

Abstract
The mossy fiber to CA3 pyramidal cell synapse (mf-CA3) provides a major source of excitation to the hippocampus. Thus far, these glutamatergic synapses are well recognized for showing a presynaptic, NMDA receptor-independent form of LTP which is expressed as a long-lasting increase of transmitter release. Here, we show that in addition to this “classical” LTP, mf-CA3 synapses can undergo a form of LTP characterized by a selective enhancement of NMDA receptor-mediated transmission. This potentiation requires coactivation of NMDA and mGlu5 receptors, and a postsynaptic calcium rise. Unlike classical LTP, expression of this novel mossy fiber LTP is due to a PKC-dependent recruitment of NMDA receptors specifically to the mf-CA3 synapse via a SNARE-dependent process. Having two mechanistically different forms of LTP may allow mf-CA3 synapses to respond with more flexibility to the changing demands of the hippocampal network.
INTRODUCTION
One of the principal inputs to the hippocampus proper is the mossy fiber (mf) pathway. Mfs are the axons of dentate granule cells which project to the proximal dendrites of CA3 pyramidal neurons and provide powerful glutamatergic synaptic excitation (Henze et al., 2000; Henze et al., 2002). Excitatory neurotransmission at the mf to CA3 pyramidal cell synapse (mf-CA3) is mediated postsynaptically by three types of ionotropic glutamate receptors: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA), N-methyl-D-aspartate (NMDA) and kainate (KA) receptors. At many excitatory synapses, activation of NMDA receptors (NMDARs) induces classical forms of long-term potentiation or depression (LTP/LTD) (Bliss and Collingridge, 1993; Malenka and Bear, 2004) that are mainly due to postsynaptic changes in AMPAR-mediated transmission (Bredt and Nicoll, 2003; Collingridge et al., 2004; Malinow and Malenka, 2002). These NMDAR-dependent, postsynaptic forms of plasticity are not expressed by the mf-CA3 synapse (Nicoll and Schmitz, 2005). Rather, these synapses are well known for expressing presynaptic forms of LTP and LTD, manifested as long-term changes in the probability of glutamate release. In addition, mf-CA3 synapses show uniquely robust frequency facilitation (Salin et al., 1996) and post-tetanic potentiation (Griffith, 1990), both presynaptic forms of short-term plasticity (Zucker and Regehr, 2002). Thus, at present, activity-dependent changes in the efficacy of the mf-CA3 synapse are mainly understood to have a presynaptic site of expression.
While mf-CA3 synapses lack the classical forms of NMDAR-dependent, postsynaptic plasticity, functional NMDARs are nonetheless present at these synapses, though their role is unclear. Initial studies using quantitative autoradiography (Monaghan et al., 1983) and high affinity fluorescent probes (Benke et al., 1993) reported a relatively low density of NMDAR binding sites in stratum lucidum, the main synaptic field of mfs. Subsequent immunohistochemical studies demonstrated NMDARs to be clearly present in s. lucidum, albeit at a lower density than AMPARs (Fritschy et al., 1998; Watanabe et al., 1998). This localization was refined by electron microscopy to the postsynaptic density of the mf-CA3 synapse (Petralia et al., 1994; Takumi et al., 1999). Furthermore, electrophysiological examination has shown that mf stimulation results in a substantial NMDAR-mediated postsynaptic current, small only in comparison to the massive AMPAR-mediated component (Jonas et al., 1993; Spruston et al., 1995; Weisskopf and Nicoll, 1995). NMDARs at the mf-CA3 synapse are reported to be modulated by endogenously released zinc (Vogt et al., 2000), and their activation has been recently associated with a transient depression of KAR-mediated transmission (Rebola et al., 2007). Beyond these data, much about the role of NMDARs at the mf-CA3 synapse remains unknown.
Although, in general, AMPARs mediate the bulk of excitatory transmission, NMDARs can also contribute to synaptic transmission (Daw et al., 1993) and neuronal excitability (Isaacson and Murphy, 2001; Sah et al., 1989). NMDARs may play a key role in the persistent activity of neural assemblies as well (Major and Tank, 2004). Because of the slow kinetics of NMDAR-EPSCs, increases in this component may contribute significantly to postsynaptic depolarization, particularly during repetitive synaptic activity. Given the high Ca2+ permeability of NMDARs and the well-known actions of Ca2+ as a second messenger, it is expected that long-term changes in NMDAR-transmission may have important functional consequences for Ca2+-dependent cellular processes, including “metaplasticity”, a change in the inducibility of synaptic plasticity (Abraham and Bear, 1996). Growing evidence from other synapses indicates that NMDAR-mediated transmission is more dynamic than originally thought (Lau and Zukin, 2007; Nong et al., 2004; Wenthold et al., 2003). However, compared to AMPARs, the mechanisms underlying NMDAR plasticity are poorly understood. In this study, we report that the mf-CA3 synapse can undergo a form of plasticity that is expressed postsynaptically as a selective, long-lasting increase in NMDAR-neurotransmission. This potentiation requires postsynaptic calcium, and is likely due to a PKC-dependent recruitment of NMDARs to the synapse. This novel form of mfLTP may provide a dynamic and potentially powerful mechanism for regulating mf-CA3 synaptic efficacy.
RESULTS
A novel form of LTP at mf-CA3 synapses expressed by NMDARs
We examined the effect of a short tetanus, 24 stimuli at 25 Hz, on the AMPAR- and the NMDAR-mediated components of the mf-CA3 EPSC (AMPA-EPSC Vhold = −60/−70 mV; NMDA-EPSC Vhold = +30/+40 mV, see methods) in 100 μM picrotoxin and 3 μM CGP55845 to block GABAA and GABAB receptors, respectively. To better isolate the mf-CA3 EPSC from recurrent associational/commissural (i.e. polysynaptic) EPSCs, we reduced cellular excitability, and thus inhibited the epileptiform activity to which the CA3 region is especially prone, using an extracellular solution containing 4 mM Ca2+ and 4 mM Mg2+. Unexpectedly, this short tetanus triggered a selective LTP of mf-CA3 NMDAR-EPSCs but not of AMPAR-EPSCs (NMDAR-EPSC: 216 ± 29% of baseline, p<0.001, AMPAR-EPSCs 105 ± 5% of baseline, p>0.3, n = 8) (Fig. 1A,B). We call this form of synaptic plasticity NMDAR mossy fiber LTP (NMDAR-mfLTP). In a separate experiment, to determine whether this potentiation was indeed selective for NMDAR-EPSCs, we tested the effect of the short-tetanus on mf-CA3 KAR-EPSCs (Castillo et al., 1997; Vignes and Collingridge, 1997). To isolate KAR-EPSCs, these experiments were performed in presence of the AMPAR selective antagonist GYKI 53655 (30 μM) in addition to picrotoxin and CGP55845 while voltage-clamping CA3 pyramidal cells to −70 mV. Under these recording conditions, we found that the same induction protocol that triggers robust NMDAR-mfLTP does not trigger any potentiation of KAR-EPSCs (101 ± 3% of baseline, n = 4 cells, p>0.5) (Fig. 1C). The selective potentiation of the NMDAR-mediated component strongly suggests a postsynaptic mechanism of expression for NMDAR-mfLTP indicating that it constitutes a novel form of plasticity at the mf-CA3 synapse.
Figure 1.
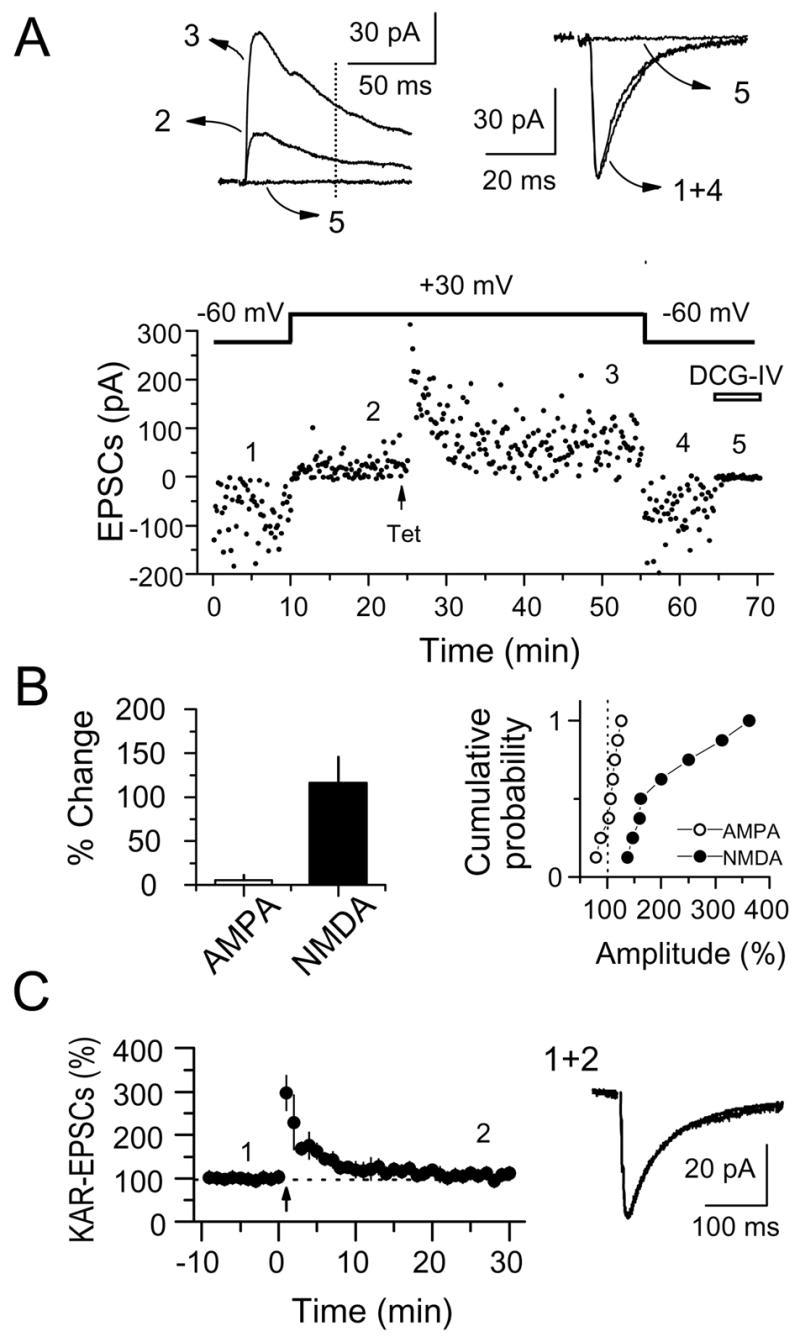
Selective LTP of NMDAR-EPSCs at mf-CA3 synapses (NMDAR-mfLTP). (A) Representative experiment in which both NMDAR- and AMPAR-EPSCs from a CA3 pyramidal cell were monitored over time. LTP was induced by a single tetanus (Tet) of 24 stimuli at 25 Hz. At the end of the experiment, the mGluR2 agonist DCG-IV (1 μM) was applied to confirm that EPSCs were indeed mediated by mossy fibers. NMDAR-EPSC amplitudes were measured 50 ms post-stimulus (vertical dotted line) to avoid the contribution of the AMPAR-mediated component. Averaged sample traces taken at times indicated by numbers are shown on top. (B) Summary plots showing the selective potentiation of NMDAR-EPSCs but not AMPAR-EPSCs (8 cells). (C) Same induction protocol (24 pulses at 25 Hz) failed to increase KAR-EPSCs (4 cells). Average traces before and after tetanus are shown on the right.
In the previous experiments, NMDAR-mfLTP was induced under rather non-physiological recording conditions (i.e. voltage-clamping at +30/+40 mV, room temperature, high divalent concentration). We next used several approaches to trigger NMDAR-mfLTP under more physiological conditions, all induced by the same tetanus used in the previous experiments (24 stimuli at 25 Hz). While monitoring NMDAR-EPSCs at Vhold = +30 mV, we delivered the induction tetanus while voltage clamping to -60 mV, close to the normal resting membrane potential of CA3 pyramidal cells. These experiments were performed in presence of 20 μM NBQX, 100 μM picrotoxin, and 3 μM CGP55845 to block AMPA/KA, GABAA and GABAB receptors respectively, yielding significant NMDAR-mfLTP (257 ± 33% of baseline, n = 4, p<0.005) (Fig. 2A). During the tetanus, a substantial inward current was observed, confirmed in separate experiments to be NMDAR-dependent by complete block with the NMDAR-selective antagonists 20 μM CPP (Fig. 2A, inset). While most of our studies were performed at room temperature (25 °C), NMDAR-mfLTP can also occur at 35 °C (208 ± 31% of baseline, n = 6, p <0.001)(Fig. 2B). NMDAR-mfLTP can also be elicited using low -more physiological- extracellular concentrations of Ca2+ (1.7 mM) and Mg2+ (1.7 mM) (275 ± 41% of baseline, n = 5, p<0.001)(Fig. 2C). In addition, we took a less invasive extracellular recording approach and monitored NMDAR-mediated field potentials (NMDAR-fEPSPs). We tracked the amplitude of synaptic responses evoked by 200 Hz bursts of three test stimuli in a low [Mg2+] extracellular solution (0.1 mM) in the continuous presence of 20 μM NBQX and 3 μM CGP55845 (picrotoxin was omitted in these experiments to avoid epileptiform activity). With these manipulations we were able to induce NMDAR-mfLTP (290 ± 40% of baseline, n = 3, p<0.005) (Fig. 2D). Finally, we monitored NMDAR-EPSPs using whole-cell recordings in current-clamp mode, with an extracellular solution containing divalent concentrations more typically used in electrophysiological experiments, namely, 2.5 mM Ca2+ and 1.3 mM Mg2+. Resting membrane potential was maintained near −65 mV by current injection. NMDAR-EPSPs were elicited with brief 25 Hz bursts of 5 stimuli in 20 μM NBQX, 100 μM picrotoxin, and 3 μM CGP55845. Under these conditions, we found significant NMDAR-mfLTP (171 ± 19% of baseline, n = 4, p<0.005) (Fig. 2E). Together, these results show that NMDAR-mfLTP is a robust phenomenon that can be induced under several near-physiological recording conditions.
Figure 2.
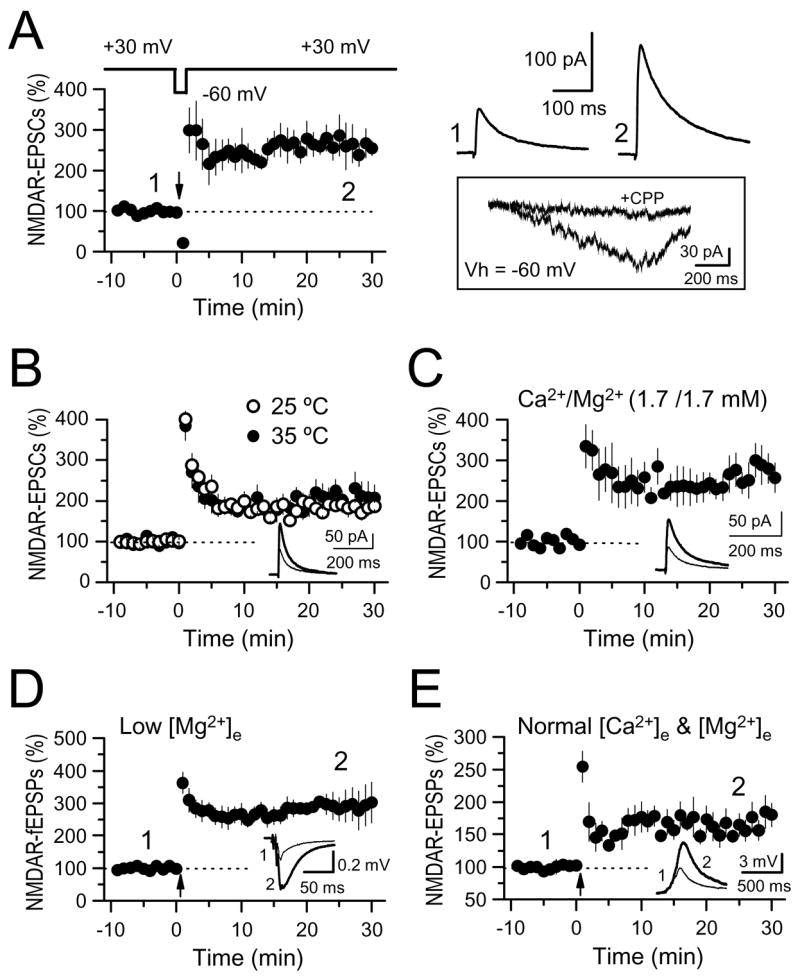
NMDAR-mfLTP can be induced under more physiological conditions. (A) mf NMDAR-EPSCs were monitored at Vh = +30 mV, but the induction tetanus was delivered at Vh = β60 mV (4 cells). Averaged sample traces taken at times indicated by numbers are shown on the top right. Below that (box) are representative traces of the NMDAR-mediated inward current induced by the tetanus (24 stimuli, 25 Hz) before and after application of 20 μM CPP. Stimulation artifacts were deleted for clarity. (B) Summary plots showing that the magnitude of NMDAR-mf LTP obtained at 25°C (6 cells) and 35°C (6 cells) is virtually identical. (C) Summary plots showing NMDAR-mfLTP while using 1.7 mM Ca2+ and 1.7 mM Mg2+ in the extracellular solution (5 cells). (D) NMDAR-mfLTP was induced while monitoring field potential amplitude (NMDAR-fEPSPs) in a 4.0 mM Ca2+, 0.1 mM Mg2+ extracellular solution (3 slices). Each fEPSP was induced with a burst of 3 stimuli at 200 Hz. (E) NMDAR-mfLTP was induced while recording NMDAR-EPSPs in current clamp mode, and in the presence of more physiological extracellular concentrations of Ca2+ and Mg2+ (2.5 and 1.3 mM, respectively) (4 cells). Each EPSP was induced with a burst of 5 stimuli at 25 Hz. Insets of B, C, D and E: averaged sample traces from before and after NMDAR-mfLTP are superimposed.
We next examined whether the induction protocol responsible for LTP of NMDAR-EPSCs at mf-CA3 synapses could trigger any form of plasticity at the associational/commissural (ac) inputs to CA3 cells (ac-CA3 synapses). In these experiments we alternately evoked mf-CA3 and ac-CA3 NMDAR-EPSCs in the same CA3 pyramidal cell. NMDAR-EPSCs were monitored in 20 μM NBQX, 100 μM picrotoxin, and 3 μM CGP55845 while voltage clamping at +30/+40 mV (these recording conditions were used throughout the rest of this study). To investigate potential heterosynaptic spread of NMDAR-mfLTP to nearby ac-CA3 synapses, we placed the ac stimulating pipettes <50 μm from s. lucidum. We found that NMDAR-LTP occurs at mf-CA3 but not at ac-CA3 synapses (mf-CA3: 189 ± 20% of baseline, p<0.001; ac-CA3: 107 ± 8% of baseline, p>0.5, n = 5) (Fig. 3A,B), Previous studies suggested that a stronger tetanus is required for NMDAR-LTP versus AMPAR-LTP at Schaffer collateral/commissural fiber inputs to CA1 pyramidal cells (Sch-CA1) (Aniksztejn and Ben-Ari, 1995; Bayazitov and Kleschevnikov, 2000; Berretta et al., 1991). However, doubling the stimulus intensity during tetanus application to ac fibers triggered no NMDAR-LTP at ac-CA3 synapses (109 ± 9% of baseline, n = 3, p>0.3, data not shown). Together, these observations indicate that NMDAR-LTP, at least when induced by a short tetanus, does not occur at, or spread to, ac-CA3 synapses. To address the question of whether spread to adjacent, naïve mf inputs on the same CA3 pyramidal cell occurs, we monitored two independent mf inputs (see Supplemental Experimental Procedures online), and only one of these inputs received the induction tetanus (Fig. 3C). We found that only those mf inputs receiving the induction tetanus underwent NMDAR-mfLTP, whereas naïve inputs showed no plasticity (Tet: 188 ± 30%, p<0.01; Naïve: 115 ± 18%, p>0.1, n = 4)(Fig. 3D), indicating that, at least under these experimental conditions, NMDAR-mfLTP is input-specific.
Figure 3.

NMDAR-mfLTP is input-specific. (A) Time-course of a representative experiment in which NMDAR-EPSCs evoked by alternating stimulation of mfs (mf, top panel) and associational-commissural fibers (ac, bottom panel) were monitored in the same CA3 pyramidal cell. Tetanic stimulation (vertical arrow: 24 stimuli, 25 Hz) induced LTP at mf-CA3 synapses only. Averaged sample traces taken at times indicated by numbers (upper right). (B) Summary plot of 5 cells recorded as in A showing robust LTP of NMDAR-EPSCs at mf-CA3 synapses only. (C) Representative experiment in which NMDAR-EPSCs were evoked by alternatively stimulating two independent mf pathways. At the time point indicated by the vertical arrow, one pathway received a tetanus of 24 stimuli at 25 Hz (Tet) whereas the naïve pathway served as control. Averaged sample traces taken at times indicated by numbers (right). (D) Summary plot of 4 cells as recorded in C showing that NMDAR-mfLTP was induced at mf-CA3 synapses that received the induction tetanus but not at naïve synapses.
To further characterize the induction of NMDAR-mfLTP, we tested several stimulation frequencies (2, 5, 25, 100 Hz) and found that, among the frequencies tested, 25 Hz triggers maximal NMDAR-mfLTP (see Fig. S1A in the Supplemental Data available with this article online). The decrease in NMDAR-mfLTP induced by the 100 Hz tetanus was associated with a reduction in the NMDAR-mediated charge transfer during tetanus application (25 Hz: 155 ± 14, n = 8; 100 Hz: 93 ± 15; n = 4, p <0.05) (Fig. S1B). As discussed below (Fig. 6A–D), this decrease in charge transfer could account for the reduction of NMDAR-mfLTP induced by high frequency bursts (i.e. 100 Hz). Having established an optimal frequency for induction, we finally explored whether NMDAR-mfLTP can be saturated by successive tetanization. To this end, we delivered three tetani separated by 20 min. We found that a second and a third bout of stimulation produced an incremental enhancement of NMDAR-mfLTP (Fig. S1C), while the magnitude of enhancement decreased with each additional bout (Fig. S1D). These results show that NMDAR-mfLTP is not saturated by a single tetanus and likely reaches its maximal magnitude with few (~3) bouts of tetanus.
Figure 6.

NMDAR-mfLTP requires the coactivation of NMDA and mGluR5 receptors, and Ca2+ release from postsynaptic IP3-sensitive stores. (A) Transiently blocking NMDARs during tetanus (7 cells) inhibited induction of NMDAR-mfLTP. The NMDAR antagonist CPP (5 μM, horizontal bar) was bath applied for 4 min to slices receiving the tetanus (Tet) and also to naïve slices (4 cells) to map the rate of CPP washout. NMDAR-mfLTP in control slices (7 cells) is superimposed for comparison. (B) Representative NMDAR-mediated currents induced by the tetanus under control conditions and in the presence of 5 μM and 20 μM CPP. Note that 5 μM CPP, a competitive antagonist, was insufficient to completely block these currents, whereas 20 μM CPP produced a complete block. (C) Same procedure as in panel (A), but using enough CPP (20 μM) to completely block NMDAR current during the tetanus (5 cells). (D) Summary graph showing that while incomplete blockade of NMDARs (measured as charge transfer) during tetanus application reduced NMDAR-mfLTP only partially, full blockade of NMDARs abolished NMDAR-mfLTP completely. White bars indicate the magnitude of LTP, black bars indicate charge transfer normalized to control conditions (e.g. in the absence of CPP). (E) Activation of mGluR5, but not mGluR1, is required for NMDAR-mfLTP. Summary plot comparing three experimental groups in which the induction tetanus was delivered in the presence of the mGluR5 antagonist MPEP (4 μM, 6 cells), the mGluR1 antagonist CPCCOEt (100 μM, 4 cells), or in interleaved control slices (n = 6 cells). (F) Effects of GDP-βS (2 mM) postsynaptic loading on NMDAR-mfLTP. GDP-βS was allowed to diffuse into CA3 cells (n = 4) at least for 30 minutes before tetanus. For comparison, NMDAR-mfLTP elicited in interleaved control experiments (n = 4) is superimposed. (G) Summary plots showing that bath application of 50 μM DHPG (in the presence of 100 μM CPCCOEt) induced weak LTP of mf NMDAR-EPSCs (Control, 8 cells). In separate set of experiments, this potentiation was occluded by prior induction of NMDAR-mfLTP (after Tet, 6 cells). (H) NMDAR-mfLTP requires Ca2+ release from IP3-sensitive Ca2+ stores. Summary plots comparing the magnitude of NMDAR-mfLTP in hippocampal slices treated with cyclopiazonic acid (CPA, 5 cells) and in interleaved control slices (5 cells). Test hippocampal slices were incubated in 30 μM CPA for at least 30 min before and continuously during recordings. (I) Summary plots showing the effects of the IP3-receptor blocker heparin (2.5 mg/ml) or the ryanodine receptor blocker ruthenium red (RR) (20 μM) on NMDAR-mfLTP. Heparin (5 cells) and RR (4 cells) were added to the recording pipette.
Postsynaptic Ca2+ is required for the induction of NMDAR-mfLTP
To investigate the role of postsynaptic Ca2+ in the induction of NMDAR-mfLTP, CA3 pyramidal cells were loaded with 20 mM BAPTA through the patch pipette. This manipulation completely abolished induction of NMDAR-mfLTP by a short tetanus of 24 stimuli at 25 Hz (control 194 ± 26% of baseline, n = 6; in BAPTA 110 ± 9% of baseline, n = 6) (Fig. 4A). A longer tetanus (125 stimuli, 25 Hz), commonly used to induce classical presynaptic mfLTP (Castillo et al., 2002; Hsia et al., 1995; Schmitz et al., 2003), was only partially blocked by intracellular BAPTA (control: 409 ± 66% of baseline, n = 6; in BAPTA: 220 ± 21% of baseline, n = 6) (Fig. 4B). Thus, while a short tetanus induces postsynaptic Ca2+-dependent NMDAR-mfLTP, a longer tetanus seems to trigger two complementary forms of LTP: BAPTA-sensitive NMDAR-mfLTP and BAPTA-resistant classical presynaptic mfLTP (Nicoll and Schmitz, 2005). This differential sensitivity to postsynaptic BAPTA strongly suggests that different protocols can induce mechanistically different forms of mfLTP. Moreover, these results suggest that both types of plasticity can coexist at the same synapse.
Figure 4.
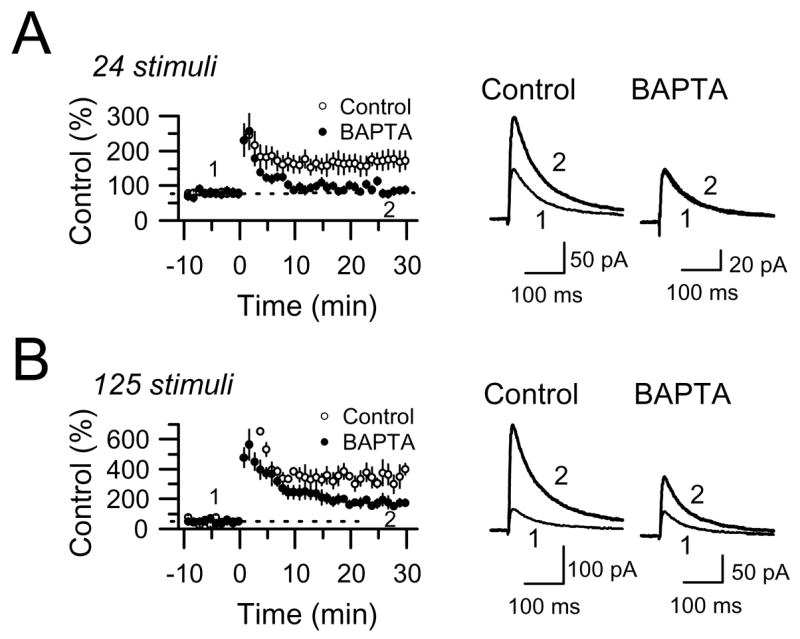
Role of postsynaptic Ca2+ in the induction of NMDAR-mfLTP. (A) Loading CA3 pyramidal cells with 20 mM BAPTA abolished NMDAR-mfLTP induced by 24 stimuli at 25 Hz (6 cells in BAPTA and 6 control cells). (B) A longer induction tetanus (125 stimuli, 25 Hz) triggered a larger potentiation with BAPTA-sensitive and BAPTA-insensitive components (6 cells in BAPTA and 6 control cells).
Unlike classical mfLTP, NMDAR-mfLTP is NOT associated with an increase in the probability of transmitter release, and is independent of RIM1α
Classical mfLTP is one of the best characterized examples of presynaptic LTP. While some controversy regarding its induction mechanism still remains, there is universal agreement that the expression of mfLTP is due to an increase in neurotransmitter release (Nicoll and Schmitz, 2005) which requires the active zone protein RIM1α (Castillo et al., 2002). Consistent with a presynaptic mechanism of expression, mfLTP is associated with a decrease in paired-pulse facilitation (PPF). Changes in PPF reliably track manipulations that affect transmitter release (Manabe et al., 1993; Zucker and Regehr, 2002). In addition, increased probability of transmitter release is usually accompanied by a decrease in the coefficient of variation (CV) (Faber and Korn, 1991; Zucker and Regehr, 2002). Remarkably, induction of NMDAR-mfLTP with a short tetanus caused no lasting change in PPF (97 ± 5%, n = 7, p>0.5) and no change in CV (control: 0.35 ± 0.02, 24 stimuli: 0.35 ± 0.03, n = 18, p>0.5). However, when CA3 cells were loaded with 20 mM BAPTA to block NMDAR-mfLTP, classical mfLTP induced by a long tetanus was accompanied by a sustained reduction in both PPF (70 ± 4% of baseline, n = 4, p<0.01) and CV (control: 0.36 ± 0.04, 125 stimuli: 0.26 ± 0.03, n = 8, p<0.01) (Fig. 5A–C). These findings strongly suggest that NMDAR-mfLTP does not involve changes in transmitter release.
Figure 5.
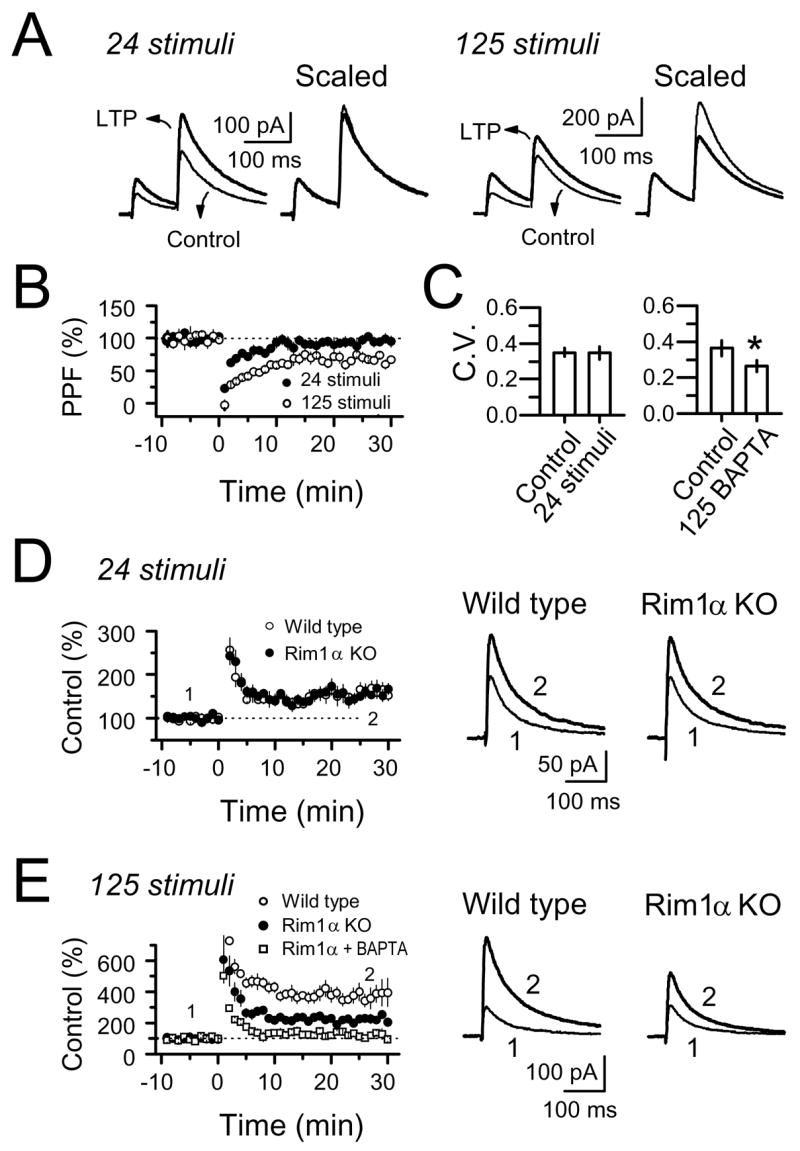
NMDAR-mfLTP (24 stimuli, 25 Hz) differs from the classical presynaptic form of LTP (125 stimuli, 25 Hz). LTP induced by 24 stimuli had no lasting effect on paired-pulse facilitation (PPF) or coefficient of variation (CV), but LTP induced by 125 stimuli significantly reduced both. (A) Averaged traces before and after LTP induced by either 24 or 125 stimuli. (B) PPF. (C) CV. (D) RIM1α deletion had no effect on NMDAR-mfLTP (4 WT mice and 4 RIM1α KO mice). (E) A longer tetanus (125 stimuli) induced robust mfLTP which included a RIM1α-dependent component (presynaptic mfLTP) and a RIM1α-independent component (NMDAR-mfLTP) (4 WT mice and 4 RIM1α KO mice). This latter component was abolished by BAPTA (4 cells/2 mice).
To further compare the expression mechanism of classical mfLTP and NMDAR-mfLTP, we tested RIM1α-KO mice, known to lack classical mfLTP (Castillo et al., 2002). We found a virtually identical magnitude of NMDAR-mfLTP in RIM1α KO and wild-type mice (WT: 161 ± 15%, n = 7 cells, 4 mice vs. KO: 160 ± 13%, n = 8 cells, 4 mice, p>0.5) (Fig. 5D). Here, as previously observed in rat hippocampal slices (Fig. 4B), a longer tetanus (125 stimuli at 25 Hz) induced a much larger LTP (~four-fold potentiation), presumably comprised of both classical and NMDAR-only parts. Only a component of this total potentiation was abolished in RIM1α KO mice (WT: 376 ± 68%, n = 7 cells, 4 mice, vs. KO: 227 ± 21%, n = 6 cells, 4 mice, p<0.005) (Fig. 5E), consistent with a RIM1α-independent mechanism for NMDAR-mfLTP. Importantly, in RIM1α KO mice, the remaining LTP induced by the longer tetanus was abolished by postsynaptic BAPTA (20 mM) (110 ± 8%, n = 4 cells, 2 mice, p>0.1) (Fig. 5E). Thus, in distinct contrast to classical presynaptic mfLTP, NMDAR-mfLTP does not require the presynaptic active zone protein RIM1α, supporting the notion that it is a novel form of plasticity at mf-CA3 synapses.
The induction of NMDAR-mfLTP requires NMDAR, mGluR5 coactivation, and calcium release from internal stores
Thus far, our findings support the following hypothesis: neurotransmitter released by repetitive stimulation of mfs activates postsynaptic receptors and triggers a Ca2+-dependent cascade of events leading to selective modification of NMDAR-mediated neurotransmission. Given that all experiments shown in Figs. 2–3 were performed in the continuous presence of 20 μM NBQX, NMDAR-mfLTP induction must be independent of AMPAR/KAR activation. Could NMDARs and/or metabotropic glutamate receptors (mGluRs) mediate induction? NMDARs are good candidates because they are strongly activated by the induction tetanus (Fig. 2), have high Ca2+ permeability, and a well-established role in the induction of other forms of long-term synaptic plasticity (Malenka and Bear, 2004; Malenka and Nicoll, 1993). Group I mGluRs (mGluR-I) which couple to phospholipase C are also good candidates as their activation by mf stimulation reportedly increases cytosolic Ca2+ in CA3 pyramidal cells (Kapur et al., 2001; Yeckel et al., 1999).
To examine whether activation of NMDARs is necessary for the induction of NMDAR-mfLTP we transiently blocked these receptors during the inducing tetanus using the competitive and selective antagonist CPP (Fig. 6A–D). After obtaining a stable NMDAR-EPSC baseline (~20 min), CPP was added to the bath for ~4 min. To speed up CPP washout and recovery of NMDAR-mediated synaptic transmission, the perfusion rate was increased (to ~4 ml/min for 20 min) immediately after the tetanus. We found that 5 μM CPP markedly reduced NMDAR-mfLTP when compared to interleaved control experiments without CPP (CPP: 123 ± 15% of baseline, n = 7: control LTP: 181 ± 16% of baseline, n = 7; p<0.005; CPP washout was not complete 30–40 min post application in naïve slices, 88 ± 10% of baseline, n = 4) (Fig. 6A). At this dose, however, CPP only produced a partial blockade of NMDAR-EPSCs during the induction tetanus (Fig. 6B); the large facilitation of glutamate release that occurs at mf-CA3 synapses during the induction tetanus is probably sufficient to out-compete 5 μM CPP. Increasing the dose of CPP to 20 μM fully blocked this tetanus-induced current (Fig. 6B) as well as NMDAR-mfLTP, although the recovery of synaptic transmission during washout in nontetanized slices was much slower (CPP, tetanized: 75 ± 5% of baseline, n = 5; CPP, nontetanized: 73 ± 10% of baseline, n = 5, p>0.5; measurements were taken 30–40 min post CPP application) (Fig. 6C). These results clearly show that the induction of NMDAR-mfLTP requires NMDAR activation.
We next examined whether mGluR-I (mGluR1 and mGluR5 subtypes) might also be required for the induction of NMDAR-mfLTP by using the selective antagonists CPCCOEt which blocks mGluR1, and MPEP which blocks mGluR5. Bath application of MPEP (4 μM) abolished NMDAR-mfLTP (MPEP: 107 ± 18% of baseline, n = 6, p>0.5; control LTP in interleaved slices: 185 ± 16% of baseline, n = 6, p<0.001) (Fig. 6E). In contrast, NMDAR-mfLTP was normal in CPCCOEt (100 μM) (CPCCOEt: 185 ± 15% of baseline, n = 4, p<0.005) (Fig. 6E). Neither of these antagonists had any significant effect on baseline transmission (data not shown). If postsynaptic mGluRs are required for NMDAR-mfLTP, interfering with the signaling cascade downstream from these receptors in CA3 pyramidal cells should also affect the magnitude of this form of plasticity. To test this possibility, we included the irreversible G protein inhibitor GDP-βS (2 mM) in the recording pipette and found that this manipulation also blocked NMDAR-mfLTP (GDP-βS 100 ± 4% of baseline, n = 4, p>0.5; control LTP in interleaved slices 196 ± 13% of baseline, n = 4, p<0.001) (Fig. 6F). Together, these results show that in addition to the NMDAR, the induction of NMDAR-mfLTP also requires activation of postsynaptic mGluR5.
To investigate whether mGluR5 activation is sufficient to induce NMDAR-mfLTP, we bath applied the mGluR-I agonist DHPG in the presence of the mGluR1 antagonist CPCCOEt (100 μM). In these experiments we also monitored ac NMDAR-EPSCs from the same CA3 pyramidal cell. We found that a 50 μM DHPG application for 10 min induced modest but significant LTP of mf NMDAR-EPSCS (134 ± 18% of baseline, n = 8, p<0.05)(Fig. 6G). In contrast, DHPG application triggered LTD of ac NMDA-EPSCs (80 ± 5% of baseline, n = 7, p<0.01)(data not shown). Thus, like the synaptically-induced NMDAR-mfLTP, the DHPG-induced potentiation of NMDAR-EPSCs selectively occurs at mf-CA3 but not ac-CA3 synapses. To investigate whether DHPG-induced potentiation mimics synaptically induced NMDAR-mfLTP, we tested for occlusion and applied DHPG 15–20 minutes after tetanus. Indeed, once NMDAR-mfLTP was established, DHPG application failed to trigger additional LTP (91 ± 2% of baseline, n = 6; p<0.05 vs. control), suggesting that both synaptically-induced and DHPG-induced phenomena share a similar mechanism. While activation of mGluRs5 alone can trigger long-lasting potentiation of mf NMDAR-mediated transmission, this induction mechanism seems to be less efficient than a burst of presynaptic activity.
mGluR5 couples to phospholipase C (PLC) via Gq, setting in motion two well known signaling cascades: the release of Ca2+ from IP3-sensitive intracellular stores, and DAG-triggered PKC activation. Due to the necessity of mGluR5 activation, we investigated the role of these downstream signaling pathways in NMDAR-mfLTP. Several studies have implicated Ca2+ stores in synaptic plasticity (reviewed in Berridge, 1998; Fitzjohn and Collingridge, 2002). Importantly, Ca2+ stores are known to contribute to the Ca2+ transients induced by repetitive activation of mfs (Kapur et al., 2001). To test the role of smooth endoplasmic reticulum (ER)-derived Ca2+ stores, we incubated hippocampal slices in 30 μM cyclopiazonic acid (CPA), a selective blocker of sarcoplasmic–endoplasmic reticulum Ca2+/ATPase pumps which is used to deplete smooth ER-derived Ca2+ stores. As shown in Fig. 6H, no NMDAR-mfLTP was observed in slices treated with CPA (CPA: 107 ± 10% of baseline, n = 5, p>0.5; LTP in interleaved control slices: 183 ± 12% of baseline, n = 5; p<0.001). To assess the relative contributions of IP3- and ryanodine receptor-mediated Ca2+ release, the respective blockers heparin (2.5 mg/ml) or ruthenium red (20 μM) were included in the recording pipette (Fig. 6I). We found that heparin markedly reduced NMDAR-mfLTP (heparin: 128 ± 20% of baseline, n = 5; control LTP in interleaved experiments: 192 ± 20% of baseline, n = 5; p<0.05), whereas ruthenium red had no effect (Ruthenium red: 196 ± 19% of baseline, n = 4). Thus, Ca2+ release from intracellular stores via IP3, but not ryanodine receptors, is also involved in NMDAR-mfLTP.
PKC activation is necessary and sufficient to trigger NMDAR-mfLTP
Phosphorylation is an important regulator of NMDAR function and trafficking (Carroll and Zukin, 2002; Chen and Roche, 2007; Kotecha and MacDonald, 2003; Lau and Zukin, 2007; Wenthold et al., 2003). Previous studies have shown that PKC facilitates NMDAR-mediated currents in Xenopus oocytes expressing recombinant NMDARs (Kelso et al., 1992; Lan et al., 2001a) and in cultured neurons (Chen and Huang, 1992; Gerber et al., 1989; Lu et al., 1999; Xiong et al., 1998). In addition, PKC blockade interferes with potentiation of NMDAR-mediated transmission in CA1 pyramidal cells (Grosshans et al., 2002), and in ventral tegmental area (VTA) dopaminergic neurons (Borgland et al., 2006). Given these results, we wondered whether NMDAR-mfLTP might be mediated by PKC activation. The fact that postsynaptic Ca2+ rise and mGluR5 activation are both required for the induction of NMDAR-mfLTP strongly suggests the involvement of a conventional (e.g. Ca2+-dependent) PKC isoform in this form of plasticity. To test this possibility, we included the selective PKC blocker chelerythrine (10 μM) in the recording pipette, a manipulation that did not noticeably affect baseline transmission (data not shown). Chelerythrine abolished NMDAR-mfLTP (chelerythrine: 113 ± 13% of baseline, n = 6, p>0.5; control LTP in interleaved experiments: 180 ± 18% of baseline, n = 5; p<0.005) (Fig. 7A).
Figure 7.
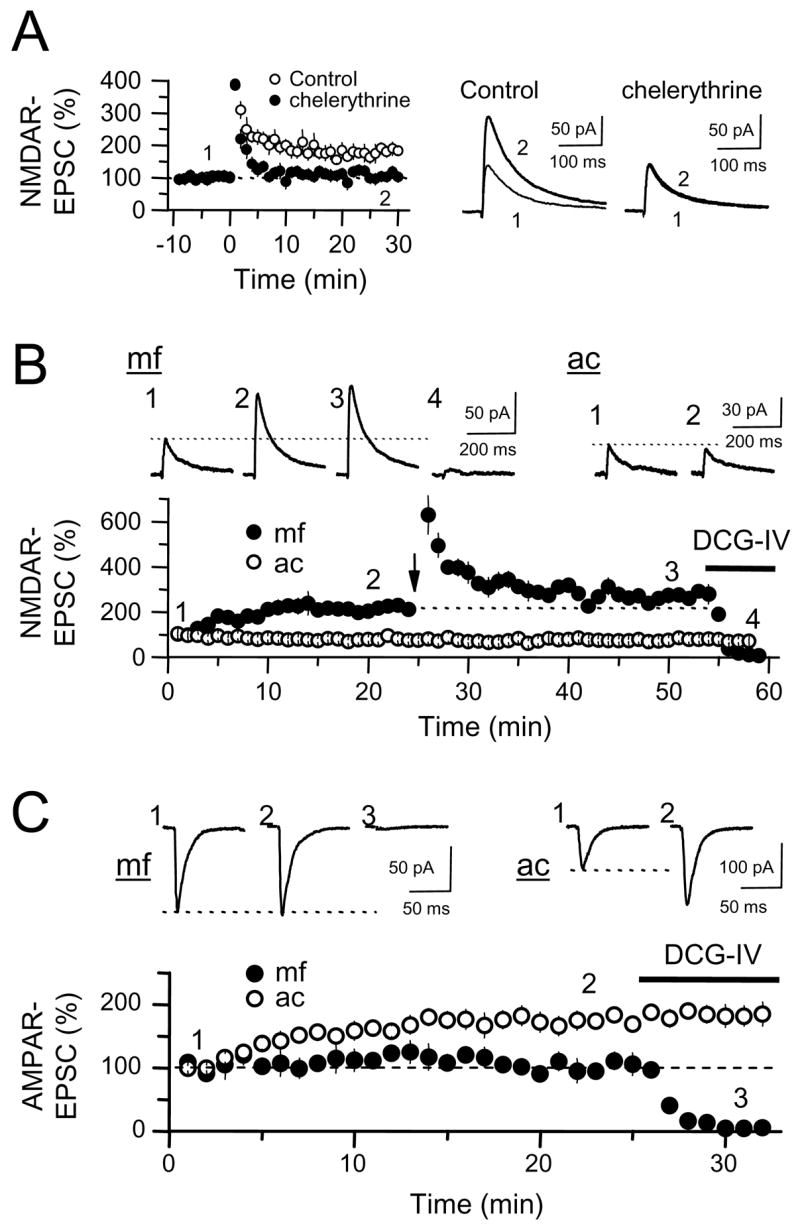
PKC activation is required to induce NMDAR-mfLTP, and it is sufficient to potentiate NMDAR-mfEPSCs. (A) Loading CA3 pyramidal cells (n = 6) with the PKC blocker chelerythrine (10 μM) abolished NMDAR-mfLTP. (B) Loading CA3 pyramidal cells with PKM, a constitutively active fragment of PKC, potentiated NMDAR-mfEPSCs (4 cells). Note that PKM potentiated mossy fiber (mf) but not associational-commissural (ac) NMDAR-EPSCs. Once PKM-mediated potentiation stabilized, subsequent tetanus (24 stimuli, 25 Hz) produced only a weak NMDAR-mfLTP, likely due to occlusion. Averaged sample traces taken at the times indicated by numbers are inset above. (C) Same procedure as in (B) while monitoring AMPAR-EPSCs. PKM potentiated the AMPAR-mediated transmission at ac-CA3 but not mf-CA3 synapses (7 cells).
We also tested whether PKC activation could mimic NMDAR-mfLTP. Previous studies have shown that loading cultured and isolated hippocampal neurons with PKM, a constitutively active fragment of PKC, facilitates NMDAR-mediated currents (Lan et al., 2001a; Lu et al., 1999; Xiong et al., 1998). We found that loading of CA3 pyramidal cells with 0.3 μM PKM potentiates mf NMDAR-EPSCs, but in the same cell this manipulation did not affect ac NMDAR-EPSCs (mf: 215 ± 12 %, p<0.0005; ac 82 ± 11 %, p>0.1; n = 4) (Fig. 7B). Furthermore, subsequent application of the induction tetanus to mfs triggered only a weak NMDAR-mfLTP (125 ± 5% of baseline post PKM-loading) (Fig. 7B), strongly suggesting that PKM-induced and synaptically-induced potentiation share a common mechanism. At the ac-CA3 synapse, while PKM did not potentiate NMDAR-EPSCs, PKM clearly potentiated AMPAR-EPSCs (185 ± 13%, n = 7, p<0.001) (Fig. 7C), indicating that the lack of effect on NMDAR-EPSCs was not an artifact of poor diffusion of PKM to the more distal ac synapses. This observation is consistent with previous studies showing PKC/M potentiation of AMPAR-EPSCs at CA3-CA1 pyramidal cell synapses (Hu et al., 1987; Ling et al., 2002). Importantly, PKM had no effect on mf-CA3 AMPAR-EPSCs in the same cells (101 ± 13%, n = 7, p>0.5) (Fig. 7C), indicating that this PKC-mediated potentiation, like synaptically-induced NMDAR-mfLTP, is selective for the NMDAR-mediated component of mf-CA3 synaptic transmission. Taken together, these results show that activation of PKC is necessary to induce NMDAR-mfLTP, and is sufficient to potentiate the NMDAR component of mf-CA3 synaptic transmission.
NMDAR-mfLTP results from postsynaptic recruitment of NMDARs via a SNARE-dependent process
The SNARE family of membrane fusion proteins is thought to play a crucial role in the postsynaptic trafficking of glutamate receptors (Lan et al., 2001a; Lledo et al., 1998; Lu et al., 2001; Luscher et al., 1999; Washbourne et al., 2004). Rapid NMDAR delivery to the cell membrane in Xenopus oocytes reportedly occurs via SNARE-dependent exocytosis (Lan et al., 2001a). If NMDAR-mfLTP results from the delivery of new receptors to the postsynaptic area, disruption of exocytosis should also disrupt potentiation. To test this possibility we first examined the effects of loading CA3 pyramidal cells with the light chains of type B botulinum toxin (BoTx), which is known to inactivate v-SNAREs (e.g. synaptobrevin), thereby preventing exocytosis and AMPA receptor surface insertion in CA1 pyramidal cells (Lledo et al., 1998; Luscher et al., 1999). We found that BoTx inhibited NMDAR-mfLTP, whereas in interleaved slices, heat-inactivated BoTx (95 °C for 30 min) had no effect (BoTx 120 ± 15% of baseline, n = 5, p>0.1; inactivated BoTx 193 ± 15% of baseline, n = 6, p<0.001) (Fig. 8A). To increase the enzymatic activity of BoTx, these experiments were performed at 35 °C. Second, we used a short peptide (11 amino acids, see methods) that mimics the C-terminal sequence of the synaptosomal-associated protein of 25 kDa (SNAP-25) and interferes with the formation of the SNARE complex (Gutierrez et al., 1997). Loading CA3 cells with this peptide also inhibited NMDAR-mfLTP, whereas a scrambled peptide had no effect (SNAP-25 peptide 108 ± 4% of baseline, n = 5, p>0.1; scrambled peptide 189 ± 15% of baseline, n = 6; p<0.001) (Fig. 8B). Importantly, intracellular perfusion with BoTx or the SNAP-25 peptide alone did not affect basal mf NMDAR-mediated synaptic transmission (30 min perfusion, data not shown, n = 3 cells for each agent), suggesting a relatively low rate of NMDAR constitutive recycling at mf-CA3 synapses. Thus, interfering with postsynaptic SNARE complex formation disrupts NMDAR-mfLTP. This finding strongly suggests that NMDAR-mfLTP is likely due to the postsynaptic insertion of NMDARs.
Figure 8.
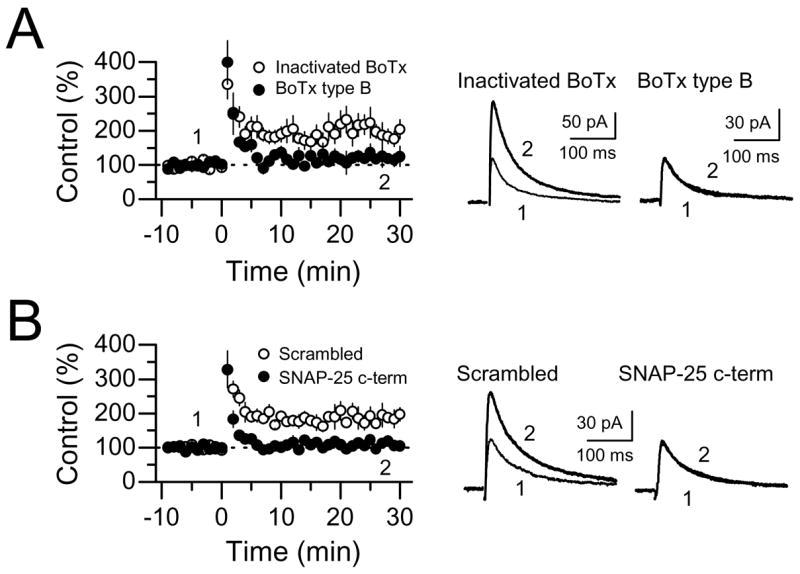
NMDAR-mfLTP requires SNARE-dependent exocytosis. (A) Summary plots showing the effects of the recombinant light chain of botulinum neurotoxin (BoTx) type B on NMDAR-mfLTP (5 cells). Heat-inactivated BoTx (100 ng/ml) was used as control (6 cells). Both BoTx and heat-inactivated BoTx were loaded into the CA3 pyramidal cell, and allowed to perfuse for at least 30 min before tetanic stimulation. (B) Summary plots comparing the effects of a short SNAP-25 interfering peptide (SNAP-25 c-term)(5 cells) and a scrambled peptide on NMDAR-mfLTP (6 cells). Averaged sample traces are shown on the right side.
DISCUSSION
Two mechanistically different forms of LTP coexist at mossy fiber to CA3 pyramidal cell synapses
Here we have identified a novel form of plasticity at mf-CA3 synapses characterized by a selective enhancement of NMDAR-mediated synaptic transmission. In contrast to classical presynaptic-mfLTP (Nicoll and Schmitz, 2005), NMDAR-mfLTP is induced and expressed postsynaptically. Thus, mf-CA3 synapses can undergo two different forms of LTP (Fig. S2): a presynaptic form which is typically induced by relatively long repetitive stimulation of mfs and is independent of postsynaptic activation (classical mfLTP), and a postsynaptic form which requires NMDAR/mGluR5 coactivation and can be triggered by a brief burst of mf activity (NMDAR-mfLTP). Previous studies support a model for classical mfLTP in which the induction tetanus causes presynaptic Ca2+ to increase and activate PKA which then enhances evoked glutamate release, probably by modifying the transmitter release machinery in a process requiring Rab3A and the active zone protein RIM1α (Nicoll and Schmitz, 2005). In contrast, NMDAR-mfLTP induction requires coactivation of NMDA and mGlu5 receptors, resulting in postsynaptic Ca2+ increase. This Ca2+ signal in conjunction with DAG, activates PKC, thus promoting NMDAR-insertion into mf-CA3 synapses via a SNARE-dependent process. Having two mechanistically different forms of LTP may allow mf-CA3 synapse to respond with more specificity and flexibility to the changing demands of the circuit.
Why has NMDAR-mfLTP escaped detection until now? This lapse is likely due to the experimental conditions commonly used by most investigators to study mf-CA3 synaptic plasticity. Early studies demonstrated that, unlike the well-characterized NMDAR-dependent LTP at Sch-CA1 synapses, activation of NMDARs is not required for the induction of mfLTP (Harris and Cotman, 1986; Zalutsky and Nicoll, 1990). Because mf responses can be easily contaminated by non-mf ones (e.g. ac-CA3 synapses) (Claiborne et al., 1993; Henze et al., 2000; Nicoll and Schmitz, 2005) which express NMDAR-dependent LTP (Zalutsky and Nicoll, 1990), most studies have included NMDAR antagonists in the recording solution in order to avoid the potential contribution of this component when studying mfLTP which precluded observation of NMDAR-mfLTP. Some studies have assessed mfLTP by monitoring NMDAR-EPSCs (Schmitz et al., 2003; Weisskopf and Nicoll, 1995). Although these studies assumed that the LTP that they observed was a strictly presynaptic phenomenon, our findings suggest that there may well have been a post-synaptic component in some cases, particularly when inducing LTP with brief bursts of presynaptic activity. Schmitz et al. (2003) have found no potentiation of mf NMDAR-EPSCs using the same induction protocol that in our study triggers NMDAR-mfLTP (e.g. 24 stimuli at 25 Hz). However, mfLTP of NMDAR-EPSCs was elicited just by doubling the number of stimuli (Schmitz et al., 2003). Minor differences in experimental procedures could account for a different induction threshold between these studies. Finally, Schmitz et al (2003) have reported that presynaptic KARs facilitate the induction of NMDAR-mfLTP. Given the postsynaptic nature of this form of plasticity, it is possible that at least part of this facilitation may actually be mediated by postsynaptic KARs.
There have been numerous reports of postsynaptic LTP expressed selectively through increased AMPAR but not NMDAR transmission (Heynen et al., 2000; Isaac et al., 1995; Kauer et al., 1988; Kullmann, 1994; Liao et al., 1995; Lu et al., 2001; Muller and Lynch, 1988; Perkel and Nicoll, 1993). However, LTP of NMDAR-mediated transmission has also been reported in several brain structures including the CA1 area of the hippocampus (Aniksztejn and Ben-Ari, 1995; Bashir et al., 1991; Bayazitov and Kleschevnikov, 2000; Berretta et al., 1991; Clark and Collingridge, 1995; Grosshans et al., 2002; Xiao et al., 1995), dentate gyrus (O’Connor et al., 1994; Xie et al., 1992) and visual cortex (Watt et al., 2004). The reason for this discrepancy is unclear, but some studies have suggested that a stronger tetanus is required to induce LTP of NMDAR versus AMPAR responses (Aniksztejn and Ben-Ari, 1995; Bayazitov and Kleschevnikov, 2000; Berretta et al., 1991). It has been recently shown that induction of AMPAR plasticity at neonatal Sch-CA1 synapses is associated with rapid changes in the subunit composition of synaptic NMDARs; such changes, however, disappear in mature synapses and do not result in any significant modification in NMDAR-EPSC amplitude (Bellone and Nicoll, 2007). Here we show that repetitive activation of mfs can induce robust LTP of NMDAR transmission at mature mf-CA3 synapses, even with a relatively weak tetanus and in the absence of mf-CA3 AMPAR plasticity. It will be interesting to know whether other mf targets (i.e. CA3 interneurons) which show NMDAR-dependent plasticity (Lei and McBain, 2002) are also capable of expressing LTP of NMDAR transmission.
Induction mechanism of NMDAR-mfLTP
Two Ca2+ sources may contribute to the induction of NMDAR-mfLTP, Ca2+ influx through NMDARs and Ca2+ release from internal stores. There is good evidence that activation of mGluR-I, which leads to PLC activation and IP3 production, is one of the most prevalent and effective means of triggering Ca2+ release from intracellular stores in CNS neurons (Ross et al., 2005). Indeed, Ca2+ stores have been found to contribute to the Ca2+ transients induced by repetitive mf activation (Kapur et al., 2001; Pozzo-Miller et al., 1996; Yeckel et al., 1999). Using protocols of mf stimulation similar to those used in our study, Kapur et al (2001) showed that brief 20 Hz bursts of presynaptic activity, by activating mGluR-I on CA3 pyramidal cells, were sufficient to induce Ca2+ release from IP3-sensitive internal stores. Consistent with these observations, we report here that the induction of NMDAR-mfLTP requires mGluR5 activation and IP3-mediated, but not ryanodine-mediated, Ca2+ release. The mGluR5 requirement for the induction of NMDAR-mfLTP is consistent with a previous report showing blockade of NMDAR-LTP at Sch-CA1 synapses in mice lacking mGluR5 (Jia et al., 1998), and an earlier study showing that pharmacological blockade of mGluR-I abolishes NMDAR-LTP in dentate gyrus (O’Connor et al., 1994). Intriguingly, Yeckel et al (1999) have reported that activation of mGluR-I in CA3 pyramidal cells and the resulting increase in postsynaptic Ca2+ plays a critical role in the induction of presynaptic mfLTP. Thus, it is possible that mGluR-I may contribute to both presynaptic and postsynaptic forms of mfLTP.
Notably, the induction of NMDAR-mfLTP not only requires mGluR5 but also NMDAR activation. This requirement fits well with previous observations showing that coactivation of mGluR5 and NMDARs potentiates NMDAR currents in cultured hippocampal neurons (Kotecha et al., 2003), and that activation of NMDARs acts synergistically with mGluR-I to enhance Ca2+ release in both cultured hippocampal cells (Rae et al., 2000) and in CA1 pyramidal neurons in acute hippocampal slices (Nakamura et al., 2002) (additional evidence for mGluR5/NMDAR interaction can be found in Kotecha and MacDonald, 2003). NMDARs have high Ca2+ permeability and reportedly contribute to the Ca2+ transients observed in thorny excrescences following mf activation (Ho et al., 2007; Reid et al., 2001). Because robust NMDAR-mfLTP could still be induced at a holding potential (e.g., +30 mV) where the Ca2+ influx is expected to be relatively small, it could be argued that Ca2+ influx through NMDARs is unlikely to be the main source of Ca2+ for induction. At positive potentials, however, significant Ca2+ influx can occur through NMDARs (Schneggenburger et al., 1993), in particular as a result of repetitive activation of these receptors during tetanus application. Even a small amount of Ca2+ influx through NMDARs may provide an important local signal for the synergistic interaction with mGluR5 (Kotecha and MacDonald, 2003). Regardless of the precise Ca2+ source, it is possible that the induction of NMDAR-mfLTP may require a significant postsynaptic Ca2+ accumulation within the large thorny excrescence, a condition that may occur only as a result of a supralinear Ca2+ increase induced by NMDAR/mGluR5 coactivation.
Postsynaptic expression of NMDAR-mfLTP
Our results strongly suggest that NMDAR-mfLTP is due to rapid recruitment of NMDARs to mf-CA3 synapses most likely by insertion of vesicle-associated NMDARs. Until recently, NMDARs were considered immobile once in the plasma membrane, especially compared to AMPARs (Bredt and Nicoll, 2003; Malinow and Malenka, 2002). However, recent studies have revealed that NMDARs can cycle rapidly into and out of synapses through several different mechanisms (reviewed in Carroll and Zukin, 2002; Collingridge et al., 2004; Lau and Zukin, 2007; Nong et al., 2004; Perez-Otano and Ehlers, 2005; Wenthold et al., 2003). First, NMDARs can be induced to redistribute in the plane of the membrane, moving from extrasynaptic to synaptic pools (Groc et al., 2004; Tovar and Westbrook, 2002). Second, there is regulated delivery of new receptors to the postsynaptic membrane via SNARE-dependent exocytosis, and this process may require synaptic activity and the activity of various protein kinases (Barria and Malinow, 2002; Grosshans et al., 2002; Lan et al., 2001a; Lan et al., 2001b; Scott et al., 2001; Skeberdis et al., 2001). Third, NMDARs can be rapidly removed from the neuronal surface and/or synapse by endocytosis (Li et al., 2002; see Morishita et al., 2005 for an alternative mechanism; Nong et al., 2003; Roche et al., 2001; Snyder et al., 2001). Because most studies addressing NMDAR trafficking have been performed in expression systems and cultured hippocampal neurons, the extent to which similar mechanisms apply in more intact preparations and in vivo is largely unknown. In this regard, the mf-CA3 synapse constitutes a model synapse where activity-dependent NMDAR trafficking can be conveniently studied, not only because of the strong NMDAR trafficking-mediated LTP observed at this synapse, but also because its proximity to the soma makes it relatively easy to access the postsynaptic compartment via the patch pipette.
By targeting two different proteins critical to the formation of the SNARE complex (with patch pipette delivery of BoNT B, which inactivates the v-SNARE synaptobrevin, and a short peptide which interferes with SNAP-25), we show that NMDAR-mfLTP requires normal SNARE-dependent exocytosis postsynaptically. Although we cannot formally exclude the contribution of reduced NMDAR endocytosis to NMDAR-mfLTP, we deem this possibility unlikely as the inhibitors of exocytosis did not substantially impact basal NMDAR-mediated mf-CA3 neurotransmission. Rather, our results are more consistent with previous observations in hippocampal cultured neurons showing that PKC, within minutes, promotes the insertion of functional NMDARs into the surface of neuronal dendrites (Lan et al., 2001a), a process that can also be triggered by activation of mGluR-I (Lan et al., 2001b). In acute hippocampal slices, the induction of LTP at CA1 synapses is reportedly associated with an increase in NMDAR surface expression in adult but not early postnatal rats (Grosshans et al., 2002). The young age of our animals might have contributed to our own inability to induce NMDAR-LTP at ac-CA3 or Sch-CA1 synapses (unpublished results from our laboratory and Fig. 3B). A recent study has shown that acute cocaine injection enhances NMDAR-mediated neurotransmission at glutamatergic synapses onto VTA dopaminergic neurons by promoting the rapid insertion of NMDARs in a PKC dependent manner (Borgland et al., 2006). This action is mediated by the activation of orexin receptors on VTA neurons which, like mGluR-I in CA3 pyramidal cells, couple to PLC (Zhu et al., 2003). These studies together with our findings at mf-CA3 synapses strengthen the hypothesis that NMDARs can undergo activity-dependent changes in intact synapses in a PKC-dependent manner.
Functional relevance
mf-CA3 synapses present an especially interesting case for NMDAR function because of their relatively high efficacy (Henze et al., 2002) and uniquely robust frequency facilitation (Salin et al., 1996). Here, even brief bursts of mf activity are sufficient to bring the postsynaptic membrane potential to action potential threshold, ensuring the relief of NMDAR Mg2+ block. Moreover, such bursts of activity also activate postsynaptic mGluR-I which mobilize Ca2+ from intracellular stores (Kapur et al., 2001). Because of the supralinear dendritic Ca2+ accumulation that follows mGluR and NMDAR coactivation (Nakamura et al., 1999; Rae et al., 2000), minor increases in Ca2+ influx through NMDARs may produce important changes in the magnitude as well as the spatial and temporal profile of Ca2+ signals. Influx of Ca2+ through NMDARs and its augmentation of release from Ca2+ stores could lead to the activation of several Ca2+-sensitive enzymes (e.g. CaMKII, PKA, PKC, mitogen-activated protein kinase, and protein phosphatases)(Berridge, 1998), and thereby modulate numerous cellular processes in CA3 pyramidal cells.
NMDAR-mfLTP could modify the input/output properties of CA3 neurons and the inducibility of synaptic plasticity. Given the slow kinetics of NMDAR-mediated synaptic responses, NMDAR-mfLTP could also alter the temporal nature of synaptic integration in CA3 pyramidal cells. In this context, it is worth noting that selective removal of NMDARs (NR1 subunit) from these cells by genetic manipulation has an important impact on memory acquisition (Nakazawa et al., 2003), associative memory recall and pattern completion (Nakazawa et al., 2002), contextual learning (Cravens et al., 2006) and trace conditioning learning (Kishimoto et al., 2006). While it is commonly assumed that NMDARs at ac-CA3 synapses mediate this effect on learning and memory, direct evidence for this possibility is lacking and NMDARs at mf-CA3 synapses may also contribute (Treves and Rolls, 1992; Tsukamoto et al., 2003). Future studies will be necessary to determine the link between these receptors and hippocampal network function.
METHODS
Hippocampal brain slice preparation
Hippocampal slices were prepared from Wistar rats (18–25 days old; Charles River) and paired RIM1α KO and WT mouse littermates (18–32 days old). RIM1α KO mice were generated as described previously (Schoch et al., 2002). All animal procedures were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. After animals were deeply anesthetized with isoflurane, they were decapitated and the brain rapidly removed into chilled cutting solution consisting of (in mM): 215 sucrose, 2.5 KCl, 20 glucose, 26 NaHCO3, 1.6 NaH2PO4, 1 CaCl2, 4 MgCl2, and 4 MgSO4. Hippocampi were dissected out and cut into 400 μm thick transverse sections on a DTK-2000 vibrating microslicer (Dosaka EM Co., Ltd., Japan). The cutting solution was slowly exchanged with artificial cerebrospinal fluid (ACSF) containing (in mM) 124 NaCl, 2.5 KCl, 10 glucose, 26 NaHCO3, 1.0 NaH2PO4, 2.5 CaCl2, and 1.3 MgCl2. Both cutting and ACSF solutions were saturated with 95% O2 and 5% CO2 (pH 7.4). The slices were incubated at room temperature for at least 1.5 hour before recording. The slices were then transferred as needed to a recording chamber and were perfused with ACSF (2 ml/min).
Electrophysiological techniques
Whole-cell recordings of CA3 pyramidal cells were obtained using standard techniques. To maximize cell health and recording stability, cells deep below the surface of the slice were recorded semi-“blind”. The recording pipette solution for voltage-clamp recordings contained (in mM) 123 cesium gluconate, 8 NaCl, 1 CaCl2, 10 EGTA, 10 HEPES, 10 glucose (pH 7.3, 290–295 mOsm), and the recording pipette resistance ranged between 3–4 MΩ. When BAPTA (20 mM) was added to internal solution, CaCl2 and EGTA were excluded and the concentration of cesium gluconate was reduced to maintain optimal osmolality (290~295 mmol/kg). Series resistance (6–15 MΩ) and input resistance were monitored throughout each voltage clamp recording with 80 ms, −4 mV steps. Recordings with >10 % change in series resistance were systematically excluded. The pipette solution for current-clamp recordings contained (in mM): 135 KMeS03, 5 KCl, 1 CaCl2, 5 EGTA-Na, 10 HEPES, 10 glucose, 5 MgATP, 0.4 Na3GTP. Resting potential ranged from −69 to −58 mV. Field potentials were recorded extracellularly with patch-type pipettes filled with 1 M NaCl and placed in the s. lucidum of CA3. To avoid polysynaptic contamination, Ca2+ and Mg2+ extracellular concentrations were increased to 4 mM unless otherwise stated. Maximal recording time after dissection was 6 hours. Recording temperature was set to 25.0 ± 0.1 °C (unless stated differently) using a TC-344B dual-channel temperature controller (Warner Instruments, Inc, Hamden, CT, USA).
Synaptic afferents were activated by monopolar stimulation delivered via a patch-type pipette broken to a tip diameter of ~10 μm and filled with external saline. This stimulating electrode was placed in the dentate gyrus cell body layer to activate mfs and in the CA3 s. radiatum to activate ac fibers. The baseline stimulation rate was 0.1 Hz for all experiments, except when short bursts were applied during baseline (Fig. 2D,E) where the inter-burst-stimulus interval was 30 s. To confirm that the activated afferents are not contaminated by ac inputs, 1 μM DCG-IV, a group II mGluR agonist that blocks mf but not ac synaptic transmission, was applied at the end of every experiment, and the data were accepted only if synaptic responses were reduced by more than 90%. The synaptic response remaining in DCG-IV was then subtracted from all previous responses before further analysis to isolate mf-specific synaptic activity. Unless otherwise noted, NMDAR-EPSCs were monitored in 20 μM NBQX, 100 μM picrotoxin, and 3 μM CGP55845 while voltage clamping to +30/+40 mV. For KAR-EPSC, the selective AMPAR antagonist GYKI 53655 (30 μM) was used instead of NBQX and cells were voltage-clamped to −60 mV. AMPAR-, NMDAR- and KAR-mediated mf-CA3 EPSCs were all evoked by single stimulation in the dentate gyrus.
Data analysis and drugs
All experiments were executed with a MultiClamp 700B (Axon Instruments Inc./Molecular Devices, Union City, CA, USA). Electrophysiological data were digitized (3–5 kHz) and analyzed on-line using custom-made software for IgorPro (Wavemetrics Inc., Lake Oswego, OR, USA). NMDAR-mfLTP magnitude was quantified by averaging synaptic responses, for 10 min periods right before and 20 min after the induction protocol. Statistical significance between means was calculated using Student’s t test. In all figures, error bars indicate ± SEM, and averaged traces include 15–30 consecutive individual responses.
NBQX, D-APV, picrotoxin, CGP 55845, DCG-IV, GYKI 53655, CPP, MPEP, CPCCOEt, ruthenium red, and cyclopiazonic acid were obtained from Tocris-Cookson Inc. (Ellisville, MO, USA). Heparin, BAPTA and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and GDP-βS was purchased from Biomol (Plymouth Meeting, PA, USA). Recombinant light chain of botulinum neurotoxin (BoTx) type B was acquired from List Biological Laboratories Inc. (Campbell, CA, USA).
Supplementary Material
Additional experimental procedures are described in Supplemental Data.
Acknowledgments
We thank the members of the Castillo laboratory for their constructive comments during the execution of this study and Dr. Kanji Takahashi, Mr. Boris Heifets and Mr. David Hunt for their critical reading of the manuscript. We also thank Dr. Suzanne Zukin (Dept. Neuroscience, Albert Einstein College of Medicine) for providing the SNAP-25 interfering peptide and the scrambled peptide control, and Dr. Thomas Südhof and Dr. Pascal Kaeser for providing RIM1α KO and WT mice. This work was supported by the US National Institutes of Health/NIDA (P.E.C.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Aniksztejn L, Ben-Ari Y. Expression of LTP by AMPA and/or NMDA receptors is determined by the extent of NMDA receptors activation during the tetanus. J Neurophysiol. 1995;74:2349–2357. doi: 10.1152/jn.1995.74.6.2349. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R. Subunit-specific NMDA receptor trafficking to synapses. Neuron. 2002;35:345–353. doi: 10.1016/s0896-6273(02)00776-6. [DOI] [PubMed] [Google Scholar]
- Bashir ZI, Alford S, Davies SN, Randall AD, Collingridge GL. Long-term potentiation of NMDA receptor-mediated synaptic transmission in the hippocampus. Nature. 1991;349:156–158. doi: 10.1038/349156a0. [DOI] [PubMed] [Google Scholar]
- Bayazitov I, Kleschevnikov A. Afferent high strength tetanizations favour potentiation of the NMDA vs. AMPA receptor-mediated component of field EPSP in CA1 hippocampal slices of rats. Brain Res. 2000;866:188–196. doi: 10.1016/s0006-8993(00)02279-4. [DOI] [PubMed] [Google Scholar]
- Bellone C, Nicoll RA. Rapid Bidirectional Switching of Synaptic NMDA Receptors. Neuron. 2007;55:779–785. doi: 10.1016/j.neuron.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Benke TA, Jones OT, Collingridge GL, Angelides KJ. N-Methyl-D-aspartate receptors are clustered and immobilized on dendrites of living cortical neurons. Proc Natl Acad Sci U S A. 1993;90:7819–7823. doi: 10.1073/pnas.90.16.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berretta N, Berton F, Bianchi R, Brunelli M, Capogna M, Francesconi W. Long-term Potentiation of NMDA Receptor-mediated EPSP in Guinea-pig Hippocampal Slices. Eur J Neurosci. 1991;3:850–854. doi: 10.1111/j.1460-9568.1991.tb00096.x. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Borgland SL, Taha SA, Sarti F, Fields HL, Bonci A. Orexin A in the VTA is critical for the induction of synaptic plasticity and behavioral sensitization to cocaine. Neuron. 2006;49:589–601. doi: 10.1016/j.neuron.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Zukin RS. NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci. 2002;25:571–577. doi: 10.1016/s0166-2236(02)02272-5. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Schoch S, Schmitz F, Sudhof TC, Malenka RC. RIM1alpha is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- Chen BS, Roche KW. Regulation of NMDA receptors by phosphorylation. Neuropharmacology. 2007;53:362–368. doi: 10.1016/j.neuropharm.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Huang LY. Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature. 1992;356:521–523. doi: 10.1038/356521a0. [DOI] [PubMed] [Google Scholar]
- Claiborne BJ, Xiang Z, Brown TH. Hippocampal circuitry complicates analysis of long-term potentiation in mossy fiber synapses. Hippocampus. 1993;3:115–121. doi: 10.1002/hipo.450030202. [DOI] [PubMed] [Google Scholar]
- Clark KA, Collingridge GL. Synaptic potentiation of dual-component excitatory postsynaptic currents in the rat hippocampus. J Physiol. 1995;482(Pt 1):39–52. doi: 10.1113/jphysiol.1995.sp020498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Cravens CJ, Vargas-Pinto N, Christian KM, Nakazawa K. CA3 NMDA receptors are crucial for rapid and automatic representation of context memory. Eur J Neurosci. 2006;24:1771–1780. doi: 10.1111/j.1460-9568.2006.05044.x. [DOI] [PubMed] [Google Scholar]
- Daw NW, Stein PS, Fox K. The role of NMDA receptors in information processing. Annu Rev Neurosci. 1993;16:207–222. doi: 10.1146/annurev.ne.16.030193.001231. [DOI] [PubMed] [Google Scholar]
- Faber DS, Korn H. Applicability of the coefficient of variation method for analyzing synaptic plasticity. Biophys J. 1991;60:1288–1294. doi: 10.1016/S0006-3495(91)82162-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzjohn SM, Collingridge GL. Calcium stores and synaptic plasticity. Cell Calcium. 2002;32:405–411. doi: 10.1016/s0143416002001999. [DOI] [PubMed] [Google Scholar]
- Fritschy JM, Weinmann O, Wenzel A, Benke D. Synapse-specific localization of NMDA and GABA(A) receptor subunits revealed by antigen-retrieval immunohistochemistry. J Comp Neurol. 1998;390:194–210. [PubMed] [Google Scholar]
- Gerber G, Kangrga I, Ryu PD, Larew JS, Randic M. Multiple effects of phorbol esters in the rat spinal dorsal horn. J Neurosci. 1989;9:3606–3617. doi: 10.1523/JNEUROSCI.09-10-03606.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith WH. Voltage-clamp analysis of posttetanic potentiation of the mossy fiber to CA3 synapse in hippocampus. J Neurophysiol. 1990;63:491–501. doi: 10.1152/jn.1990.63.3.491. [DOI] [PubMed] [Google Scholar]
- Groc L, Heine M, Cognet L, Brickley K, Stephenson FA, Lounis B, Choquet D. Differential activity-dependent regulation of the lateral mobilities of AMPA and NMDA receptors. Nat Neurosci. 2004;7:695–696. doi: 10.1038/nn1270. [DOI] [PubMed] [Google Scholar]
- Grosshans DR, Clayton DA, Coultrap SJ, Browning MD. LTP leads to rapid surface expression of NMDA but not AMPA receptors in adult rat CA1. Nat Neurosci. 2002;5:27–33. doi: 10.1038/nn779. [DOI] [PubMed] [Google Scholar]
- Gutierrez LM, Viniegra S, Rueda J, Ferrer-Montiel AV, Canaves JM, Montal M. A peptide that mimics the C-terminal sequence of SNAP-25 inhibits secretory vesicle docking in chromaffin cells. J Biol Chem. 1997;272:2634–2639. doi: 10.1074/jbc.272.5.2634. [DOI] [PubMed] [Google Scholar]
- Harris EW, Cotman CW. Long-term potentiation of guinea pig mossy fiber responses is not blocked by N-methyl D-aspartate antagonists. Neurosci Lett. 1986;70:132–137. doi: 10.1016/0304-3940(86)90451-9. [DOI] [PubMed] [Google Scholar]
- Henze DA, Urban NN, Barrionuevo G. The multifarious hippocampal mossy fiber pathway: a review. Neuroscience. 2000;98:407–427. doi: 10.1016/s0306-4522(00)00146-9. [DOI] [PubMed] [Google Scholar]
- Henze DA, Wittner L, Buzsaki G. Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci. 2002;5:790–795. doi: 10.1038/nn887. [DOI] [PubMed] [Google Scholar]
- Heynen AJ, Quinlan EM, Bae DC, Bear MF. Bidirectional, activity-dependent regulation of glutamate receptors in the adult hippocampus in vivo. Neuron. 2000;28:527–536. doi: 10.1016/s0896-6273(00)00130-6. [DOI] [PubMed] [Google Scholar]
- Ho MT, Pelkey KA, Topolnik L, Petralia RS, Takamiya K, Xia J, Huganir RL, Lacaille JC, McBain CJ. Developmental Expression of Ca2+-Permeable AMPA Receptors Underlies Depolarization-Induced Long-Term Depression at Mossy Fiber CA3 Pyramid Synapses. J Neurosci. 2007;27:11651–11662. doi: 10.1523/JNEUROSCI.2671-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsia AY, Salin PA, Castillo PE, Aiba A, Abeliovich A, Tonegawa S, Nicoll RA. Evidence against a role for metabotropic glutamate receptors in mossy fiber LTP: the use of mutant mice and pharmacological antagonists. Neuropharmacology. 1995;34:1567–1572. doi: 10.1016/0028-3908(95)00115-m. [DOI] [PubMed] [Google Scholar]
- Hu GY, Hvalby O, Walaas SI, Albert KA, Skjeflo P, Andersen P, Greengard P. Protein kinase C injection into hippocampal pyramidal cells elicits features of long term potentiation. Nature. 1987;328:426–429. doi: 10.1038/328426a0. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Murphy GJ. Glutamate-mediated extrasynaptic inhibition: direct coupling of NMDA receptors to Ca(2+)-activated K+ channels. Neuron. 2001;31:1027–1034. doi: 10.1016/s0896-6273(01)00428-7. [DOI] [PubMed] [Google Scholar]
- Jia Z, Lu Y, Henderson J, Taverna F, Romano C, Abramow-Newerly W, Wojtowicz JM, Roder J. Selective abolition of the NMDA component of long-term potentiation in mice lacking mGluR5. Learn Mem. 1998;5:331–343. [PMC free article] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur A, Yeckel M, Johnston D. Hippocampal mossy fiber activity evokes Ca2+ release in CA3 pyramidal neurons via a metabotropic glutamate receptor pathway. Neuroscience. 2001;107:59–69. doi: 10.1016/s0306-4522(01)00293-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- Kelso SR, Nelson TE, Leonard JP. Protein kinase C-mediated enhancement of NMDA currents by metabotropic glutamate receptors in Xenopus oocytes. J Physiol. 1992;449:705–718. doi: 10.1113/jphysiol.1992.sp019110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto Y, Nakazawa K, Tonegawa S, Kirino Y, Kano M. Hippocampal CA3 NMDA receptors are crucial for adaptive timing of trace eyeblink conditioned response. J Neurosci. 2006;26:1562–1570. doi: 10.1523/JNEUROSCI.4142-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha SA, Jackson MF, Al-Mahrouki A, Roder JC, Orser BA, MacDonald JF. Co-stimulation of mGluR5 and N-methyl-D-aspartate receptors is required for potentiation of excitatory synaptic transmission in hippocampal neurons. J Biol Chem. 2003;278:27742–27749. doi: 10.1074/jbc.M301946200. [DOI] [PubMed] [Google Scholar]
- Kotecha SA, MacDonald JF. Signaling molecules and receptor transduction cascades that regulate NMDA receptor-mediated synaptic transmission. Int Rev Neurobiol. 2003;54:51–106. doi: 10.1016/s0074-7742(03)54003-x. [DOI] [PubMed] [Google Scholar]
- Kullmann DM. Amplitude fluctuations of dual-component EPSCs in hippocampal pyramidal cells: implications for long-term potentiation. Neuron. 1994;12:1111–1120. doi: 10.1016/0896-6273(94)90318-2. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS. Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci. 2001a;4:382–390. doi: 10.1038/86028. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Zheng X, Bennett MV, Zukin RS. Activation of metabotropic glutamate receptor 1 accelerates NMDA receptor trafficking. J Neurosci. 2001b;21:6058–6068. doi: 10.1523/JNEUROSCI.21-16-06058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- Lei S, McBain CJ. Distinct NMDA receptors provide differential modes of transmission at mossy fiber-interneuron synapses. Neuron. 2002;33:921–933. doi: 10.1016/s0896-6273(02)00608-6. [DOI] [PubMed] [Google Scholar]
- Li B, Chen N, Luo T, Otsu Y, Murphy TH, Raymond LA. Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat Neurosci. 2002;5:833–834. doi: 10.1038/nn912. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Ling DS, Benardo LS, Serrano PA, Blace N, Kelly MT, Crary JF, Sacktor TC. Protein kinase Mzeta is necessary and sufficient for LTP maintenance. Nat Neurosci. 2002;5:295–296. doi: 10.1038/nn829. [DOI] [PubMed] [Google Scholar]
- Lledo PM, Zhang X, Sudhof TC, Malenka RC, Nicoll RA. Postsynaptic membrane fusion and long-term potentiation. Science. 1998;279:399–403. doi: 10.1126/science.279.5349.399. [DOI] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT. Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron. 2001;29:243–254. doi: 10.1016/s0896-6273(01)00194-5. [DOI] [PubMed] [Google Scholar]
- Lu WY, Xiong ZG, Lei S, Orser BA, Dudek E, Browning MD, MacDonald JF. G-protein-coupled receptors act via protein kinase C and Src to regulate NMDA receptors. Nat Neurosci. 1999;2:331–338. doi: 10.1038/7243. [DOI] [PubMed] [Google Scholar]
- Luscher C, Xia H, Beattie EC, Carroll RC, von Zastrow M, Malenka RC, Nicoll RA. Role of AMPA receptor cycling in synaptic transmission and plasticity. Neuron. 1999;24:649–658. doi: 10.1016/s0896-6273(00)81119-8. [DOI] [PubMed] [Google Scholar]
- Major G, Tank D. Persistent neural activity: prevalence and mechanisms. Curr Opin Neurobiol. 2004;14:675–684. doi: 10.1016/j.conb.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. NMDA-receptor-dependent synaptic plasticity: multiple forms and mechanisms. Trends Neurosci. 1993;16:521–527. doi: 10.1016/0166-2236(93)90197-t. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Monaghan DT, Holets VR, Toy DW, Cotman CW. Anatomical distributions of four pharmacologically distinct 3H-L-glutamate binding sites. Nature. 1983;306:176–179. doi: 10.1038/306176a0. [DOI] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka RC. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Muller D, Lynch G. Long-term potentiation differentially affects two components of synaptic responses in hippocampus. Proc Natl Acad Sci U S A. 1988;85:9346–9350. doi: 10.1073/pnas.85.23.9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Barbara JG, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lasser-Ross N, Nakamura K, Ross WN. Spatial segregation and interaction of calcium signalling mechanisms in rat hippocampal CA1 pyramidal neurons. J Physiol. 2002;543:465–480. doi: 10.1113/jphysiol.2002.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Quirk MC, Chitwood RA, Watanabe M, Yeckel MF, Sun LD, Kato A, Carr CA, Johnston D, Wilson MA, Tonegawa S. Requirement for hippocampal CA3 NMDA receptors in associative memory recall. Science. 2002;297:211–218. doi: 10.1126/science.1071795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Sun LD, Quirk MC, Rondi-Reig L, Wilson MA, Tonegawa S. Hippocampal CA3 NMDA receptors are crucial for memory acquisition of one-time experience. Neuron. 2003;38:305–315. doi: 10.1016/s0896-6273(03)00165-x. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Ju W, Kalia LV, Ahmadian G, Wang YT, Salter MW. Glycine binding primes NMDA receptor internalization. Nature. 2003;422:302–307. doi: 10.1038/nature01497. [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Salter MW. NMDA receptors are movin’ in. Curr Opin Neurobiol. 2004;14:353–361. doi: 10.1016/j.conb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- O’Connor JJ, Rowan MJ, Anwyl R. Long-lasting enhancement of NMDA receptor-mediated synaptic transmission by metabotropic glutamate receptor activation. Nature. 1994;367:557–559. doi: 10.1038/367557a0. [DOI] [PubMed] [Google Scholar]
- Perez-Otano I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 2005;28:229–238. doi: 10.1016/j.tins.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Perkel DJ, Nicoll RA. Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation. J Physiol. 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Yokotani N, Wenthold RJ. Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti-peptide antibody. J Neurosci. 1994;14:667–696. doi: 10.1523/JNEUROSCI.14-02-00667.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzo-Miller LD, Petrozzino JJ, Golarai G, Connor JA. Ca2+ release from intracellular stores induced by afferent stimulation of CA3 pyramidal neurons in hippocampal slices. J Neurophysiol. 1996;76:554–562. doi: 10.1152/jn.1996.76.1.554. [DOI] [PubMed] [Google Scholar]
- Rae MG, Martin DJ, Collingridge GL, Irving AJ. Role of Ca2+ stores in metabotropic L-glutamate receptor-mediated supralinear Ca2+ signaling in rat hippocampal neurons. J Neurosci. 2000;20:8628–8636. doi: 10.1523/JNEUROSCI.20-23-08628.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Sachidhanandam S, Perrais D, Cunha RA, Mulle C. Short-term plasticity of kainate receptor-mediated EPSCs induced by NMDA receptors at hippocampal mossy fiber synapses. J Neurosci. 2007;27:3987–3993. doi: 10.1523/JNEUROSCI.5182-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Fabian-Fine R, Fine A. Postsynaptic calcium transients evoked by activation of individual hippocampal mossy fiber synapses. J Neurosci. 2001;21:2206–2214. doi: 10.1523/JNEUROSCI.21-07-02206.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KW, Standley S, McCallum J, Dune Ly C, Ehlers MD, Wenthold RJ. Molecular determinants of NMDA receptor internalization. Nat Neurosci. 2001;4:794–802. doi: 10.1038/90498. [DOI] [PubMed] [Google Scholar]
- Ross WN, Nakamura T, Watanabe S, Larkum M, Lasser-Ross N. Synaptically activated ca2+ release from internal stores in CNS neurons. Cell Mol Neurobiol. 2005;25:283–295. doi: 10.1007/s10571-005-3060-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Hestrin S, Nicoll RA. Tonic activation of NMDA receptors by ambient glutamate enhances excitability of neurons. Science. 1989;246:815–818. doi: 10.1126/science.2573153. [DOI] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Breustedt J, Nicoll RA. Presynaptic kainate receptors impart an associative property to hippocampal mossy fiber long-term potentiation. Nat Neurosci. 2003;6:1058–1063. doi: 10.1038/nn1116. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Zhou Z, Konnerth A, Neher E. Fractional contribution of calcium to the cation current through glutamate receptor channels. Neuron. 1993;11:133–143. doi: 10.1016/0896-6273(93)90277-x. [DOI] [PubMed] [Google Scholar]
- Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Sudhof TC. RIM1alpha forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–326. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- Scott DB, Blanpied TA, Swanson GT, Zhang C, Ehlers MD. An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J Neurosci. 2001;21:3063–3072. doi: 10.1523/JNEUROSCI.21-09-03063.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeberdis VA, Lan J, Opitz T, Zheng X, Bennett MV, Zukin RS. mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C. Neuropharmacology. 2001;40:856–865. doi: 10.1016/s0028-3908(01)00005-3. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Spruston N, Jonas P, Sakmann B. Dendritic glutamate receptor channels in rat hippocampal CA3 and CA1 pyramidal neurons. J Physiol. 1995;482(Pt 2):325–352. doi: 10.1113/jphysiol.1995.sp020521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi Y, Ramirez-Leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nat Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. Mobile NMDA receptors at hippocampal synapses. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- Treves A, Rolls ET. Computational constraints suggest the need for two distinct input systems to the hippocampal CA3 network. Hippocampus. 1992;2:189–199. doi: 10.1002/hipo.450020209. [DOI] [PubMed] [Google Scholar]
- Tsukamoto M, Yasui T, Yamada MK, Nishiyama N, Matsuki N, Ikegaya Y. Mossy fibre synaptic NMDA receptors trigger non-Hebbian long-term potentiation at entorhino-CA3 synapses in the rat. J Physiol. 2003;546:665–675. doi: 10.1113/jphysiol.2002.033803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- Vogt K, Mellor J, Tong G, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–196. doi: 10.1016/s0896-6273(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Liu XB, Jones EG, McAllister AK. Cycling of NMDA receptors during trafficking in neurons before synapse formation. J Neurosci. 2004;24:8253–8264. doi: 10.1523/JNEUROSCI.2555-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Fukaya M, Sakimura K, Manabe T, Mishina M, Inoue Y. Selective scarcity of NMDA receptor channel subunits in the stratum lucidum (mossy fibre-recipient layer) of the mouse hippocampal CA3 subfield. Eur J Neurosci. 1998;10:478–487. doi: 10.1046/j.1460-9568.1998.00063.x. [DOI] [PubMed] [Google Scholar]
- Watt AJ, Sjostrom PJ, Hausser M, Nelson SB, Turrigiano GG. A proportional but slower NMDA potentiation follows AMPA potentiation in LTP. Nat Neurosci. 2004;7:518–524. doi: 10.1038/nn1220. [DOI] [PubMed] [Google Scholar]
- Weisskopf MG, Nicoll RA. Presynaptic changes during mossy fibre LTP revealed by NMDA receptor-mediated synaptic responses. Nature. 1995;376:256–259. doi: 10.1038/376256a0. [DOI] [PubMed] [Google Scholar]
- Wenthold RJ, Prybylowski K, Standley S, Sans N, Petralia RS. Trafficking of NMDA receptors. Annu Rev Pharmacol Toxicol. 2003;43:335–358. doi: 10.1146/annurev.pharmtox.43.100901.135803. [DOI] [PubMed] [Google Scholar]
- Xiao MY, Karpefors M, Niu YP, Wigstrom H. The complementary nature of long-term depression and potentiation revealed by dual component excitatory postsynaptic potentials in hippocampal slices from young rats. Neuroscience. 1995;68:625–635. doi: 10.1016/0306-4522(95)00173-g. [DOI] [PubMed] [Google Scholar]
- Xie X, Berger TW, Barrionuevo G. Isolated NMDA receptor-mediated synaptic responses express both LTP and LTD. J Neurophysiol. 1992;67:1009–1013. doi: 10.1152/jn.1992.67.4.1009. [DOI] [PubMed] [Google Scholar]
- Xiong ZG, Raouf R, Lu WY, Wang LY, Orser BA, Dudek EM, Browning MD, MacDonald JF. Regulation of N-methyl-D-aspartate receptor function by constitutively active protein kinase C. Mol Pharmacol. 1998;54:1055–1063. [PubMed] [Google Scholar]
- Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalutsky RA, Nicoll RA. Comparison of two forms of long-term potentiation in single hippocampal neurons. Science. 1990;248:1619–1624. doi: 10.1126/science.2114039. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Miwa Y, Yamanaka A, Yada T, Shibahara M, Abe Y, Sakurai T, Goto K. Orexin receptor type-1 couples exclusively to pertussis toxin-insensitive G-proteins, while orexin receptor type-2 couples to both pertussis toxin-sensitive and -insensitive G-proteins. J Pharmacol Sci. 2003;92:259–266. doi: 10.1254/jphs.92.259. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional experimental procedures are described in Supplemental Data.