

Abstract
K-channels and valinomycin molecules share the exquisite ability to select K+ over Na+ ions. Highly selective K-channels maintain a special local environment around their binding sites devoid of competing hydrogen bond donor groups, which enables spontaneous transfer of K+ from states of low coordinations in water into states of over-coordination by 8 carbonyl ligands. In such a phase-activated state, electrostatic interactions from these 8-fold binding sites, constrained to maintain high coordinations, result in K+/Na+ selectivity with no need for a specific cavity size. Under such conditions, however, direct coordination from 5 or 6 carbonyl ligands does not result in selectivity. Yet, valinomycin molecules achieve selectivity by providing only 6 carbonyl ligands. Does valinomycin use additional coordinating ligands from the solvent or does it have special structural features not present in K-channels? Quantum chemical investigations undertaken here demonstrate that valinomycin selectivity is due to cavity size constraints that physically prevent it from collapsing onto the smaller Na+ ion. Valinomycin enforces these constraints using a combination of intra-molecular hydrogen bonds and other structural features, including its specific ring size and the spacing between its connected ligands. Results from these investigations also provide a consistent explanation for the experimental data available for valinomycin’s ion-complexation properties in solvents of varying polarity. Together, investigations on these two systems reveal how nature, despite being popular for its parsimony in recycling functional motifs, can use different combinations of phase, coordination number, cavity size, and rigidity (constraints) to achieve K+/Na+ selectivity.
Keywords: Ion coordination, Quantum chemistry, Molecular association, Solvation phase, Ligand binding
Natural systems possess the remarkable ability to distinguish between two equivalently charged ions, Na+ and K+, which differ in size by less than 0.4 Å. As a consequence of this ability, virtually all cells can transport these ions selectively and regulate their concentration gradients across membranes.1 Preferential binding of one of these ions over the other also affects activities of several other globular protein2 and RNA3 enzymes. Selective ion permeation and binding ultimately enable a wide variety of high-level physiological tasks to be accomplished; from nutrient uptake and volume control in cells, to generation and propagation of nerve signals, maintenance of rhythmic heart rates, vision and dialysis in eukaryotes.
Among bio-molecules that differentiate between these two ions, some selectively bind K+, while others bind Na+. What drives selectivity in favor of a particular ion? Equilibrium thermodynamics dictates that transfer of a particular ion A from one solvation phase m, say water, to another solvation phase p, say protein, is favorable when the ion’s solvation free energy in phase p is lower than its value in phase m,
| [1] |
where ΔGA(m) is the free energy change for ion A in phase m relative to the gas phase. Selective partitioning of ion A over another ion B into phase p is favorable when the free energy change for transfer from phase m to p is lower for A relative to B,
| [2] |
While these equations describe the thermodynamic driving forces required for selectivity, they do not indicate the determining factors. The determinants of A/B (K+/Na+ or Na+/K+) selectivity are the chemical and structural features present in phase p that alter the free energy difference between these ions with respect to their difference in phase m.
The pursuit to understand the determinants of K+ over Na+ selectivity began several decades ago, and now with the advent of new initiatives to engineer biomimetic nano-devices for addressing critical issues concerning dialysis, desalination and power generation (see for example Ref. 4), such efforts are being undertaken with an added impetus. Early investigations led to the formulation of two main concepts: one built around host-guest steric relationships,5, 6 where the host molecule maintains a specific cavity size that fits one ion better than the other, while the other built around host-guest chemistry,7, 8 where specific electric field strengths of the host molecule ligands solvate one ion better than the other. These concepts revolutionized the field of ion channel selectivity, especially concerning equivalently charged ions. Until a few years ago, however, lack of theoretical and experimental data limited testing of these mechanisms to determine how well they explain selectivity in biomolecules.
Here we first briefly review recent investigations of potassium (K−) channels to determine how current ideas regarding K+/Na+ selectivity relate to the two older mechanisms. Next we describe how recent results on K-channel mechanism raise critical questions regarding the mechanism of K+/Na+ selectivity in valinomycin, a tiny ionophore (169 atoms) in comparison to K-channels. Finally, we present our investigations of valinomycin ion selectivity using quantum chemical (QC) methods.
When the seminal work of Mackinnon and coworkers led to the determination of the first crystal structure of a strongly selective K-channel,9 K+/Na+ selectivity was attributed to a host-guest steric mechanism.6,9–12 As illustrated in figure 1(a), the crystal structure revealed that the channel selectivity filter coordinates K+ ions using 8 carbonyl ligands. K+ ions were thought10, 11 to partition spontaneously into such highly coordinated 8-fold binding sites because these sites mimicked coordination of K+ ions in aqueous phase.13, 14 This notion of ion partitioning, however, no longer holds in light of new experimental15, 16 and theoretical17–20 data, which show that the coordination number of K+ in water is much lower than 8, and the probability that a K+ ion simultaneously coordinates with 8 water molecules in aqueous phase is negligible. At the same time, highly selective ion discrimination was attributed to binding sites that formed rigid cavities matching K+, but not the smaller Na+, ion size. Several subsequent molecular dynamics (MD) simulations,21–23 however, demonstrated that, in accord with experimental B-factors from the crystallographic data, fluctuations of the coordinating carbonyl-oxygens were larger than the size difference between Na+ and K+ ions, thus challenging the validity of the host-guest steric mechanism of selectivity. In light of this realization, recent theoretical work based on free energy perturbation (FEP) calculations24 demonstrated how the host-guest chemistry model could instead be invoked to explain K+/Na+ selectivity in K-channels.
Figure 1.

Structural differences between K+ binding sites in strongly selective K-channels and valinomycin. (a) Partial view of the x-ray structure9 of a representative K-channel, KcsA, illustrating a K+ ion occupying the S2 site of the selectivity filter in a state of high coordination by 8 carbonyl oxygens. (b) X-ray structure36 of valinomycin illustrating a K+ ion coordinated by 6 carbonyl oxygens. K+ ions are drawn as green spheres and all other atoms as sticks with oxygens in red, carbons in yellow and nitrogens in blue. Dashed lines connecting the ion and the oxygen atoms represent coordination with K+.
Certain critical experimental and theoretical observations, however, still remained unexplained.20, 25 For instance, how does K+ partition between low coordination states in bulk water and a high 8-fold coordination state in the filter? Why should the chemically identical binding sites S1, S2 and S3 in strongly selective KcsA channels have strikingly different degrees of computed26, 27 selectivities? Why should specific mutations28, 29 in the region adjacent to these binding sites render the filter non-selective? Why should NaK channels30 have reduced selectivity despite sharing two structurally and chemically identical binding sites with strongly selective K-channels?
Recent QC-based studies20 helped reconcile these seemingly disparate observations by identifying the roles of the chemical and structural properties of the solvation phase in modulating ion coordination architectures, and the importance of maintaining specific high numbers of coordinating ligands in the binding sites for selectivity. Because the spatial region proximal to the binding sites in strongly selective K-channels is devoid of competing hydrogen bond donors the protein solvation environment cannot directly and favorably interact with the coordinating carbonyl oxygen atoms. Within such a local “quasi-liquid” pocket, electrostatically equivalent to a low dielectric phase (ε < 3), the thermodynamic probability for formation of an 8-fold K+ coordination complex with carbonyl ligands increases to a value that no longer obstructs K+ ion partitioning between water and the binding sites. At the same time, the free energy associated with transferring a Na+ ion from liquid water to the same 8-fold carbonyl binding site is unfavorable, resulting in K+/Na+ selectivity. Furthermore, as also inferred from previous MD simulations,24 such an 8-coordinate K+-selective binding site does not require restrictions on its cavity size. Nevertheless, if the binding site were fully flexible, occupation by Na+ ions could reduce the number of coordinating ligands to 5 or 6 and consequently eliminate selectivity.20, 31 Therefore, although a specific cavity size is not required, some form of rigidity20 or topological constraint,31 which could be supplied by structural and chemical properties of the solvation phase or the binding site itself, is necessary to prevent distortion32 of the binding sites upon Na+ occupation. In particular, rigidity is needed in strongly selective K-channels to enforce direct ion coordination by high numbers (>6) of carbonyl ligands.
Factors that disturb the quasi-liquid environment,20 via structural or chemical modifications, can lead to distortions in binding site coordination architectures, which can in turn result in a complete loss of K+/Na+ selectivity. For example, hydrogen bond donor side chains introduced next to the selectivity filter binding sites can compete with the permeating ion for direct favorable interactions with carbonyl oxygens, and thus reduce the probability for formation of binding sites with high numbers of coordinating ligands.20 Fewer ligands coordinating the ions in such a binding site result in significantly reduced selectivity, as was demonstrated recently using thermodynamic calculations.20 Experimental observations of reduced selectivity with mutations that introduce hydrogen bond donor groups near K-channel binding sites also lend support to such a mechanism. For example, when tryptophan, a hydrogen bond donor, replaces serine at the S177 position in GIRK2, selectivity is substantially reduced.28, 29 Note that while serine is also a hydrogen bond donor, its distant location on the transmembrane helix and shorter length in comparison to tryptophan may provide it with fewer opportunities to physically approach the carbonyl oxygens of the binding sites. In a different example from the comprehensive sequence alignment of K-channels,33 weakly selective K-channels carry hydrogen bond donors in the form of arginines in proximity to their selectivity filters, and these side chains are completely absent in all strongly selective K-channels. Note that arginine side chains are normally implicated in salt-bridge formation. In the absence of proximal negative charges, however, they can also engage in hydrogen bond formation.
Since water is also a hydrogen bond donor that can disrupt the quasi-liquid environment, exposure of K-channel binding sites to different amounts of intra- or extra-cellular water can naturally be expected to affect the degrees of their selectivity. For example, binding site S2 in the highly selective KcsA channel is the site least exposed to water.9 Even though it is chemically identical to sites S1 and S3, all-atom MD simulations24,27,34 predict a higher selectivity of −5 kcal/mol for this site, as compared to a −2 kcal/mol selectivity for the other sites. On the same note, water penetration into the protein matrix behind the selectivity filter can also be expected to reduce selectivity. Such water penetration has been reported in several MD simulations, and has also been seen in x-ray studies. This is perhaps one reason why the computed selectivity of approximately −10 kcal/mol for an isolated binding site20 is higher than the measured selectivity of K-channels. In another example, the NaK channel shows weak selectivity and, in contrast to highly selective K-channels, all its ion binding sites30 are exposed to water. Note that increased flexibility20 or reduced topological constraints31 in the NaK binding sites, compared with KcsA, can also contribute to its reduced selectivity. Increased flexibility is evident from the recent MD simulation data34 on the NaK channel where its binding sites are seen to adopt substantially different coordination structures around the two ions relative to KcsA.
Exposure of ion binding sites to water can reduce their selectivities via two different mechanisms. In a host-guest chemistry mechanism, water molecules on average replace some of the carbonyl ligands during ion complexation and thus reduce the electric field density to values that exhibit lowered K+/Na+ selectivity, as explored recently using reduced models.34 The reason weaker-field water molecules would preferentially replace some of the stronger-field carbonyl ligands in a channel binding site is not simply because more waters are present near those binding sites, but because water molecules are energetically easier to extract from aqueous phase in comparison with carbonyl ligands.20 In an alternative mechanism, the exposure to water could result in an overall drop in binding site coordination numbers, without ligand substitution, which also reduces selectivity.20, 31 Regardless of the mechanistic details, the degree of exposure of binding sites to water indeed determines the absolute potency of a given carbonyl binding site to carry out K+/Na+ discrimination.
In summary, recent work strongly suggests that K-channels use a “phase-activated” mechanism20 where the local environment around their binding sites is tuned to sustain high coordination numbers (> 6) around K+ ions, which otherwise are rarely observed in liquid water. When such high coordinations are enforced, using some form of rigidity20 or topological constraint31 provided by properties of the protein solvation phase or binding site, and combined with the field strength of carbonyl ligands, they create the electrical scenario necessary for rapid and selective K+ partitioning20 with no need to maintain specific cavity sizes.20,24,31
Note that in these and other simulation studies,20,31,34,35 the general strategy to investigate the effects of structure on ion selectivity was to utilize representative models of ion binding sites, without an explicit description of the rest of the protein matrix. Such representative models were constructed only after a systematic analysis20 of the effects of both numbers and ligand chemistries on ion partitioning and selectivity. Results of these simulations were then analyzed in the context of known experimental and theoretical data, which consequently led to an improved understanding of the role of the protein matrix20, 31 in driving ion selectivity. This is not a shortcoming of the approach. Instead, it is part of a strategy essential for understanding the individual and collective roles of the physiologically relevant degrees of freedom available to ion binding sites, an apparently impossible task if the remaining protein matrix is explicitly incorporated, as done in standard MD simulations or in wet-lab experiments. In addition, such a strategy provides an opportunity to utilize quantum chemical methods that excel at describing complex interactions, such as those occurring between an ion and its coordinating ligands.
In view of this K-channel mechanism, which depends on the electrostatic and structural properties of the local environment along with constraints that enforce high ion coordination (> 6), how does valinomycin achieve K+/Na+ selectivity by providing only 6 carbonyl ligands for ion coordination?
Valinomycin is a tiny ionophore that has, over the past decades, helped provide a wealth of information on bio-mechanisms at the molecular level. Chemically, it is a cyclic depsipeptide built from a 3-fold repetition of the alternating amino- and hydroxy-acid residues L-Valine, D-α-hydroxyvaleric acid, D-valine, and L-lactic acid, [d-Val-d-HyV-l-Val-l-Lac]3. Its structure, in the absence of bound cations, is solvent dependent. When bound to K+, however, it adopts a configuration wherein its amino- and hydroxy-acid side-chains always point outward creating a hydrophobic exterior, while the 6 carbonyl oxygens from the hydroxy-acid residues point inward forming a cavity for ion complexation,36 as illustrated in figure 1(b). Such a structural configuration allows valinomycin to transport the bound K+ ions across cellular membranes. Because of this selective ion transport property in combination with its small size, valinomycin has found important applications in numerous areas,36 including investigations of oxidative phosphorylation reactions in biosystems, as ion-selective components in liquid-membrane electrodes, and in synthesis of antilipolytic agents, insecticides and nematodicides.
Several independent experiments36–42 have demonstrated that valinomycin selects between K+ and Na+ ions in low as well as high dielectric solvents. For example, salt extraction equilibrium measurements show41 that its selectivity ΔΔΔGNa+→K+ (ε → Val,ε) of −7.6 kcal/mol is almost independent of solvent dielectric coefficients ε in the range 2 (hexane) through 9 (dichloromethane). In addition, permeability ratio measurements in lipid membranes result in a selectivity of ~ −6 kcal/mol41, 42. In methanol, which has a higher dielectric coefficient of 33, complexation constant experiments40 find that valinomycin prefers binding K+ to Na+ ions by −5.4 kcal/mol. In water, which has an even higher dielectric coefficient value of 80, valinomycin still selects for K+, but its degree of selectivity has not been quantified in this medium.36, 39 This ability of valinomycin to select K+ over Na+ in solvents of varying polarity indicates that the core mechanism underlying valinomycin’s selectivity does not rely on interaction with solvent and thus should be amenable to evaluation in the gas phase. Nevertheless, solvent can be expected to modify its degree of selectivity.
To gain insight about the mechanism of selectivity in valinomycin, we first investigate how additional ligation from solvent molecules, which raise the coordination numbers of K+ or Na+ ions to the high values found in K-channels, can alter its degree of selectivity. We choose water as a representative additional ligand because water extraction from aqueous solution and addition to the ion-complexed valinomycin molecule simulates the physiologically most relevant scenario that occurs in lipid membranes. We compute the free energy change ΔΔgn(ε) associated with extracting n waters from aqueous phase (aq.) and adding them as extra ligands to K+ when it is already complexed with valinomycin [K+ .Val] in a dielectric phase ε,
| [3] |
We use a molecular association statistical theory43, 44 to determine the values of ΔΔgn(ε), an approach identical to the one used previously for investigating selective ion partitioning in K-channels.20 The ion along with its directly coordinating ligands, which in this case consist of the valinomycin molecule and the n ligating waters, are treated quantum chemically using density functional theory (DFT) and the B3LYP45, 46 functional (implemented in the Gaussian03 package47). The only difference in parameters, as compared to those used previously,20 is with respect to the choice of basis set functions. Here all optimization and frequency calculations are carried out using a 6-311+G(d) basis set for K+, and a 6-31G(d) basis set for the remaining atoms, which reproduces the experimental vibrational spectra of valinomycin.48 For computational feasibility, the hydrophobic side chains of the valinomycin molecule are clipped and replaced with methyl groups. Note, however, that the resulting decrease in the number of atoms in valinomycin (from 169 to 114) does not affect the high frequency vibrational spectra of the molecule,48 implying that the computed reaction free energies will be minimally affected.
The structural and energetic effects of increasing the coordination of K+ bound to valinomycin [K+ .Val] by n=1 and n=2 water molecules are illustrated in figure 2. Additional ligation by water significantly destabilizes the [K+ .Val] complex, which implies that the contribution of these higher coordination complexes to the free energy difference between the K+- and Na+-complexed states will be negligible. In a separate set of calculations, attempts to optimize water molecules around a [Na+ .Val] complex such that they increase coordination of Na+ ions to 7 or 8 failed, implying that the 7- and 8-fold Na+ complexes are also energetically less favorable. Therefore in a physiologically relevant scenario, additional ligation by water will not alter valinomycin’s selectivity. Thus, unlike K-channels, valinomycin achieves K+/Na+ selectivity utilizing only 6 carbonyl ligands.
Figure 2.
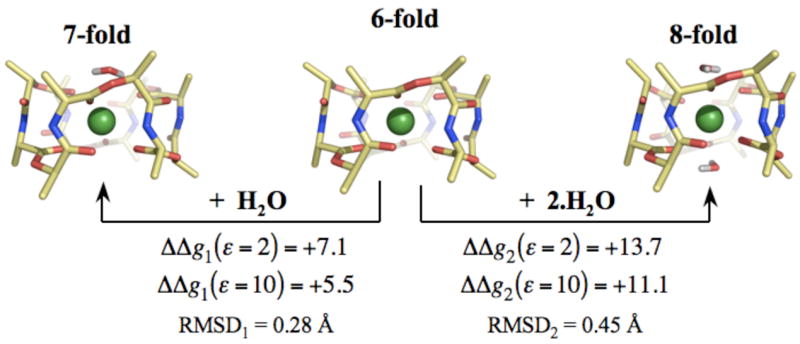
Structural and energetic changes associated with increasing the coordination of K+ ions bound to valinomycin. K+ ions complexed to valinomycin are in direct coordination with 6 carbonyl ligands. Increasing their coordination to 7 and 8 using n=1 and n=2 water molecules destabilizes the K+ complex. ΔΔgn(ε) denotes this change in complexation energy, as determined from the reaction given by equation 3. ΔΔgn(ε) are provided for two different values of ε, which are representative of the dielectric constant of the lipid membranes. Note that calculations using higher values of ε, such as ε=80, also result in a greater stability of the 6-fold coordination. RMSDn reflects the change in the backbone structure of K+ bound valinomycin due to complexation by n water molecules.
To understand the core mechanism behind valinomycin’s ability to select between K+ and Na+ ions using only 6 carbonyl ligands, we first compare the gas phase QC optimized structures of its Na+ and K+ complexes. The two structures, as illustrated in figure 3, are not in the absolute sense “isosteric”,49 but nevertheless closely resemble each other. The root mean square deviation (RMSD) between the structures is only 0.14 Å, a result consistent with early circular dichroism (CD) data in low dielectric media.39 In both cases, the ions prefer positions in the center of the cavity created by the 6 carbonyl oxygens, consistent with previous classical MD studies.50 The 6 oxygens are arranged in a skewed-triangular-prism geometry (not octahedral), with 3 oxygens on either side of the ions. Note that this arrangement is typical for optimum 6-fold Na+ and K+ ion interactions with carbonyl ligands (see Fig. S1 in supplementary text). The average Na+-oxygen distance in valinomycin is smaller than the average K+-oxygen distance, but by only 0.15 Å. This is consistent with results from chromatography experiments50 where the Na+ complex was found to have a cross-sectional area slightly smaller than that of the K+ complex.
Figure 3.
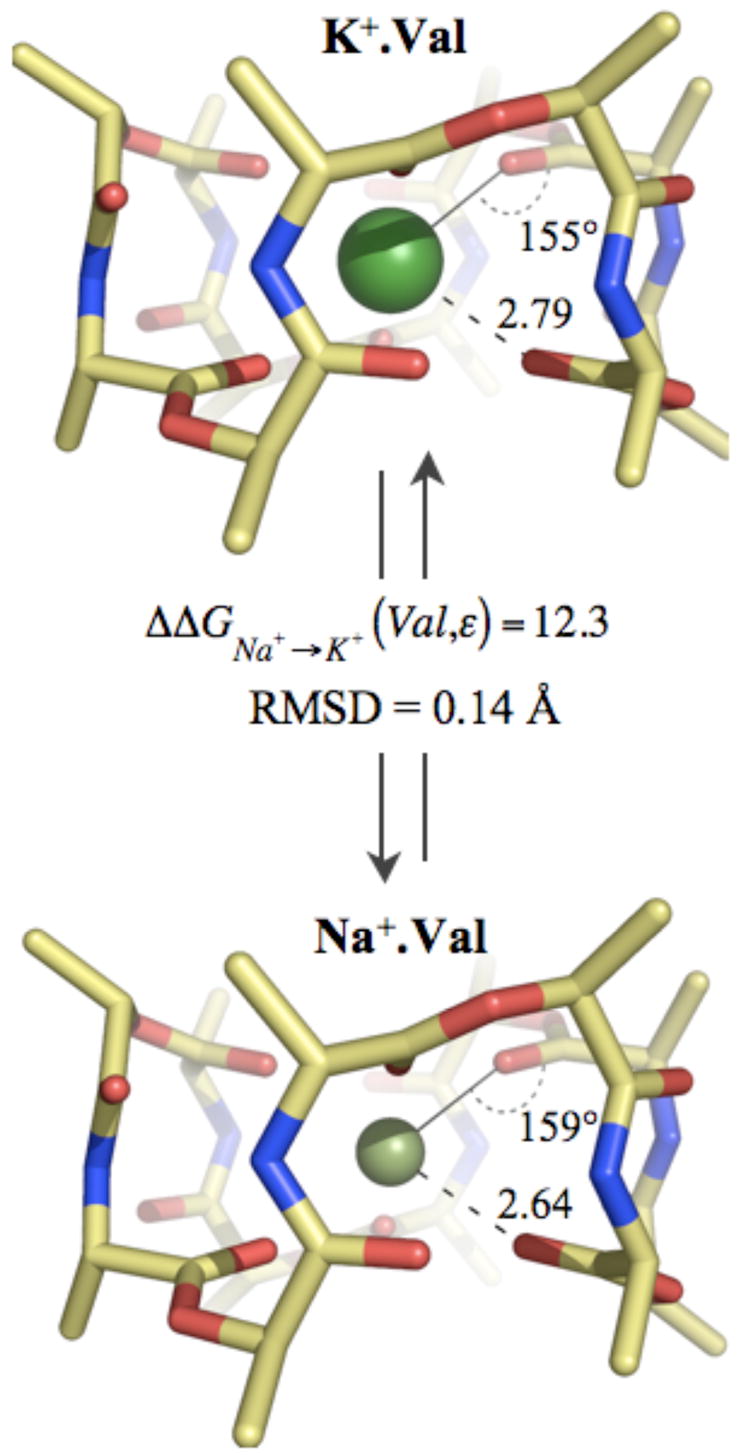
Optimized (DFT/B3LYP) structures of Na+ and K+ complexes of valinomycin. Average ion-oxygen distances are indicated in Ångström units. Average angles formed between the ion and the carbonyl ligands (I+-O-C) are also shown.
Free energies computed using these optimized ion-complexed valinomycin structures in the gas phase result in a more stable Na+ complex relative to the K+ complex, with ΔΔGNa+→K+ (Val,ε = 1) of 12.3 kcal/mol. When the stability due to the reaction field from a dielectric media (ε < 80) is added (computed using the APBS package51), the magnitude of this relative stability ΔΔGNa+→K+ (Val,ε) increases by less than 0.05 kcal/mol; a result that naturally emerges from the structural and electrical (see Fig. S2 of the supplementary text) similarities of the two valinomycin complexes.
This relative stability between the two valinomycin complexes is, however, much less than the relative stability of the ions ΔΔGNa+→K+ (m) in relevant liquid media m where valinomycin selectivity measurements have been conducted. In water, our computed20 solvation energy of Na+ is ~ 21 kcal/mol more favorable than K+. In methanol, which has a smaller dielectric coefficient than water (ε = 33), the relative solvation energy of Na+ compared to K+ still remains close to 21 kcal/mol, as computed here using the same methodology described previously.20 These results are consistent with experimental data,52 which in fact show that ΔΔGNa+→K+ (m) remains invariant across these and a range of relevant solvents, including dichloromethane and other alcohols. This implies that transfer of these ions from any of these solvents m into valinomycin will result in a K+/Na+ selectivity ΔΔΔGNa+→K+ (m → Val) of approximately −9 kcal/mol. This describes the core selectivity of valinomycin, independent from structural changes effected by solvent on its Na+ and K+ complexes. Consequences of structural changes brought upon by interaction with solvent will be considered, but a little later.
We now investigate the mechanism that gives rise to valinomycin’s core selectivity, or in other words we investigate why the relative stabilities of ions in valinomycin complexes are smaller than their values in liquid media, ΔΔGNa+→K+ (Val) ≪ 21 kcal/mol. Note from table 1 that this behavior is not typical of 6-fold coordinations in gas phase. In fact, a 6-fold coordination complex involving all carbonyl ligands results in a relative free energy difference ΔΔGNa+→K+ close to 21 kcal/mol. Therefore, the specific carbonyl chemistry of the ligands in valinomycin is not responsible for its K+/Na+ selectivity, as postulated otherwise.8,24,34 Note also from table 1 that the average Na+-oxygen distance of 2.64 Å in valinomycin is larger than the distance of ~2.4 Å that Na+ prefers when it interacts with 6 fully flexible monodentate or partially flexible bidentate ligands. This increase in ion-ligand distance is seen in spite of the skewed triangular prism geometry that Na+ prefers when coordinating with 6 ligands. K+ ion, on the other hand, binds to valinomycin at an average distance similar to what it prefers when it forms 6-fold complexes with other ligands. This observation raises two questions. First, is such an increase in ligation distance sufficient to reduce the relative stability of the ions in valinomycin from ~21 to ~12 kcal/mol? Indeed it is, as can be verified using Coulomb’s equation and the ion-ligand data given in figure 3 and figure S2 in the supplementary text. Second, why is the Na+ ligation distance in valinomycin larger than its preferred length? In other words, what restrains valinomycin from collapsing onto the smaller Na+ ion? There are three main interactions in proteins, or in this case a depsipeptide that define 3-dimensional structure: hydrophobic forces, salt-bridges, and hydrogen bonds. Among these, hydrogen bonds are the only form of interaction relevant to such a structural feature in valinomycin as there are six hydrogen bonds present in its optimum Na+ and K+ complexes. In addition, since valinomycin is a cyclic molecule, constraints that might arise from its specific ring size also matter, as explored in the past.37, 53
Table 1.
Computed (DFT/B3LYP) structural and thermo-chemical properties of 6-fold ion-oxygen clusters in gas phase. Ion-oxygen distances Na+-O and K+-O are in Ångström units and ΔΔGNa+→K+ (ε = 1) are the free energy differences between the Na+ and K+ complexes in gas phase.
To investigate the contribution of hydrogen bonds to valinomycin’s selectivity, we turn them off in a manner that can be emulated in experiments. As illustrated in figure 4, we start with the K+ optimized valinomycin configuration, and substitute all six of its hydrogen bond acceptor carbonyl oxygens =O with non-hydrogen-bonding =CH2 groups. A QC re-optimization in the presence of K+ noticeably alters its backbone configuration (RMSD = 0.65 Å), but the 6-fold skewed-triangular-prism coordination geometry is retained, and the average K+-oxygen distance increases by only 0.1 Å. A QC re-optimization in the presence of Na+, however, results in an altogether different structure. The molecule adopts an elliptical shape and coordinates the Na+ ion using only 4 carbonyl oxygens. The four coordinating oxygens are closer than in the native hydrogen-bonded complex, at an average distance of 2.33 Å, while the remaining two carbonyl oxygens fall away to a distance of 5.1 Å. The intra-molecular hydrogen bonding in valinomycin, therefore, plays a more important role in driving the structure of its Na+ complex, as compared to its K+ complex. These changes in coordination configurations consequently result in a significant increase in the relative stability ΔΔGNa+→K+ (mVal) of the mutated Na+ compared to K+ valinomycin complex by 5.5 kcal/mol. The hydrogen bonds, therefore, significantly contribute to K+/Na+ selectivity because they make valinomycin less flexible and prevent its ligands from adopting an energetically more stable configuration around the smaller Na+ ion.
Figure 4.
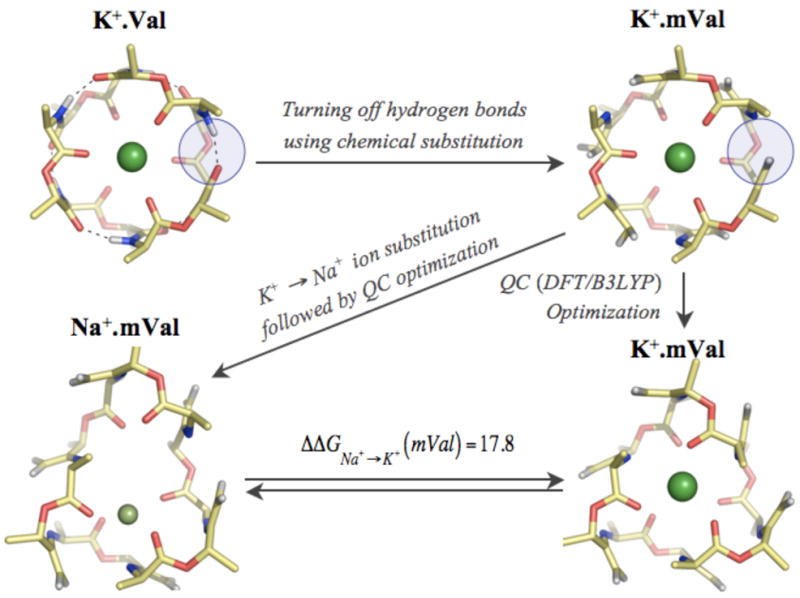
Structural and energetic effects of turning off the six intra-molecular hydrogen bonds in valinomycin. The hydrogen bonds, represented as dashed lines, were turned off using chemical substitutions involving replacement of proton acceptor atoms =O with =CH2 groups. A representative substitution is highlighted inside blue circles. QC optimization following these substitutions resulted in a K+ complexed structure K+.mVal with a backbone atom RMSD of 0.65 Å, and a K+-O distance of 2.88 Å. In the case of Na+ complex, Na+.mVal, these substitutions resulted in 4 of the 6 carbonyl oxygens to coordinate Na+ ions at smaller distances of 2.33 Å, as compared to 2.64 Å in the native Na+.Val structure. The remaining two carbonyl oxygens are at a distance of 5.12 Å from the Na+ ion. The absence of hydrogen bonds also resulted in increasing the free energy difference between the Na+ and K+ complexes from 12.3 to 17.8 kcal/mol.
The results above also indicate that some residual K+/Na+ selectivity (~ −3 kcal/mol) is retained even in the complete absence of intra-molecular hydrogen bonds, suggesting the presence of other structural features in valinomycin that contribute to its selectivity. Note from figure 4 that, in the absence of hydrogen bonds, valinomycin prefers a 4-fold coordination around Na+ ions. Valinomycin’s residual selectivity for K+ is surprising given previous gas phase investigations with formamide (carbonyl) ligands20 that show 4-fold coordination of Na+ is energetically more stable than its corresponding 6-fold coordination by 3.0 kcal/mol. In a similar calculation, we find that a 4-fold coordination from 2 glycine dipeptide molecules (carbonyl ligands) is also energetically more stable than its corresponding 6-fold coordination by 2.5 kcal/mol. Adding to this the observation that a 6-fold coordination from flexible carbonyl ligands leads to no K+/Na+ selectivity, the 4-fold coordination of Na+ in mutated valinomycin should have led to a reversal of ion selectivity. The reason valinomycin retains K+/Na+ selectivity is because this 4-fold geometry, in which all the 4 ligands are cluttered on one side of the Na+ ion (figure 4), is energetically less stable than the preferred 4-fold geometries in gas phase, such as the tetrahedral or planar18 geometries. These 4 ligands from valinomycin are unable to adopt such preferred geometries because of the combined restraints in valinomycin from structural features such as its specific ring size and the spacing between its connected ligands.
These results on the effects of intra-molecular hydrogen bonding, in fact, also suggest how interaction with solvent, other than by direct coordination, can alter valinomycin’s core selectivity. It is well known that thermal fluctuations cause local opening and closing of hydrogen bonds.54–57 In the event of their exposure to solvent, as in the case of valinomycin, solvent molecules can compete for hydrogen bonding and decrease the stability of backbone hydrogen bonds. The higher the polarity of the solvent, the more disruptive its effect will be on valinomycin’s intra-molecular hydrogen bonding. This is also evident from valinomycin’s structure in the absence of bound cations,36 where the number of intra-molecular hydrogen bonds are known to decrease with increase in solvent polarity. Therefore, we can expect that even when valinomycin is complexed with ions, its propensity to maintain its hydrogen bonds will decrease with increase in solvent polarity.
Increase in solvent polarity will therefore affect valinomycin in at least three ways. First, the reaction free energy associated with ion complexation to valinomycin will change. In addition to a higher energetic penalty associated with extracting valinomycin from the solvent phase, the decrease in its intra-molecular hydrogen bonding will also contribute to an overall reduction (to less favorable values) in its ion complexation free energy. At the same time, however, increased flexibility will allow valinomycin to wrap itself around an ion in an energetically more favorable configuration. In addition, there will be a gain in configurational entropy. This gain, however, is expected to be less than 1 kcal/mol as the contribution from entropy when n out of 6 hydrogen bonds are broken is given by k.T.log((6−n)!n!/6!). Experimental binding constant data collected as a function of increasing polarity8,39,41,58 in fact reveal a rough pattern wherein ion complexation energy decreases with increased solvent polarity, suggesting that the former two effects override the latter two competing effects. This specific dependence of ion complexation energy on solvent polarity could be vital for valinomycin to transport K+ ions across lipid membranes as valinomycin would be weakly bound to K+ at the high dielectric lipid-water interface for spontaneous ion uptake and release, and strongly bound to K+ when inside the low dielectric lipid membrane for transport.
Second, as the solvent polarity is increased, the approximate “isostericity” observed between the Na+ and K+ complexes in gas phase will decrease. Based on the results above, the configuration of the K+ complex will not change as significantly as that of the Na+ complex, which will adopt a more elliptical shape. CD and optical rotary dispersion (ORD) data39 collected in solvents with varying polarity, in fact, lend direct support to such a trend. In addition, recent MD simulations of valinomycin carried out in ethanol24, 59 show that it adopts a 6-fold coordination around K+, but a lower 4-fold coordination around Na+ ion. These simulations, however, were carried out in the absence of polarization, which has been argued to be important for describing ion binding in valinomycin8 (also see ab initio data in table S1 of supplementary text).
Finally, a characteristic dependence of K+/Na+ selectivity on solvent polarity will emerge. In low polarity solvents, where all hydrogen bonds are maintained and the Na+ and K+ structures remain approximately isosteric, the K+/Na+ selectivity will attain a constant high value of selectivity similar to its core selectivity. This idea was in fact conceptualized49 in the late 1960s and eventually proved41 by carrying out salt extraction equilibrium experiments in low (ε < 9) polarity solvents. In higher polarity solvents, where the proclivity to maintain all six hydrogen bonds is lower, valinomycin will be able to adopt a relatively more stable configuration around the Na+ ion. In such a scenario, selectivity can be expected to decrease, but further investigation is still required. Nonetheless, valinomycin selectivity will not vanish even in very high polarity solvents due to the intrinsic contribution of −3 kcal/mol from its other structural restraints that are not affected by solvent polarity.
Together, we find that valinomycin is able to use only 6 carbonyl ligands to achieve K+/Na+ selectivity because it can physically prevent all of its 6 ligands from simultaneously collapsing onto the smaller Na+ ion. In other words, it creates a specific cavity size for ion binding that matches the K+, but not the smaller Na+, ion size. Valinomycin enforces such constraints through a combination of intra-molecular hydrogen bonds and other structural features, including its specific ring size and the spacing between its connected ligands.
This host-guest steric mechanism of valinomycin is, however, entirely different from K-channels, which use a different variety of topological constraint. In the absence of cavity size constraints, K-channels utilize constraints on over-coordination by > 6 carbonyl ligands. In addition to any rigidity supplied by the binding site structure, K-channels also require a special local solvation phase around their binding sites that is devoid of competing hydrogen bond donors to sustain such over-coordination.20 One of the key advantages of the K-channel mechanism is that it allows the degree of selectivity to be easily tuned via mutations in the protein solvation environment surrounding the binding sites. In fact, nature appears to have capitalized on this feature to create strong, intermediate as well as weakly selective K-channels without major backbone structural rearrangements.30, 33 The main advantage of valinomycin’s mechanism over the K-channel mechanism is that valinomycin can tolerate the presence of hydrogen bond donors in its local neighborhood, as is evident from its persistent selectivity in solvents of varying polarity. In addition, its complexation energy with K+ is very small in liquid water.8,39,41,58 This currently makes it a choice mechanism for nano-device technology. Cyclodextrins, for example, encoded with valinomycin’s structural features can be plugged into the pores of large channel proteins like α-hemolysins60 to create artificial K+/Na+ selective channels for devices intended for various purposes, including power generation, drug delivery and dialysis.
In general, these and all previous investigations on K-channels and valinomycin together reveal how nature has utilized different combinations of phase, coordination number, cavity size, rigidity, and chemistry to construct widely different mechanisms for K+/Na+ selectivity. These investigations have, however, merely uncovered what lies beneath the surface of an iceberg. Much remains to be discovered before a comprehensive picture of selective ion partitioning mechanisms in biological systems is achieved.
Supplementary Material
Acknowledgments
We would like to thank Dr. Polavarapu for providing us with the optimized coordinates of valinomycin in its uncomplexed propeller configuration and C.P. Beamis for critically reading this manuscript. We would also like to thank Dr. Jakobsson and the National Center for Supercomputing Applications at UIUC for providing us with compute time on their IBM p690 shared memory cluster. A total of ~70,000 hours of compute time was utilized for this work.
This work was supported in part by the DOE under Contract DE-AC04-94Al85000 and in part by the National Institutes of Health through the NIH Roadmap for Medical Research. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the U.S. DOE.
Non-standard Abbreviations
- QC
Quantum Chemical
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hille B. Ionic channels of excitable membranes. 3. Sinauer Associates, Sunderland; Mass: 2001. [Google Scholar]
- 2.Page MJ, Di Cera E. Role of Na+ and K+ in Enzyme Function. Physiol Rev. 2006;86:1049–1092. doi: 10.1152/physrev.00008.2006. [DOI] [PubMed] [Google Scholar]
- 3.Draper DE, Misra VK. RNA shows its metal. Nature Struc Biol. 1998;5:927–930. doi: 10.1038/2901. [DOI] [PubMed] [Google Scholar]
- 4.NIH. Center for Design of Biomimetic Nanoconductors. URL: http://www.nanoconductor.org.
- 5.Pauling L. Nature of forces between large molecules of biological interest. Nature. 1948;161:707–709. doi: 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- 6.Bezanilla F, Armstrong CM. Negative Conductance Caused by Entry of Sodium and Cesium Ions into the Potassium Channels of Squid Axons. J Gen Physiol. 1972;60:588–608. doi: 10.1085/jgp.60.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenman G. In: Symposium on Membrane Transport and Metabolism. Kleinzeller A, Kotyk A, editors. Academic Press NY; 1961. [Google Scholar]
- 8.Aqvist J, Alvarez O, Eisenman G. Ion-Selective Properties of a Small Ionophore in Methanol Studied by Free Energy Perturbation Simulations. J Phys Chem. 1992;96:10019–10025. [Google Scholar]
- 9.Doyle DA, Cabral JM, Pfuetzner RA, Kuo AL, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 10.Miller C. See potassium run. Nature. 2001;414:23–24. doi: 10.1038/35102126. [DOI] [PubMed] [Google Scholar]
- 11.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–8. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 12.Gouaux E, MacKinnon R. Principles of selective ion transport in channels and pumps. Science. 2005;310:1461–1465. doi: 10.1126/science.1113666. [DOI] [PubMed] [Google Scholar]
- 13.Ohtomo N, Arakawa K. Bull Chem Soc Jpn. 1980;53:1789. [Google Scholar]
- 14.Tongraar A, Liedl KR, Rode BM. Born-Oppenheimer ab Initio QM/MM Dynamics Simulations of Na+ and K+ in Water: From Structure Making to Structure Breaking Effects. J Phys Chem A. 1998;102:10340–10347. [Google Scholar]
- 15.Neilson GW, Mason PE, Ramos S, Sullivan D. Neutron and X-ray scattering studies of hydration in aqueous solutions. Philos Trans R Soc London Ser A. 2001;359:1575–1591. [Google Scholar]
- 16.Soper AK, Weckstrom K. Ion solvation and water structure in potassium halide aqueous solutions. Biophys Chem. 2006;124:180–191. doi: 10.1016/j.bpc.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Ramaniah L, Bernasconi M, Parrinello M. Ab initio molecular-dynamics simulation of K+ solvation in water. J Chem Phys. 1999;111:1587–1591. [Google Scholar]
- 18.Rempe SB, Asthagiri D, Pratt LR. Inner shell definition and absolute hydration free energy of K+(aq) on the basis of quasi-chemical theory and ab initio molecular dynamics. Phys Chem Chem Phys. 2004;6:1966–1969. [Google Scholar]
- 19.Varma S, Rempe SB. Coordination numbers of alkali metal ions in aqueous solutions. Biophys Chem. 2006;124:192–199. doi: 10.1016/j.bpc.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 20.Varma S, Rempe SB. Tuning ion coordination architectures to enable selective partitioning. Biophys J. 2007;93:1093–1099. doi: 10.1529/biophysj.107.107482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guidoni L, Torre V, Carloni P. Potassium and sodium binding in the outer mouth of the K. Biochemistry. 1999;38:8599–8604. doi: 10.1021/bi990540c. [DOI] [PubMed] [Google Scholar]
- 22.Berneche S, Roux B. Molecular Dynamics of the KcsA K+ Channel in a Bilayer Membrane. Biophys J. 2000;78:2900–2917. doi: 10.1016/S0006-3495(00)76831-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shrivastava IH, Tieleman DP, Biggin PC, Sansom MS. K+ versus Na+ Ions in a K Channel Selectivity Filter: A Simulation Study. Biophys J. 2002;83:633–645. doi: 10.1016/s0006-3495(02)75197-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noskov SY, Berneche S, Roux B. Control of ion selectivity in potassium channels by electrostatic and dynamic properties of carbonyl ligands. Nature. 2004;431:830–4. doi: 10.1038/nature02943. [DOI] [PubMed] [Google Scholar]
- 25.Jordan PC. New and Notable: Tuning a Potassium Channel – The Caress of the Surroundings. Biophys J. 2007;93:1091–1092. doi: 10.1529/biophysj.107.110205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aqvist J, Luzhkov V. Ion permeation mechanism of the potassium channel. Nature. 2000;404:881–884. doi: 10.1038/35009114. [DOI] [PubMed] [Google Scholar]
- 27.Berneche S, Roux B. Energetics of ion conduction through the K+ channel. Nature. 2001;414:73–77. doi: 10.1038/35102067. [DOI] [PubMed] [Google Scholar]
- 28.Bichet D, Lin YF, Ibarra CA, Huang CS, Yi BA, Jan YN, Jan LY. Evolving potassium channels by means of yeast selection reveals structural elements important for selectivity. Proc Natl Acad Sci USA. 2004;101:4441–4446. doi: 10.1073/pnas.0401195101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bichet D, Grabe M, Jan YN, Jan LY. Electrostatic interactions in the channel cavity as an important determinant of potassium channel selectivity. Proc Natl Acad Sci USA. 2006;103:14355–14360. doi: 10.1073/pnas.0606660103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi N, Ye S, Alam A, Chen L, Jiang Y. Atomic structure of a Na+-and K+ -conducting channel. Nature. 2006;440:570–574. doi: 10.1038/nature04508. [DOI] [PubMed] [Google Scholar]
- 31.Bostick D, Brooks CL., III Selectivity in K+ channels is due to topological control of the permeant ion’s coordinated state. Proc Natl Acad Sci U S A. 2007;104:9260–9265. doi: 10.1073/pnas.0700554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valiyaveetil F, Leonetti M, Muir T, MacKinnon R. Ion selectivity in a semisynthetic K+ channel locked in the conductive conformation. Science. 2006;314:1004–1007. doi: 10.1126/science.1133415. [DOI] [PubMed] [Google Scholar]
- 33.Shealy RT, Murphy AD, Ramarathnam R, Jakobsson E, Subramaniam S. Sequence-function analysis of the K+-selective family of ion channels using a comprehensive alignment and the KcsA channel structure. Biophys J. 2003;84:2929–2942. doi: 10.1016/S0006-3495(03)70020-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noskov SY, Roux B. Importance of Hydration and Dynamics on the Selectivity of the KcsA and NaK Channels. J Gen Physiol. 2007;129:135–143. doi: 10.1085/jgp.200609633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asthagiri D, Pratt LR, Paulaitis ME. Role of fluctuations in a snug-fit mechanism of KcsA channel selectivity. J Chem Phys. 2006;125:24701–24706. doi: 10.1063/1.2205853. [DOI] [PubMed] [Google Scholar]
- 36.Dobler M. Ionophores and their structures. John Wiley & Sons, Inc; 1981. [Google Scholar]
- 37.Shemyakin MM, Ovchinnikov YA, Ivanov VT, Antonov VK, Vinogradova EI, Shkrob AM, Malenkov GG, Evstratov AV, Laine IA, Melink EI, Ryabova ID. Cyclodepsipeptides as Chemical Tools for Studying Ionic Transport Through Membranes. J Membr Biol. 1969;1:402–430. doi: 10.1007/BF01869790. [DOI] [PubMed] [Google Scholar]
- 38.Lauger P. Carrier-Mediated Ion Transport: Electrical relaxation experiments give insight into the kinetics of ion transport through artificial lipid membrane. Science. 1972;178:24–30. doi: 10.1126/science.178.4056.24. [DOI] [PubMed] [Google Scholar]
- 39.Rose MC, Henkens RW. Stability of sodium and potassium complexes of valinomycin. Biochim Biophys Acta. 1974;372:426–435. [Google Scholar]
- 40.Grell E, Funck T, Eggers F. In: Membranes Membranes. 3. Eisenman G, editor. Dekker NY; 1975. [PubMed] [Google Scholar]
- 41.Eisenman G, Aqvist J, Alvarez O. Free Energies underlying Ion Binding and Transport in Protein Channels: Free Energy Perturbation Simulations of Ion Binding and Selectivity for Valinomycin. J Chem Soc Faraday Trans. 1991;87:2099–2109. [Google Scholar]
- 42.Eisenman G, Alvarez O. In: Ionic Selectivity of Proteins: Lessons from Molecular Dynamics Simulations on Valinomycin Biomembrane Structure & Function - The State of the Art. Gaber BP, Easwaran KRK, editors. Academic Press; 1992. [Google Scholar]
- 43.Widom B. Potential-Distribution Theory and the Statistical Mechanics of Fluids. J Phys Chem. 1982;86:869–872. [Google Scholar]
- 44.Pratt LR, LaViolette RA. Quasi-chemical Theories of Associated Liquids. Mol Phys. 1998;95:909–915. [Google Scholar]
- 45.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 46.Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 47.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 48.Wang F, Zhao C, Polavarapu PL. A Study of the Conformations of Valinomycin in Solution Phase. Biopolymers. 2004;75:85–93. doi: 10.1002/bip.20103. [DOI] [PubMed] [Google Scholar]
- 49.Eisenman G, Ciani S, Szabo G. The Effects of the Macrotetralide Actin Antibiotics on the Equilibrium Extraction of Alkali Metal Salts into Organic Solvent. J Membr Biol. 1969;1:294–345. doi: 10.1007/BF01869787. [DOI] [PubMed] [Google Scholar]
- 50.Wyttenbach T, Batka JJ, Jr, Gidden J, Bowers MT. Host/guest conformations of biological systems: valinomycin/alkali ions. Intr J Mass Spec. 1999;193:143–152. [Google Scholar]
- 51.Baker AN, Sept D, Joseph S, Holst M, McCammon AJ. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Marcus Y. Thermodynamic functions of transfer of single ions from water to nonaqueous and mixed solvents: Part 1 - Gibbs free energies of transfer to nonaqueous solvents. Pure & Appl Chem. 1983;55:977–1021. [Google Scholar]
- 53.Ivanov VT, Ovchinnikov YA. Conformational states and biological activity of cyclic peptides. Tetra Rep. 1975;31:2177–2209. [Google Scholar]
- 54.Daggett V, Levitt M. Molecular dynamics simulations of helix denaturation. J Mol Biol. 1992;223:1121–1138. doi: 10.1016/0022-2836(92)90264-k. [DOI] [PubMed] [Google Scholar]
- 55.Avbelj F, Luo P, Baldwin RL. Energetics of the interaction between water and the helical peptide group and its role in determining helix propensities. Proc Natl Acad Sci USA. 2000;97:10786–10791. doi: 10.1073/pnas.200343197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Garcia AE, Sanbonmatsu KY. α-Helical stabilization by side chain shielding of backbone hydrogen bonds. Proc Natl Acad Sci USA. 2002;99:2782–2787. doi: 10.1073/pnas.042496899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varma S, Jakobsson E. The cPLA2 C2α Domain in Solution: Structure and Dynamics of its CA2+-activated and Cation-free States. Biophys J. 2007;92:966–976. doi: 10.1529/biophysj.106.091850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feinstein MB, Felsenfeld H. The Detection of Ionophorous Antibiotic-Cation Complexes in Water with Fluorescent Probes. Proc Natl Acad Sci USA. 1971;68:2037–2041. doi: 10.1073/pnas.68.9.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noskov SY, Roux B. Ion selectivity in potassium channels. Biophys Chem. 2006;124:279–291. doi: 10.1016/j.bpc.2006.05.033. [DOI] [PubMed] [Google Scholar]
- 60.Bayley H, Jayasinghe L. Functional engineered channels and pores (Review) Mol Membr Biol. 2004;21:209–20. doi: 10.1080/09687680410001716853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.