

Abstract
Hydroxymandelate synthase (HMS) catalyzes the committed step in the formation of para-hydroxyphenylglycine, a recurrent substructure of polycyclic non-ribosomal peptide antibiotics such as vancomycin. HMS uses the same substrates as 4-hydroxyphenylpyruvate dioxygenase (HPPD), 4-hydroxyphenylpyruvate (HPP) and O2, and also conducts a dioxygenation reaction. The difference between the two lies in the insertion of the second oxygen atom, HMS directing this atom onto the benzylic carbon of the substrate while HPPD hydroxylates the aromatic C1 carbon. We have shown that HMS will bind NTBC, a herbicide/therapeutic whose mode of action is based on the inhibition of HPPD. This occurs despite the difference in residues at the active site of HMS from those known to contact the inhibitor in HPPD. Moreover, the minimal kinetic mechanism for association of NTBC to HMS differs only slightly from that observed with HPPD. The primary difference is that three charge-transfer species are observed to accumulate during association. The first reversible complex forms with a weak dissociation constant of 520 µM, the subsequent two charge-transfer complexes form with rate constants of 2.7 s−1 and 0.67 s−1. As was the case for HPPD, the final complex has the most intense charge-transfer, is not observed to dissociate, and is unreactive towards dioxygen.
Keywords: Hydroxymandelate, Hydroxyphenylpyruvate, Dioxygenase, Dioxygen, Ferrous, Non-Heme
INTRODUCTION
Hydroxymandelate Synthase (HMS) catalyzes an α-keto acid dependent dioxygenase reaction, forming 4-hydroxymandelate (HMA) from 4-hydroxyphenylpyuruvate (HPP) in what appears to be an analogous reaction to that catalyzed by 4-hydroxyphenylpyuruvate dioxygenase (HPPD). HPPD forms homogentisate (2,5-dihydroxyphenylacetic acid, HG) by hydroxylation of the aromatic ring, inducing a 1,2-migration, while HMS hydroxylates instead the benzylic carbon (Scheme 1). HPPD has been studied quite extensively as it is a target of di- and triketone molecules that act as herbicides and therapeutics [1]. In plants these molecules prevent the synthesis of HG, which is precursor to the essential plant cofactors, plastoquinone and tocopherol [2–4]. In mammals, the inhibtion of HPPD can be used to completely alleviate the debilitating and/or lethal symptoms of specific inborn defects in tyrosine catabolism [5–8].
Scheme 1.
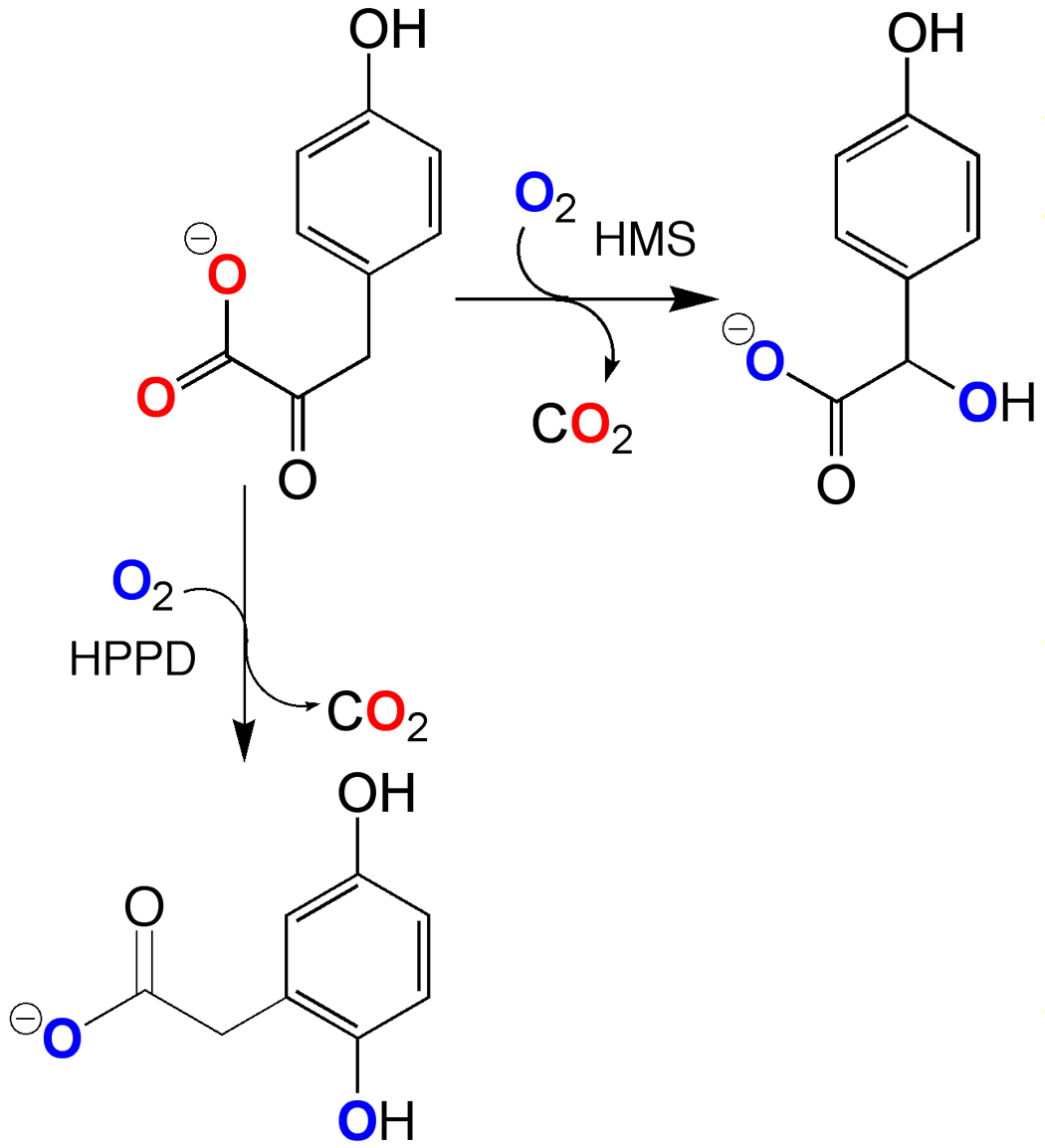
HMS is a relatively recent discovery [9, 10] that functions in a small number of bacteria to assist in the production of para-hydroxyphenylglycine, a recurrent substructure of non-ribosomal peptide antibiotics such as vancomycin and chloroeremomycin [11]. HMS has the same fold as HPPD [12]. The enzyme's tertiary structure can be divided into two domains, an N-terminal domain and a C-terminal domain that are topologically very similar. This fold was first observed for a number of extradiol type dioxygenases [13] and suggests that HMS and HPPD evolved from an extradiol dioxygenase precursor. The C-terminal domain is the catalytic portion of the structure and HMS and HPPD have significant identity and similarity in this domain. There are however, 24 residues in the C-terminus that are conserved only in the HMS primary structures. These are candidate residues critical to the delivery of the second oxygen atom. Recent mutagenesis studies have attempted to switch the catalytic specificity of HPPD to form HMA based largely on sequence comparison and structural insight. The conversion of a phenylalanine conserved only in HPPD structures (F364 in Figure 1) to valine or isoleucine, the latter of which is conserved in HMS, resulted in the formation of HMA in a fraction of total turnover [14, 15]. This phenylalanine residue had also been implicated as vital to catalysis in HPPD as it was one of two phenylalanine residues observed to sandwich the aromatic ring of the inhibitor 2-[2-nitro-4-(triflouromethyl)benzoyl]-1,3-cyclohexanedione (NTBC) that has a qualitative similarity to the structure of the substrate, HPP [16] (Figure 1). If the substrate phenol were stacked similarly, these phenyl rings could play a role in stabilizing a cationic ring species during electrophilic aromatic hydroxylation. Moreover, the complex of HPPD•Fe(II)•NTBC is ostensibly non-dissociable [17] even though the inhibitor makes no hydrogen bonding or ionic interactions with the enzyme. The only significant energetic contact observed is the bidentate contact made by the 5' and 7' oxygens of NTBC with the active site metal ion. It has been surmised recently that this contact alone does not account for the stability of the complex [18]. This has lead to the conclusion that the phenylalanines that stack with the relatively electron deficient aromatic ring of the inhibitor may contribute significant energy to the HPPD•Fe(II)•NTBC complex [16, 18]. On this basis we would therefore minimally predict that HMS would at least show diminished affinity for known HPPD inhibitors. In an added complication to this issue, the crystal structure of HMS derived from crystals grown in the presence of HPP indicates that although the density for the ligand is inconclusively assigned to either the substrate or product (HMA) it clearly adopts an entirely different orientation and conformation to that predicted by the HPPD inhibitor complex structures [12] (Figure 1). In this research note, we investigate whether HMS is subject to inhibition by NTBC, the paradigm example of molecules designed to inhibit HPPD.
Figure 1.

Comparison of the ligand binding sites in HPPD and HMS. The HPPD figure is derived from the crystal structure of the HPPD•Fe(II)•NTBC complex (1T47). NTBC is depicted in blue, while the predicted binding of HPP based on this structure is shown in red. The HMS portion of the figure depicts the HMS•Co(II)•HMA complex (insert PDB code). HMA is depicted in yellow and the predicted binding of HPP based on this structure is shown in red.
EXPERIMENTAL
Materials
HPP was purchased from Sigma-Aldrich Co. NTBC was a gift from Swedish Orphan Pharmaceuticals. The sodium salt of HEPES buffer was purchased from ACROS. HMS from Amycolatopsis orientalis was purified and concentrated according to previously published methods [12].
Extinction Coefficients
The extinction coefficient of apo-AoHMS was calculated to be 20 525 M−1 cm−1 at 280 nm by the method of Pace [19]. The molar extinction coefficient of NTBC was 20 550 M−1 cm−1 at 257 nm as previously reported [17]. The molar extinction coefficient of HPP was 3 400 M−1cm−1 at 276 nm at pH 7.0 [20].
Stopped-flow Measurements of Inhibitor Binding
The binding of NTBC was monitored under anaerobic conditions by stopped-flow spectrophotometry observing the accumulation of charge-transfer absorbance [17]. Anaerobic HMS•Fe(II) complex (48.3 µM final) was mixed with a range of pseudo-first-order concentrations of inhibitor (0.5 –3 mM final) that were each made anaerobic by sparging with argon for 10 min. Once the two reactants were mixed, accumulation of metal-to-inhibitor charge-transfer absorbance was monitored. For maximal time resolution and accurate measurement of rate constants, a single wavelength (450 nm) was initially monitored. The single wavelength data sets were fit to a linear combination of three exponentials to obtain rate constants and these values were used as initial estimates for subsequent diode-array data sets. To obtain the spectrum for each transient observed, UV/visible spectra (300–700 nm) were collected over the same time frame using a photo-diode-array attachment. Initial enzyme and inhibitor absorbance contributions were subtracted from the deconvoluted spectra of the charge-transfer species to obtain pure charge-transfer spectra for the inhibitor complexes.
Stability of The HMS•Fe(II)•NTBC complex
The degree of reversibility of inhibitor association was observed by monitoring the absorbance bands of the HMS•Fe(II)•NTBC complex arising from charge-transfer when exposed to a high dioxygen concentration. A solution of 200 µM HMS was made anaerobic in a 4 cm pathlength anaerobic cuvette by 45 exchanges of vacuum and argon. After the sample was made anaerobic, 195 µM Fe(II) (final) and 200 µM inhibitor (final) were each added from separate side arms to form 195 µM HMS•Fe(II)•NTBC complex. The solution was then exposed to 100 % oxygen and spectra were taken on a Cary 3 UV/Vis spectrophotometer over an 8 hr period at 4 °C.
RESULTS
Inhibitor Binding
The kinetics of formation of the HMS•Fe(II)•NTBC charge-transfer absorbance feature was monitored using a stopped-flow spectrophotometer by mixing limiting anaerobic HMS•Fe(II) complex with pseudo-first-order inhibitor concentrations (Figure 2). Unlike the association of NTBC with HPPD, HMS complexation exhibits three metal-centered binding events. The absorbance traces observed were fit to the sum of three exponentials. The magnitude of the observed rate constant for the first phase had a linear dependence in NTBC concentration. When the observed rate constant for this phase was plotted against NTBC concentration, the line obtained had a non-zero y-intercept (Figure 2 A & B). This suggests accumulation of an initial freely dissociable complex (Kd = 0.52±0.21 mM) that had an association rate constant (k1) of 1×105 ± 7×103 M−1s−1 and a dissociation rate constant (k−1) of 54 ± 22 s−1. The two subsequent phases involve a decrease in charge-transfer band intensity followed by an increase. Neither of these phases exhibited a dependence of the observed rate constant on the NTBC concentration (Figure 2C). The first of these concentration independent phases (k2) had an average rate constant of 2.68 ± 0.33 s−1 and the second (k3) had a rate constant of 0.69 ± 0.05 s−1. These data are consistent with the model depicted in Scheme 2. Deconvolution of photo-diode-array data sets acquired using a single high concentration of NTBC revealed the accumulation of the three absorbance bands, each having maxima around 450 nm. Subtracting the initial absorbances of the enzyme and inhibitor reveals that, other than the value of the extinction coefficient, no distinctive spectrophotometric difference between the three species can be observed in the limited spectral acquisition window (Figure 2D).
Figure 2.

The kinetics of complexation of the inhibitor NTBC with HMS. (A) Kinetic traces for the association of NTBC with the active site ferrous ion of HMS. Anearobic HMS•Fe(II) complex (48.3 µM final) was mixed with a range of pseudo-first-order concentrations of inhibitor (0.5 –5 mM final). Traces in ascending order are for final NTBC concentrations of 500 µM, 1000 µM, 2000 µM, 3000 µM, 4000 µM, 5000 µM. Each trace was fit to a sum of three exponentials according to Scheme 2 to determine rate constants. Traces have been offset from one another for the sake of clarity and otherwise converge to identical end-points. (B) The dependence of the first rate constant (kobs) on the concentration of NTBC (NB: error bars for each point are included). (C) The dependence of the second and third rate constants (k2, k3,) on the concentration of NTBC. Horizontal lines are average values for each rate constant. (D) Deconvoluted charge-transfer absorbance spectra that arise during NTBC binding events. Spectra are numbered in the order they are observed.
Scheme 2.

No decrease in the intensity of the charge-transfer band was observed over an eight hour period when the complex was exposed to ~2 mM dioxygen (data not shown). The holoenzyme (HMS.Fe(II) is prone to oxidation to the ferric form by dioxygen with a rate constant of 39 M−1s−1 (data not shown). Since it has been established that inhibitors of this type do not interact with the ferric form of these enzymes [17], this indicates that the dissociation rate constant for NTBC cannot be measured in the time frame of the methods used and that the complex is unreactive with dioxygen preventing oxidation of the active site ferrous ion, consistent with what had been observed for the HPPD.Fe(II).NTBC complex [17].
DISCUSSION
The association of known multi-ketone inhibitors such as NTBC with the active site metal ion of HPPD was believed to mimic the bidentate binding of the α-keto acid substrate, HPP [16, 18] (Figure 1). While NTBC does not posses an α-keto acid, it does contain a 1,3- diketone that forms bidentate contact with the active site metal ion [16]. When NTBC is added under anaerobic conditions to the HPPD•Fe(II) complex, a charge-transfer absorbance band at 450 nm was observed [17]. Calculations based on the structure of the HPPD•Fe(II)•NTBC inhibitor complex suggest that the bidentate association of the 1,3-diketone of NTBC does not contribute substantially to the binding energy [18]. All other interactions of the inhibitor with the enzyme appear to be Van der Waals in origin in that no salt bridges or hydrogen bonds are observed. So the energetic basis of tight binding of the inhibitor NTBC remains unknown. However, the HPPD•Fe(II)•NTBC complex structure indicates that the 2-nitro-4- trifluoromethylphenyl substituent of NTBC is sandwiched in a staggered arrangement between two conserved phenylalanines and this has been proposed to be one of the key binding interactions that may also facilitate the aromatic hydroxylation of the substrate phenol if it were bound in this position [1, 18] (Figure 1). One of these phenylalanines (F364) can only stack in this fashion when the C-terminal helix on which it resides pivots out of the active site by 40°. Sequence alignments indicate that the equivalent residue in HMS is an isoleucine [14]. The recently published structure of the HMS product complex may provide hints as to the binding position of HPP in this enzyme and confirms the importance of the isoleucine at this position to phenolic localization [12]. However, the predicted orientation of the substrate α-keto acid in this structure is ~120° from that predicted from the HPPD•Fe(II)•NTBC crystal structure. Therefore the kinetics of binding of HMS with triketone inhibitors affords an opportunity to assess whether the isoleucine will perturb inhibitor binding and/or if a phenylalanine at this position is a requirement for tight inhibitor association.
Overall the complexation of HMS with NTBC presented here is quite similar to that observed for HPPD [17, 21]. Like the HPPD•Fe(II)•NTBC complex, the HMS•Fe(II)•NTBC complex is uncreative with dioxygen. Magentic circular dichroism spectroscopy of the HPPD•Fe(II)•NTBC complex has indicated that the metal ion is a mixture of 5 and 6-coordinate species [18] as was observed in the X-ray crystal structure of the complex where it was also observed that access to the metal ion is unimpeded by any active site residue [16]. The suppression of dioxygen reactivity, therefore, does not result solely from coordination number or steric hinderance but, for reasons we do not yet understand, is an intrinsic property of the metal ion when bound to the inhibitor. Both HMS and HPPD exhibit three complexation events in the presence of NTBC. Moreover, the intensity of the charge-transfer absorbance is maximal in the final species observed, as was the case for HPPD. We had previously suggested that this was a result of the lewis acidity of the metal ion facilitating the deprotonation of the hydroxyl of the exocyclic enol form of the inibitor that dominates (ca. 96%) in aqueous solution at pH 7.0 [17]. The final complex is then proposed to be the bidentate association of the enolate form of NTBC with the ferrous ion, though it is not clear that this species is expected to yield charge-transfer absorbance bands of greater intensity a priori. The principle difference is that the initial weak, reversible complex observed in HMS has charge-transfer absorbance transitions whereas no transitions of this type are observed in the initial complex with HPPD. Therefore, three metal centered binding events occur to give an ostensibly irreversible complex of NTBC with HMS•Fe(II). No evidence is presented here for the structural origin of the individual species observed during binding, however they do suggest that slow, well delineated conformational changes are required to form the resting inhibitor complex. The HMS inhibitor complex is highly stable despite the lack of one of the two phenylalanines expected to sandwich the 2-nitro-4- trifluoromethylphenyl group. This suggests that π-stacking with this phenylalanine is not a primary determinant of the stability of the inhibitor complex and shape complementarity must allow for a net Van der Waals interaction energy that prevents NTBC and similar inhibitors from dissociating at a measurable rate.
ACKNOWLEDGEMENTS
Our laboratory has collaborated with Ed Solomon and his graduate students Michael Neidig and Christina Brown in the study of HPPD. As a direct result of this collaboration, we know vastly more about the substrate and inhibitor complexes of this curious enzyme. The great strength of the Solomon group is an ethic that drives meticulous, exhaustive data interpretation. We are indebted to Ed's friendly professionalism and his rare interpretive abilities.
†This research was supported in part by a National Institutes of Health Grant (DK59551) to G.R.M. and by a UWM Research Growth Initiative Grant to G.R.M.
ABREVIATIONS
- HMS
hydroxymandelate synthase
- HPPD
(4-hydroxyphenyl)pyruvate dioxygenase
- HMA
hydroxymandelate
- HPP
(4-hydroxyphenyl)pyruvate
- PHPG
para-hydroxyphenylglycine
- HEPES
(N-(2-hydroxyethyl) piperizine-N’-(2-ethane sulfonic acid))
- NTBC
2-(2-nitro-4-trifluoromethylbenzoyl)-1,3-cyclohexanedione
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Moran GR. Arch Biochem Biophys. 2005;433:117–128. doi: 10.1016/j.abb.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell G, Bartlett DW, Fraser TE, Hawkes TR, Holt DC, Townson JK, Wichert RA. Pest Manag Sci. 2001;57:120–128. doi: 10.1002/1526-4998(200102)57:2<120::AID-PS254>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 3.Hall MG, Wilks MF, Provan WM, Eksborg S, Lumholtz B. Br J Clin Pharmacol. 2001;52:169–177. doi: 10.1046/j.0306-5251.2001.01421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sutton P, Richards C, Buren L, Glasgow L. Pest Manag Sci. 2002;58:981–984. doi: 10.1002/ps.554. [DOI] [PubMed] [Google Scholar]
- 5.Lindblad B, Lindstedt S, Steen G. Proc Natl Acad Sci U S A. 1977;74:4641–4645. doi: 10.1073/pnas.74.10.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russo PA, Mitchell GA, Tanguay RM. Pediatr Dev Pathol. 2001;4:212–221. doi: 10.1007/s100240010146. [DOI] [PubMed] [Google Scholar]
- 7.Phornphutkul C, Introne WJ, Perry MB, Bernardini I, Murphey MD, Fitzpatrick DL, Anderson PD, Huizing M, Anikster Y, Gerber LH, Gahl WA, Engl N. J Med. 2002;347:2111–2121. doi: 10.1056/NEJMoa021736. [DOI] [PubMed] [Google Scholar]
- 8.Lindstedt S, Holme E, Lock EA, Hjalmarson O, Strandvik B. Lancet. 1992;340:813–817. doi: 10.1016/0140-6736(92)92685-9. [DOI] [PubMed] [Google Scholar]
- 9.Choroba OW, Williams DH, Spencer JB. J Am Chem Soc. 2000;122:5389–5390. [Google Scholar]
- 10.Li TL, Choroba OW, Charles EH, Sandercock AM, Williams DH, Spencer JB. Chemical communications (Cambridge, England) 2001:1752–1753. doi: 10.1039/b103548g. [DOI] [PubMed] [Google Scholar]
- 11.Hubbard BK, Thomas MG, Walsh CT. Chem Biol. 2000;7:931–942. doi: 10.1016/s1074-5521(00)00043-0. [DOI] [PubMed] [Google Scholar]
- 12.Brownlee J, He P, Moran GR, Harrison DHT. Biochemistry submitted. 2007 [Google Scholar]
- 13.Han S, Eltis LD, Timmis KN, Muchmore SW, Bolin JT. Science. 1995;270:976–980. doi: 10.1126/science.270.5238.976. [DOI] [PubMed] [Google Scholar]
- 14.Gunsior M, Ravel J, Challis GL, Townsend CA. Biochemistry. 2004;43:663–674. doi: 10.1021/bi035762w. [DOI] [PubMed] [Google Scholar]
- 15.O'Hare HM, Huang F, Holding A, Choroba OW, Spencer JB. FEBS Lett. 2006;580:3445–3450. doi: 10.1016/j.febslet.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 16.Brownlee J, Kayunta J-W, Harrison DHT, Moran GR. Biochemistry. 2004;43:6370–6377. doi: 10.1021/bi049317s. [DOI] [PubMed] [Google Scholar]
- 17.Kavana M, Moran GR. Biochemistry. 2003;42:10238–10245. doi: 10.1021/bi034658b. [DOI] [PubMed] [Google Scholar]
- 18.Neidig ML, Decker A, Kavana M, Moran GR, Solomon EI. Biochem Biophys Res Commun. 2005;338:206–214. doi: 10.1016/j.bbrc.2005.08.242. [DOI] [PubMed] [Google Scholar]
- 19.Pace NC, Vajdos F, Fee L, Grimsley G, Gray T. Prot.Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson-Winters K, Purpero VM, Kavana M, Nelson T, Moran GR. Biochemistry. 2003;42:2072–2080. doi: 10.1021/bi026499m. [DOI] [PubMed] [Google Scholar]
- 21.Purpero VM, Moran GR. Biochemistry. 2006;45:6044–6055. doi: 10.1021/bi052409c. [DOI] [PubMed] [Google Scholar]