two millimeter microdialysis probes were inserted through the guides so the membrane was contained entirely within the hippocampus (BR-2, 30-kilodalton MWCO membrane, Bioanalytical Systems). Microdialysis perfusion buffer was artificial CSF containing 0.15% bovine serum albumin that was filtered through a 0.1μM membrane. Flow rate was a constant 1.0μl/min. Samples were collected every 30–60 minutes with a refrigerated fraction collector into polypropylene tubes and assessed for Aβ by ELISA at the completion of each experiment. Basal levels of ISF Aβ were defined as the mean concentration of Aβ over 6-8 hours preceding drug treatment. For each animal, all Aβ levels were normalized to the basal Aβ concentration. Once basal ISF Aβ levels were established, Tg2576 mice were treated with a variety of compounds by reverse microdialysis. Compounds or vehicle were diluted in 0.15% BSA-aCSF perfusion buffer.
Murine ISF Aβ40 levels were assessed using a similar protocol, except longer sample times of 12 hours at a 0.5μl/min flow rate were necessary to concentrate ISF samples. For each mouse, the basal concentration of Aβ was established over 12 hours of collection followed by 12 hour treatment with either dynamin-DN, picrotoxin, or co-treatment with dynamin-DN and picrotoxin. In the latter group, dynamin-DN was pre-treated for one hour prior to co-treatment with picrotoxin.
EEG Recording
To record EEG activity, bipolar recording electrodes (Teflon coated, stainless steel wire, 0.0055” coated OD, A–M Systems, Carlsborg, WA) were attached to the shaft of the microdialysis guide cannula with Elmer’s Super-Fast Epoxy Resin (Elmer’s, Columbus, OH). Electrodes extended ~1 mm from the tip of the guide, so that the electrode tip was located at the center of the 2 mm microdialysis membrane. EEG activity was assessed using a P511K A.C. Pre-amplifier (Grass Instruments, Quincy, MA), digitized with a DigiData 1322A Data Acquisition System (Axon Instruments, Union City, CA), and recorded digitally using pClamp 9.2 (Axon Instruments). EEG amplitude standard deviation and frequency power spectrum was assessed for 15 minute epochs for four hours prior treatment and during four of dynamin-DN treatment.
Evoked field potential recordings
Microdialysis was performed as described previously, however implant coordinates were altered to coincide with optimized perforant pathway stimulation. The stimulating electrode (model SNE-200X-35, Rhodes Medical, Summerland, CA) was implanted at bregma –4.3 mm, 2.9mm lateral to midline, at ~1.2–1.6 mm below dura. Recording electrodes were designed similar to EEG recording electrodes except the electrode tips extended approximately 0.5mm beyond the tip of the microdialysis probe. Guide cannula were inserted at coordinates –2.5 mm, 1.3 mm lateral to midline, and 1.5–1.9 mm below dura. The precise location of the recording and stimulating electrodes was optimized based on the fEPSP waveform produced during placement. Two bone screws were used; one had a grounding wire attached. The cannula/electrode assembly was then cemented to the skull using a binary dental cement (Bioanalytical Systems). The DigiData 1322A and stimulus isolator (World Precision Instruments, Sarasota, FL) was used to deliver evoked potentials.
Field EPSPs were evoked by a constant current stimulus (0.1ms duration) that produced a 50% max slope as determined by an input-output curve. The slope of the initial fEPSP was assessed using pClamp version 9.2 software. Single fEPSPs were assessed at a frequency of 0.017 Hz at various times before and during drug treatments. In each animal, dynamin-DN was treated by reverse microdialysis for four hours; then the perfusion buffer was changed to contain TTX for one hour. Trains of fEPSPs were assessed at 1Hz just prior to treatment and after four hours of dynamin-DN administration.
Aβ Elimination Half-life
ISF Aβ half-life was determined as described (Cirrito et al., 2003). Microdialysis probes were inserted as described previously. Basal levels of ISF Aβ were established for four hours, followed by administration of dynamin-DN or vehicle via reverse microdialysis for the remainder of the experiment. Four hours after treatment began, LY411575, a potent, blood-brain barrier permeable γ-secretase inhibitor (Eli Lilly and Co, Indianapolis, IN), was administered subcutaneously at 3mg/kg body weight in corn oil. Samples were collected every 30 minutes for an additional one hour, then assessed for Aβ1-x by ELISA.
Statistical analysis
Data in figures represent mean ± SEM. All statistical analysis was performed using Prism version 5.01 for Windows (GraphPad, San Diego, CA). Statistical analysis was performed using a one-way ANOVA with Bonferroni post-test. Values were accepted as significant if p ≤ 0.05.
Supplementary Material
Acknowledgments
We would like to thank Patrick C. May (Eli Lilly and Co.) for the generous gifts of LY411575 and LY341495. This work was supported by National Institutes of Health grants AG13956 (D.M.H.), DA07261 (J.R.C.), AG029524 (J.R.C.), NS54174 (S.M.), MH78823 (S.M.), and AG027924 (G.B.) as well as by the American Health Assistance Foundation (S.M.), Alzheimer’s Association Zenith Award (D.M.H.), Cure Alzheimer’s disease (D.M.H.), a NIH Neuroscience Blueprint Grant P30 NS057105 to Washington University, and Eli Lilly and Company.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, et al. Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A. 2005;102:13461–13466. doi: 10.1073/pnas.0503689102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci U S A. 1993;90:2092–2096. doi: 10.1073/pnas.90.5.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Cam JA, Zerbinatti CV, Knisely JM, Hecimovic S, Li Y, Bu G. The low density lipoprotein receptor-related protein 1B retains beta-amyloid precursor protein at the cell surface and reduces amyloid-beta peptide production. J Biol Chem. 2004;279:29639–29646. doi: 10.1074/jbc.M313893200. [DOI] [PubMed] [Google Scholar]
- Cam JA, Zerbinatti CV, Li Y, Bu G. Rapid endocytosis of the low density lipoprotein receptor-related protein modulates cell surface distribution and processing of the beta-amyloid precursor protein. J Biol Chem. 2005;280:15464–15470. doi: 10.1074/jbc.M500613200. [DOI] [PubMed] [Google Scholar]
- Carey RM, Balcz BA, Lopez-Coviella I, Slack BE. Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid beta protein. BMC cell biology. 2005;6:30. doi: 10.1186/1471-2121-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic Activity Regulates Interstitial Fluid Amyloid-beta Levels In Vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Csernansky JG, Dong H, Fagan AM, Wang L, Xiong C, Holtzman DM, Morris JC. Plasma cortisol and progression of dementia in subjects with Alzheimer-type dementia. The American journal of psychiatry. 2006;163:2164–2169. doi: 10.1176/appi.ajp.163.12.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Dodson SE, Gearing M, Lippa CF, Montine TJ, Levey AI, Lah JJ. LR11/SorLA expression is reduced in sporadic Alzheimer disease but not in familial Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:866–872. doi: 10.1097/01.jnen.0000228205.19915.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Brasnjo G, Hayashi M, Wolfel M, Collesi C, Giovedi S, Raimondi A, Gong LW, Ariel P, Paradise S, et al. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science. 2007;316:570–574. doi: 10.1126/science.1140621. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Relkin NR, Sweeney D, Munoz DG, Mackenzie IR, Gandy S. Increased apolipoprotein E epsilon 4 in epilepsy with senile plaques. Ann Neurol. 1997;41:402–404. doi: 10.1002/ana.410410317. [DOI] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, et al. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. The Journal of cell biology. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Youkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikin AF, Annaert WG, Takei K, De Camilli P, Jahn R, Greengard P, Buxbaum JD. Alzheimer amyloid protein precursor is localized in nerve terminal preparations to Rab5-containing vesicular organelles distinct from those implicated in the synaptic vesicle pathway. J Biol Chem. 1996;271:31783–31786. doi: 10.1074/jbc.271.50.31783. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP Processing and Synaptic Function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kang JE, Cirrito JR, Dong H, Csernansky JG, Holtzman DM. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc Natl Acad Sci U S A. 2007;104:10673–10678. doi: 10.1073/pnas.0700148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo EH, Squazzo SL. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J Biol Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Koo EH, Squazzo SL, Selkoe DJ, Koo CH. Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J Cell Sci . 1996;109(Pt 5):991–998. doi: 10.1242/jcs.109.5.991. [DOI] [PubMed] [Google Scholar]
- Lah JJ, Levey AI. Endogenous presenilin-1 targets to endocytic rather than biosynthetic compartments. Mol Cell Neurosci. 2000;16:111–126. doi: 10.1006/mcne.2000.0861. [DOI] [PubMed] [Google Scholar]
- Lazarov O, Lee M, Peterson DA, Sisodia SS. Evidence that synaptically released beta-amyloid accumulates as extracellular deposits in the hippocampus of transgenic mice. J Neurosci. 2002;22:9785–9793. doi: 10.1523/JNEUROSCI.22-22-09785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesne S, Ali C, Gabriel C, Croci N, MacKenzie ET, Glabe CG, Plotkine M, Marchand-Verrecchia C, Vivien D, Buisson A. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J Neurosci. 2005;25:9367–9377. doi: 10.1523/JNEUROSCI.0849-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A, Somogyi P, Nusser Z. Reduction of excitatory postsynaptic responses by persistently active metabotropic glutamate receptors in the hippocampus. J Neurophysiol. 2003;89:1910–1919. doi: 10.1152/jn.00842.2002. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Miller LA. Senile plaques in temporal lobe epilepsy. Acta Neuropathol (Berl) 1994;87:504–510. doi: 10.1007/BF00294177. [DOI] [PubMed] [Google Scholar]
- Marks B, McMahon HT. Calcium triggers calcineurin-dependent synaptic vesicle recycling in mammalian nerve terminals. Curr Biol. 1998;8:740–749. doi: 10.1016/s0960-9822(98)70297-0. [DOI] [PubMed] [Google Scholar]
- Marquez-Sterling NR, Lo AC, Sisodia SS, Koo EH. Trafficking of cell-surface beta-amyloid precursor protein: evidence that a sorting intermediate participates in synaptic vesicle recycling. J Neurosci. 1997;17:140–151. doi: 10.1523/JNEUROSCI.17-01-00140.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maycox PR, Link E, Reetz A, Morris SA, Jahn R. Clathrin-coated vesicles in nervous tissue are involved primarily in synaptic vesicle recycling. The Journal of cell biology. 1992;118:1379–1388. doi: 10.1083/jcb.118.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton BL. Evidence for two physiologically distinct perforant pathways to the fascia dentata. Brain Res. 1980;199:1–19. doi: 10.1016/0006-8993(80)90226-7. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, Stalder M, Herzig MC, Kaeser SA, Kohler E, Pfeifer M, Boncristiano S, Mathews PM, Mercken M, Abramowski D, et al. Extracellular amyloid formation and associated pathology in neural grafts. Nat Neurosci. 2003;6:370–377. doi: 10.1038/nn1022. [DOI] [PubMed] [Google Scholar]
- Newton AJ, Kirchhausen T, Murthy VN. Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc Natl Acad Sci U S A. 2006;103:17955–17960. doi: 10.1073/pnas.0606212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitsch RM, Deng M, Tennis M, Schoenfeld D, Growdon JH. The selective muscarinic M1 agonist AF102B decreases levels of total Abeta in cerebrospinal fluid of patients with Alzheimer's disease. Ann Neurol. 2000;48:913–918. [PubMed] [Google Scholar]
- Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P. Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem. 1993;268:608–612. [PubMed] [Google Scholar]
- Offe K, Dodson SE, Shoemaker JT, Fritz JJ, Gearing M, Levey AI, Lah JJ. The lipoprotein receptor LR11 regulates amyloid beta production and amyloid precursor protein traffic in endosomal compartments. J Neurosci. 2006;26:1596–1603. doi: 10.1523/JNEUROSCI.4946-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, Katayama T, Baldwin CT, Cheng R, Hasegawa H, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007;39:168–177. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Price DL, Koliatsos VE. Disruption of corticocortical connections ameliorates amyloid burden in terminal fields in a transgenic model of Abeta amyloidosis. J Neurosci. 2002;22:9794–9799. doi: 10.1523/JNEUROSCI.22-22-09794.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Teplow DB, Siman R, Greenberg B, Sisodia SS. Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “beta-secretase” site occurs in the golgi apparatus. J Biol Chem. 1996;271:9390–9397. doi: 10.1074/jbc.271.16.9390. [DOI] [PubMed] [Google Scholar]
- Ting JT, Kelley BG, Lambert TJ, Cook DG, Sullivan JM. Amyloid precursor protein overexpression depresses excitatory transmission through both presynaptic and postsynaptic mechanisms. Proc Natl Acad Sci U S A. 2007;104:353–358. doi: 10.1073/pnas.0608807104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J Physiol. 2006;572:477–492. doi: 10.1113/jphysiol.2005.103754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- Wang LH, Rothberg KG, Anderson RG. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. The Journal of cell biology. 1993;123:1107–1117. doi: 10.1083/jcb.123.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenk MR, Pellegrini L, Klenchin VA, Di Paolo G, Chang S, Daniell L, Arioka M, Martin TF, De Camilli P. PIP kinase Igamma is the major PI(4,5)P(2) synthesizing enzyme at the synapse. Neuron. 2001;32:79–88. doi: 10.1016/s0896-6273(01)00456-1. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Barnes LL, Bennett DA, Li Y, Bienias JL, Mendes de Leon CF, Evans DA. Proneness to psychological distress and risk of Alzheimer disease in a biracial community. Neurology. 2005;64:380–382. doi: 10.1212/01.WNL.0000149525.53525.E7. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Evans DA, Bienias JL, Mendes de Leon CF, Schneider JA, Bennett DA. Proneness to psychological distress is associated with risk of Alzheimer's disease. Neurology. 2003;61:1479–1485. doi: 10.1212/01.wnl.0000096167.56734.59. [DOI] [PubMed] [Google Scholar]
- Yin KJ, Cirrito JR, Yan P, Hu X, Xiao Q, Pan X, Bateman R, Song H, Hsu FF, Turk J, et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J Neurosci. 2006;26:10939–10948. doi: 10.1523/JNEUROSCI.2085-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.