

Abstract
The structure of the rare-cutting restriction endonuclease NotI, which recognizes the 8 basepair target 5′-GCGGCCGC-3′, has been solved with and without bound DNA. Because of its specificity (recognizing a site that occurs once per 65kb), NotI is used to generate large genomic fragments and to map DNA methylation status. NotI contains a unique metal-binding fold, found in a variety of putative endonucleases, occupied by an iron atom coordinated within a tetrahedral Cys4 motif. This domain positions nearby structural elements for DNA recognition, and serves a structural role. While recognition of the central six basepairs of the target is accomplished via a saturated hydrogen bond network typical of restriction enzymes, the most peripheral basepairs are engaged in a single direct contact in the major groove, reflecting reduced pressure to recognize those positions. NotI may represent an evolutionary intermediate between mobile endonucleases (which recognize longer target sites) and canonical restriction endonucleases.
The primary functions of restriction endonucleases (REases) appear to be the defense of their bacterial hosts against invasion by foreign DNAand their own selfish propagation in bacterial genomes (Kobayashi, 2001; Pingoud et al., 2005). The bacterial restriction-modification (R-M) system is made up of coordinated restriction and methyltransferase enzymes which are specific for a common DNA sequence. Through their pairwise activities, invasive DNA is digested by the endonuclease while the bacterial genome is protected by methylation of adenine or cytosine bases in the same site (Bickle and Kruger, 1993). Restriction endonucleases are found in many forms, ranging from canonical 'Type IIP' enzymes (the simplest being separate homodimeric endonucleases and methyltransferases that each recognize the same palindromic DNA target sequence) to complex Type I and III enzyme machines (where both activities are coupled in large multiprotein assemblages that are driven by ATP-dependent motors) (Bourniquel and Bickle, 2002). Regardless of their complexity, the majority of DNA sequences that are recognized by restriction-modification enzymes are short (often ranging from 4 to 6 basepairs) thus ensuring the presence of target sites even in small invading phage genomes.
A "traditional" 6 basepair recognition site (for example, 5′-GAATTC-3′, cleaved by EcoRI) would be found on average once every 4,096 bases of random DNA sequence, ensuring its presence within a typical phage genome. However, bacterial genomes also contain a variety of less common 'rare' cutters, including type IIP enzymes, that recognize sites of 8 basepairs or longer. Such endonucleases may be generated as a result of their presence in host genomes with skewed base composition. They provide useful tools for genome mapping technologies, and may also provide useful insight into past evolutionary events that might link invasive DNA elements (such as homing endonucleases) with modern restriction endonuclease systems.
The type IIP restriction enzyme NotI from Nocardia otitidis-caviarum is a homodimer which recognizes the 8 basepair DNA sequence 5’- GC/GGCCGC -3’ and cleaves both strands of DNA to create 5’, 4-base cohesive overhangs (Qiang and Schildkraut, 1987). Under low salt conditions, the enzyme displays noncognate star activity, also cleaving the DNA sequence 5′ - AC/GGCCGT - 3′ (Samuelson et al., 2006). The endonuclease has been subjected to a variety of selection experiments for altered DNA recognition and cleavage properties, yielding forms of the enzyme with reduced DNA target fidelity (Samuelson et al., 2006) and a chimeric endonuclease targeted to the NotI recognition site (Zhang et al., 2007).
Because the recognition sequence of NotI is comprised solely of G:C base pairs, it is frequently located in regions of eukaryotic genomes known as CpG islands. These islands are often situated at the promoter regions of genes and are hotspots for DNA methylation that can effect gene expression (Rush and Plass, 2002). Modification of the NotI DNA target site by either the M.NotI methylase (which methylates cytosine N4 nitrogens at one or more unknown positions in the site) or by M.EagI (which acts at cytosine C5 carbons, again at unknown positions) block cleavage by NotI (X.Y Xu, unpublished data). Additionally, NotI cleavage assays using synthetic DNA substrates indicate that methylation of the cytosine C5 carbon at either position 2 or 6 of the target site blocks DNA cleavage (Roberts et al., 2007). Due to both its rare-cutting properties and its demonstrated methylation sensitivity, NotI is commonly used for the introduction of radiolabeled landmarks in the 'Restriction Landmark Genomic Scanning' (RLGS) method, which has become a common technique for the study of aberrant DNA methylation patterns in tumor cell lines (Smiraglia and Plass, 2002).
Here we present crystal structures of the NotI restriction enzyme, both in its unbound conformation (2.8Å resolution) and bound to its cognate DNA recognition sequence (2.5Å). The crystal structures reveal the presence of a novel Cys4 containing protein fold appended to the N-terminus of the core PD…(D/E)xK nuclease domain. This fold is occupied by an iron atom and appears to position adjacent structural elements on the endonuclease surface for DNA recognition; examination of bacterial genome sequences indicates that this fold is found within many putative endonuclease genes. Comparison of the structure of NotI to an unrelated 8-basepair cutter (SdaI) and its sequence to that of a closely related 6-basepair cutter (EagI) provides insight into the evolution of rare-cutting behavior in restriction endonuclease systems.
Results
Overall protein structure
The Not I endonuclease is a homodimer containing 383 residues per protein chain (42.4 kilodaltons per subunit) (Figure 1). Each protein subunit contains a single Type II restriction endonuclease fold, consisting of a central β-sheet, two α-helices, and the canonical PD…(D/E)xK catalytic motif. This domain is flanked by large structural elements at both its N- and C-termini, and is also interrupted four separate times by a variety of distinct structural elements that embellish its surface. These structural elaborations are variously involved in structural stabilization, protein dimerization, and positioning of DNA-binding regions. The core endonuclease domain spans residues 88 to 324, consisting of a β-sheet that is wrapped around a central α-helix (α4). This domain displays the topology "…α4 − β4↓… α6 − β5↓…β8↑ − β9↓ … β10↓… β12↓ − β13↑…" where the dotted lines denote the positions of structural insertions.
Figure 1. The structure of NotI restriction endonuclease.
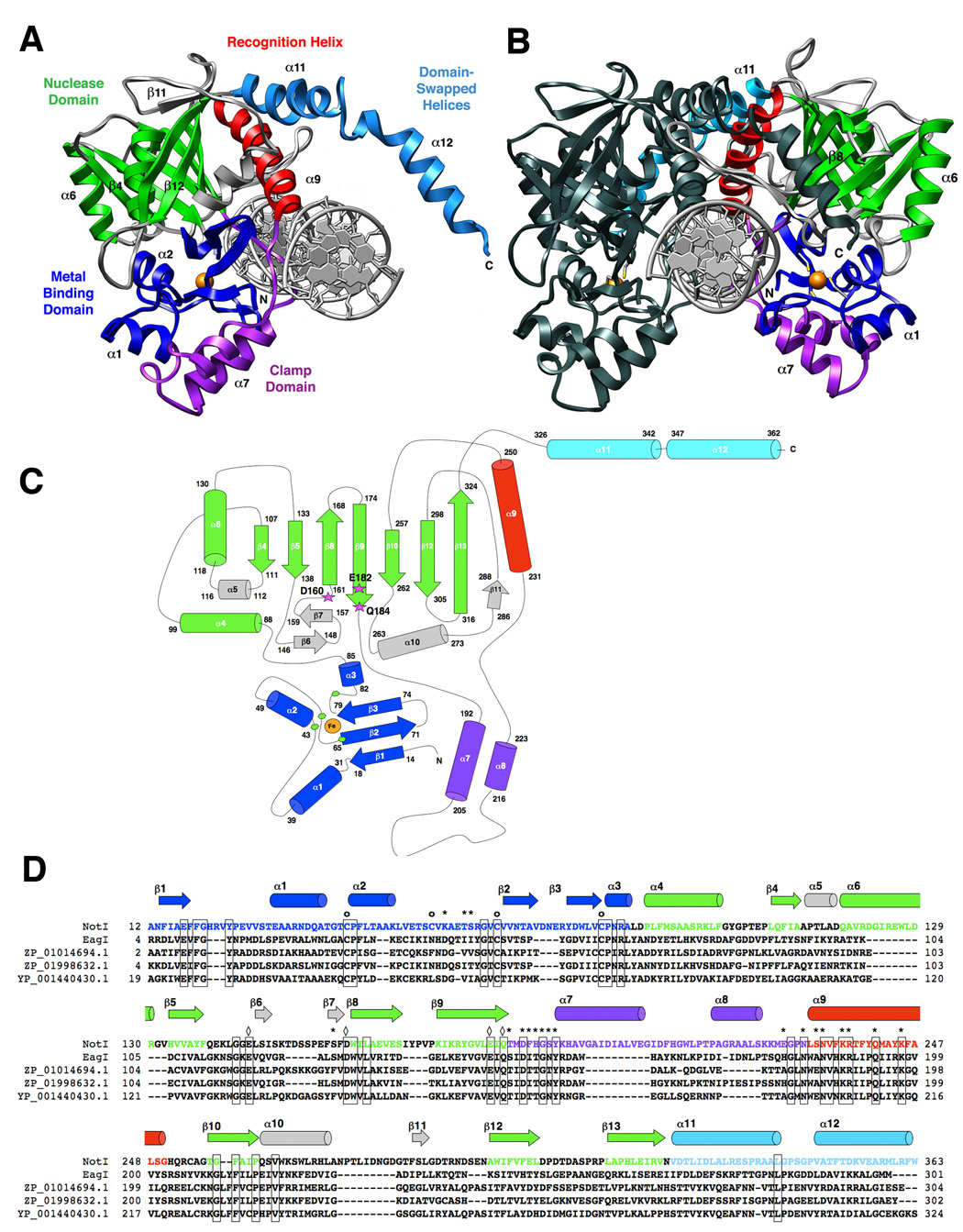
The protein is colored according to individual structural domains and motifs; the same color scheme is used in all panels. The N-terminal 85 residues (blue) form a unique metal-binding domain that uses a four-cysteine coordination scheme; this domain is followed by the PD…D/E-xK catalytic domain (green). An elaboration on this fold, extending from between two β-stands, presents a two-helix bundle “clamp” domain (purple) that wraps around the metal-bound domain. This region, along with an adjacent recognition helix (red) contributes the majority of direct contacts between the protein and bound DNA. Finally, two helices extend from the C-terminal end of the protein and form a domain-swapped dimerization motif (cyan). Additional structural elaborations on the nuclease core are colored gray. Panel A: A single monomer of NotI with the full-length (22bp) palindromic DNA target site. The bound iron ion is shown as an orange sphere, and select secondary structural elements are labeled by number for reference. Panel B: The homodimeric endonuclease bound to its cognate DNA target site. One subunit is colored as described above (the opposite protein chain colored relative to panel a); the second subunit is colored uniformly dark for clarity and contrast. The cysteine sidechains of the Fe-Cys4 iron-binding motif are shown as sticks and colored by element. Panel C: Topology of the NotI protein fold. Residues at boundaries of secondary structural elements are numbered; active site residues are indicated with magenta stars and bold labels; The bound iron is shown as an orange circle, and iron ligands (Cys 42, 55, 65, and 81) are represented by green hexagons. Individual secondary structural elements are numbered in sequential order. Panel D: Sequence homology between NotI, EagI (GenBank EU 371940, which recognizes and cuts a six-base site corresponding to the central six positions of the 8-base NotI target site), and homologues ZP_01014694.1 from Rhodobacterales bacterium HTCC2654, ZP_01998632.1 from Beggiatoa sp. PS, and YP_001440430.1 from Enterobacter sakazakii ATCC BAA_894. Iron-binding residues (of which three out of four are conserved in EagI) are indicated with circles (o); DNA-binding residues (of which ten out of nineteen are conserved in EagI) are indicated with asterisks (*); and catalytic residues of the PD…(D/E)xK endonuclease motif (two-metal ion mechanism) are indicated with diamonds (◊) (all are conserved).
The endonuclease homodimer measures approximately 100Å × 50Å × 50Å. The nearest atomic neighbors between individual catalytic domains are separated by over 20 angstroms distance. Dimerization of the protein is facilitated by two separate elaborations on the core fold (Figure 1 and Figure 2), burying a surface area of approximately 3400 Å2. A domain-swapped, C-terminal helical region (spanning residues 326 to 362) extends from each nuclease domain to make extensive packing contacts across one end of the neighboring protein subunit, and additional contacts across the dimer interface are made between a single turn of an alpha helix (α9) and its symmetry mate (α 9′). This helix is part of a large structural insertion between strands β9 and β10 of each nuclease domain. Several residues in helices α9 and α9′ also make contact with the DNA target site; however biochemical studies (Qiang and Schildkraut, 1987; Sznyter and Brooks, 1988) and the crystal structure of the free enzyme both indicate that DNA binding is not required for dimerization of the protein.
Figure 2. Homodimeric NotI restriction endonuclease.
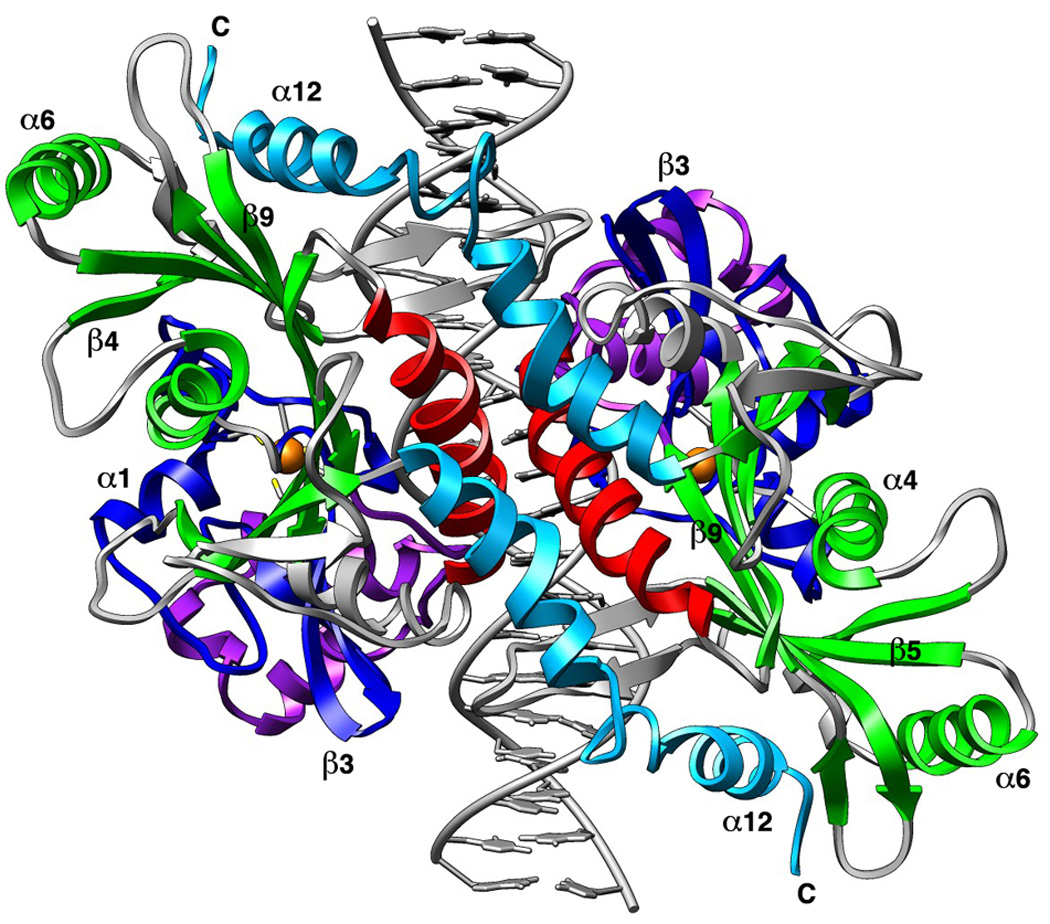
Domains are colored as described in Figure 1. Select secondary structural elements are numbered for reference. The NotI dimer interface is formed by the domain-swapped alpha helices (cyan) and the DNA recognition helices (red).
The N-terminal end of the protein (residues 1 through 85) forms a compact metal-bound fold, consisting of a three-stranded antiparallel β-sheet interrupted and flanked by three short α -helices (Figure 3). The topology of this domain is β1↑ − α1 − α2 − β2↓ − β3↑ − α3. Four cysteine residues (Cys 42, 55, 65, and 81), all located in loops connecting the secondary structure of the domain, are arranged in a tetrahedral arrangement around a single bound metal ion. X-ray fluorescence scans indicate the presence of bound iron in the protein crystal (Supplementary Fig. S4). A search for structural homologues of this domain using the DALI server (Holm and Sander, 1996) does not indicate any related folds among the structures currently in the PDB database; the closest match is the structure 2IVW (corresponding to an obviously unrelated fold in the N. meningitidis lipoprotein PilP) with a Z-score of 0.6. The tetrahedral coordination of the iron atom by four cysteine residues is similar to the ligation scheme exhibited by a number of Fe-Cys4 electron transfer proteins such as rubredoxin.
Figure 3. The NotI metal binding domain.
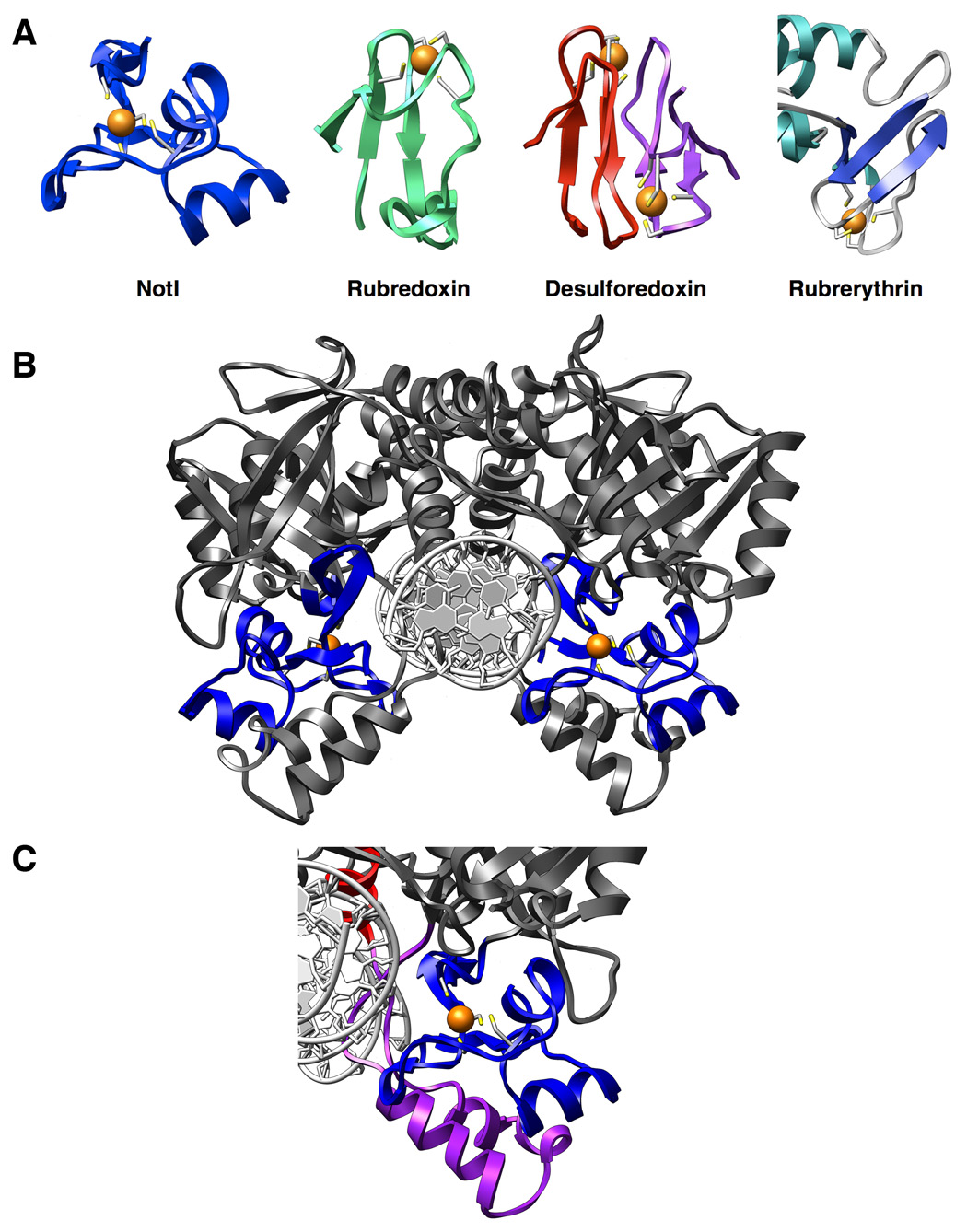
Panel A: FeCys4 Proteins - Rubredoxin (6rxn), desulforedoxin (1dxg), and rubrerythrin (1ryt) are three redox enzymes which contain the Fe-Cys4 iron binding motif, often referred to as the “rubredoxin-type” fold. Panel B: Homodimeric view of NotI with metal-binding domains highlighted in blue. Bound irons are shown as orange spheres and coordinating cysteine residues are colored by element and represented as sticks. The metal-binding domain is sandwiched between the nuclease core of the protein and the clamp domain (see panel c). Panel C: Iron-binding domain (blue) flanked by the clamp domain (purple). The iron-binding domain appears to serve an essential structural role in positioning of the clamp domain for contact with the DNA. The base of the DNA recognition helix making additional contacts to the DNA is also shown (red).
The N-terminal metal-binding domain of NotI is tightly associated with the nuclease domain and held in place by a “clamp” created by two helices (α7 and α8) that forms a large structural insertion between β-strands 9 and 10 of the nuclease domain. These two helices are part of a larger elaboration of the endonuclease domain at this insertion point, which also includes the alpha helix (α9) involved in dimerization and DNA binding, as described above. The metal ion is located approximately 15 Å from the scissile phosphate. It does not appear to be coupled to the enzyme's active site through polarizable side chains or solvent molecules in a manner that would facilitate participation in phosphoryl hydrolysis, either indirectly via redox effects or directly as a charged center that might promote proton transfer. Three residues within this protein domain (located in a loop preceding β-strand β2) make nonspecific contacts to the DNA backbone.
DNA binding and recognition
Comparison of the bound and unbound structures of NotI demonstrate a small but significant conformational change, corresponding to closure of the two protein subunits around the DNA duplex (Figure 4). This closure consists of a rigid body motion of the N-terminal metal-bound domain and its surrounding clamp region, described above, in both subunits. Very little movement is observed across the dimer interface and between the catalytic core regions of the enzyme. Three separate protein tethers simultaneously act as extended hinge regions: residues 86–87 (the linker between the N-terminal domain and the endonuclease catalytic core) and the linkers flanking the N- and C-terminal ends of the 'clamp' (residues 185 to191 and 224 to 230). As a result of this movement, a large number of residues in the latter two regions are inserted into the major groove of the target DNA to make several base-specific contacts.
Figure 4. Conformational change due to DNA-binding.
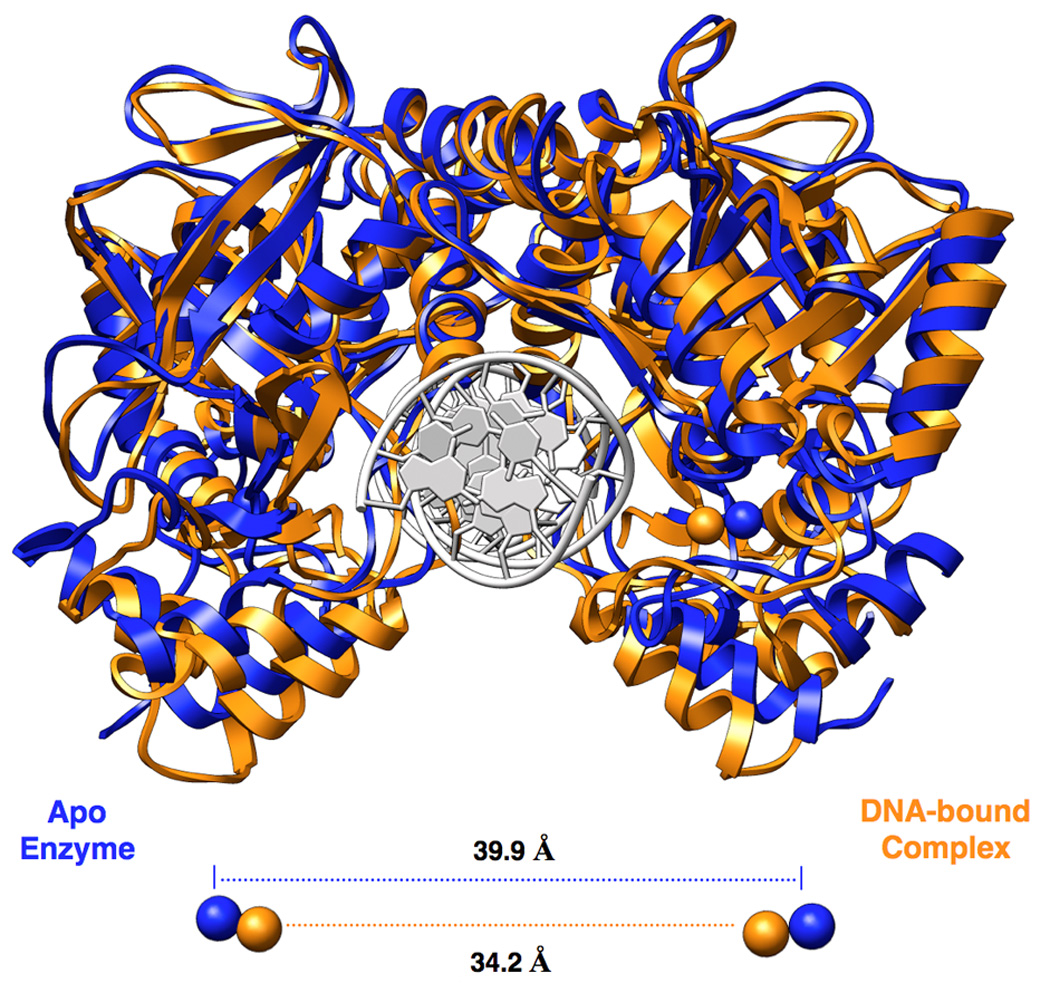
Superposition of apo (blue) and DNA-bound (orange) NotI structures for visualization of the conformational change which takes place upon binding of DNA. A decrease in distance between iron atoms of approximately 5.7Å is observed between the two structures, illustrating the motion taking place in the metal-binding and clamp domains. Less movement is observed in the nuclease core and dimerization regions of the structure. The DNA from the DNA-bound structure is colored white and shown for reference.
The DNA duplex in the bound complex is notable for its relatively unperturbed B-form conformation (Figure 1 and Figure 2, Supplementary Fig. S5). The individual values of 'roll' and 'twist' angles at each base step throughout the site (that can result in basepair unstacking due to rotation of nucleotide bases about axes orthogonal to the DNA helix) remain within +/− 6 degrees of B-form DNA, and the corresponding set of 'twist' angles (representing over- or under-winding of individual base steps) remains within 5 degrees of the average value for B-DNA. In comparison, the homodimeric restriction endonuclease EcoRV (which recognizes a 6 basepair site) and the homodimeric homing endonuclease I-CreI (which recognizes a 22 basepair target site) both display large departures from average DNA conformational parameters (Supplementary Fig. S5).
The NotI enzyme recognizes its 8 basepair target sequence by making an extensive network of direct and water-mediated interactions to both the DNA backbone and to the hydrogen bond donors and acceptors of individual nucleotide bases (Figure 5a). A total of nineteen amino acids residues from each protein subunit make contacts to the bound DNA: seventeen through direct hydrogen bonds by protein side chains and two through similar contacts by the protein backbone. These residues are contributed by several different regions of the protein, including six residues from helix α9 described above (the "DNA recognition helix" that is also involved in dimerization), three residues from a surface loop within the N-terminal iron-binding domain, nine residues from the mobile hinge regions that flank the "clamp", and one residue from a short β-strand on the surface of the nuclease domain (Figure 1). Eight of these residues are responsible for making specific hydrogen-bonding interactions to the DNA-bases; two of those interactions are water-mediated. The remaining 11 residues make contacts to the central six phosphate groups of each DNA strand, including at least four solvent-mediated contacts.
Figure 5. DNA Contacts.
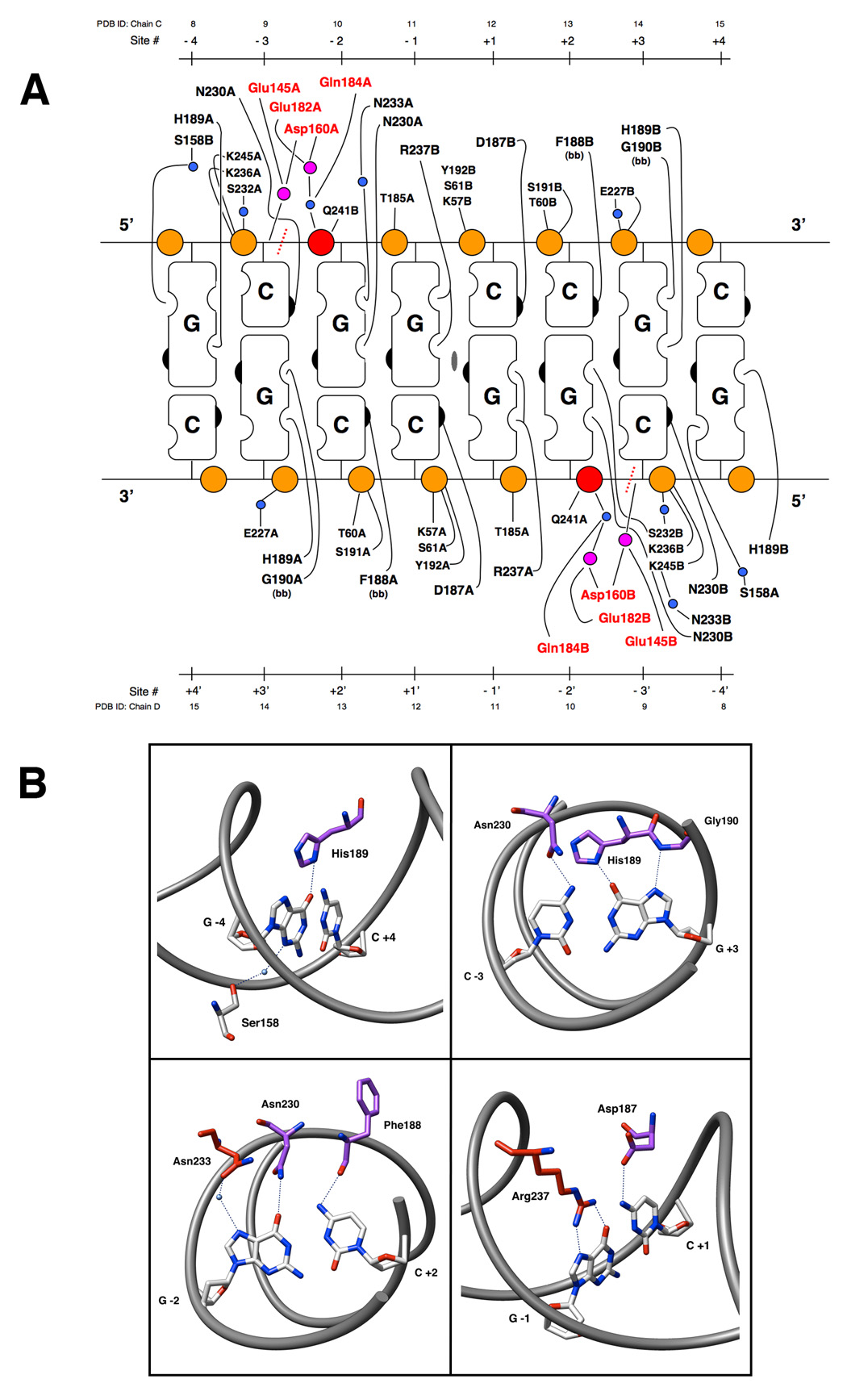
Panel A: Contacts make by the NotI restriction endonuclease to its 8 basepair DNA recognition sequence. Orange circles represent the phosphate groups of the DNA sugar-phosphate backbone. The scissile phosphats are colored red. Mounds and indents on the cartoon blocks represent the hydrogen bond donors and acceptors, respectively, of the individual nucleotide bases. The right hand side of the bases represents the major groove of the DNA and the left side represents the minor groove. The 2-fold rotation axis in the center of the cartoon designates the center of symmetry for the palindromic target DNA sequence. Small blue circles represent water molecules, and magenta circles are calcium ions. Residues from the active site are colored red. The site of DNA cleavage is designated by a dashed red line. The bases of the recognition sequence are numbered both outward from the center of symmetry (designated Site #) as well as by their PDB ID from the model coordinates. Panel B: Specific contacts made to each basepair of the NotI recognition sequence. Protein sidechain stick representations are colored based on their domain of origin: clamp domain (purple) or DNA recognition helix (red). Important atoms of bases and sidechains are colored by element. Hydrogen bonding interactions are represented by dashed lines. Waters are shown as blue spheres.
Contacts to the central six basepairs of the entirely G:C containing target site are fully saturated throughout the DNA major groove, as is commonly observed for most restriction endonuclease/DNA complexes (Figure 5b). Each cytosine extracyclic amine is contacted by a carbonyl oxygen from the protein backbone or an Asn/Asp side chain, and each guanine is engaged by two proton donating groups (one guanine in each half-site by a pair of contacts to a single arginine residue; the other two by individual contacts to Asn or His side chains or backbone nitrogens). In contrast, the most distal G:C basepair (positions +/−4) are read out through a single contact in the major groove (between a histidine side chain and the guanine extracyclic carbonyl oxygen) and a single solvent-mediated contact to the same base in the minor groove. No contacts are made to the cytosine base of this position (Supplementary Fig. S6).
Active Site
The DNA strands are uncleaved in the cocrystal structure. Electron density and atomic contacts consistent with a pair of bound metal ions (modeled as calcium, which is present in the crystallization buffer) are located near the scissile phosphate of each DNA strand (Figure 6a). One ion is loosely coordinated to the 3′ oxygen that becomes the leaving group upon phosphoryl hydrolysis (distance = 3.1 Å), and is more tightly coordinated by carboxylate oxygens contributed by two neighboring acidic residues: Glu 145 (2.9 Å) and Asp 160 (2.6 Å). The bond angles through the calcium between each of the three oxygen ligands are near 90°. Additional solvent molecules that would be necessary to complete the coordination of the calcium ion in each active site are not resolved at the resolution of this structure, but there is adequate room and appropriate chemical neighbors to accommodate a fully solvated metal ion at this position.
Figure 6. The NotI active site.
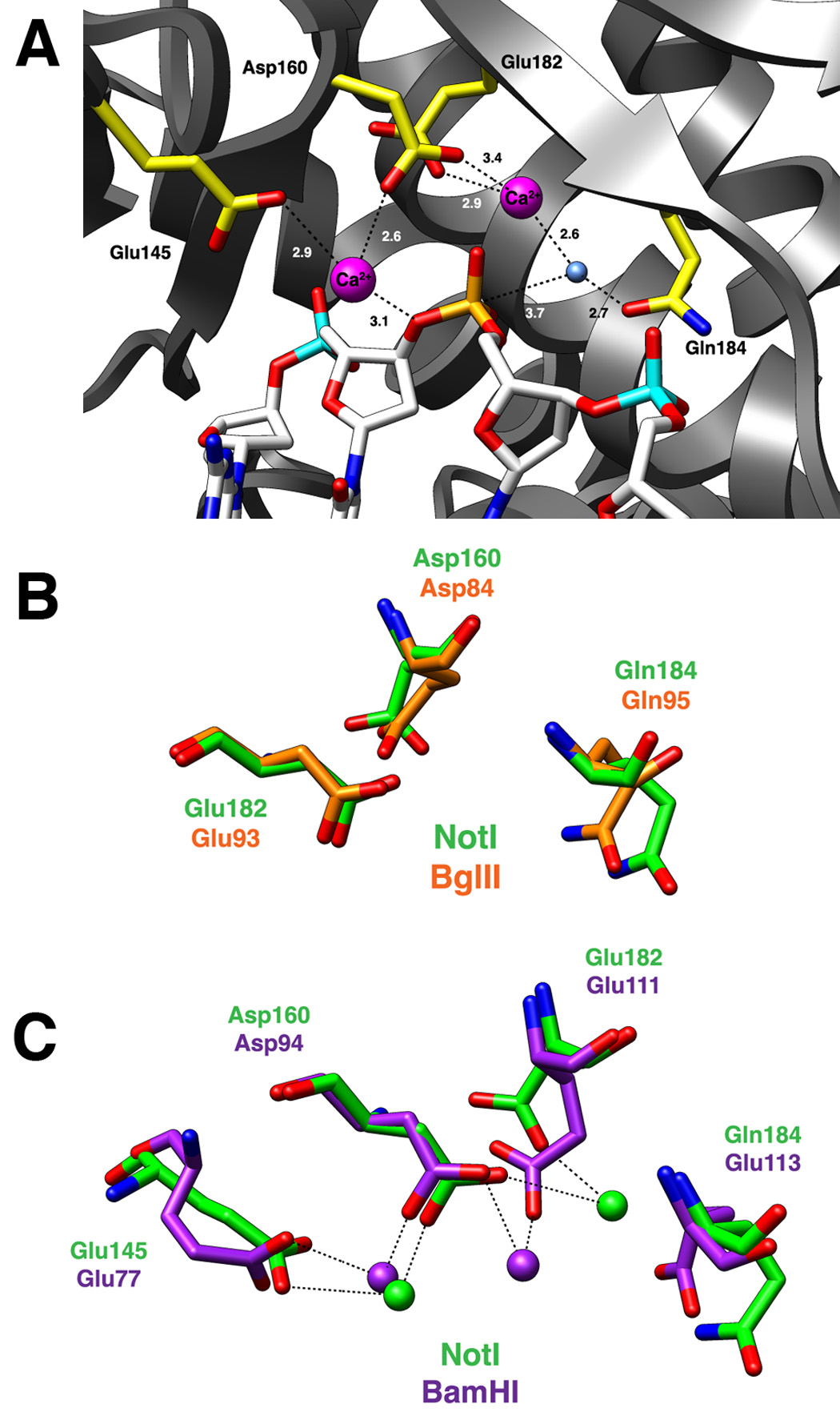
Panel A: Active site residues Glu145, Asp160, Glu182, and Gln184 are colored yellow. The bound DNA is colored white with the scissile phosphate designated orange. The nucleophilic water molecule is shown as a blue sphere and two calcium ions are depicted as magenta spheres. Dashed lines represent interactions between atoms in the active site and numbers indicate distances in angstroms (as average values calculated between the two active sites of the NotI homodimer). Panel B: Superposition of active site residues of the PD…(D/E)xK active site motif from NotI (green) and BglII (orange). Both REases have a glutamine in the general base position typically occupied by a lysine residue. Panel C: Superposition of the two-metal ion active sites of NotI (green) and BamHI (purple). Asp160/Asp94, Glu182/Glu111, and Gln184/Glu113 are residues belonging to the canonical PD…(D/E)xK nuclease motif, while the 3rd acidic residue, Glu145/Glu77, helps coordinate a second metal ion in the active site. The position of this 3rd acidic residue is conserved between the NotI and BamHI enzymes.
A second bound metal is located near the nonbridging oxygens of the scissile phosphate and is coordinated by a third acidic residue (Glu 182). A well ordered water molecule is coordinated by this second metal ion and by a neighboring glutamine side chain (Gln 184) and occupies a position appropriate for in-line attack and displacement of the 3′ oxygen leaving group (distance to the phosphate 3.7 Å; angle through the phosphate ~ 160°).
The active site chemistry is similar to that observed in the BglII restriction endonuclease (Figure 6b) and fulfills the PD…(D/E)xK catalytic motif using the arrangement F159D160…E182xQ184. Asp 160 and Glu 182 have been identified as catalytic residues by mutational analyses (Zhang et al., 2007). The NotI active site represents the third example of a glutamine residue (Gln 184) occupying the site commonly reserved for a lysine residue in the PD…(D/E)xK motif, joining BglII and BstYI. Additionally, an essential glutamine residue fills a similar structural position in the LAGLIDADG homing endonuclease I-CreI (Chevalier et al., 2001) and its closest relatives. In NotI, a single lysine residue (Lys 245) is found in the vicinity of the active site; however this residue is engaged in a tight interaction with the DNA backbone at an adjacent phosphate group.
Discussion
Elaborations on restriction endonuclease folds that alter their properties
The universe of restriction-modification systems encompasses a wide variety of oligomeric architectures, domain organizations, structural and functional elaborations, and variations of catalytic mechanisms, ranging from ATPase driven, multi-subunit assemblages (Type I and III endonucleases) to the simplest forms of free-standing enzymes (Type II endonucleases) (Bourniquel and Bickle, 2002; Pingoud et al., 2005).
Although a significant fraction of restriction-modification systems are Type I or III molecular machines, the majority of restriction enzymes that have been biochemically characterized to date belong to the Type II superfamily (Pingoud et al., 2005). Due to the extensive diversity found within the Type II REases, they are further categorized into subclasses based on their recognition sequence, cleavage pattern, domain organization, quaternary structure, and requirements for binding and cleavage (Roberts et al., 2003). Canonical Type IIP restriction endonucleases, including NotI, are homodimers that recognize palindromic DNA target sequences and cleave both strands of the DNA within or immediately adjacent to their recognition sequence. Their only mechanistic requirement is bound magnesium ion(s), although exceptions have been identified (Grazulis et al., 2005).
To achieve their considerable variation in structure and mechanism, restriction endonucleases have sampled and incorporated various amounts of structural elaboration of the nuclease core fold. These elaborations range from extension of DNA-interacting regions beyond the nuclease core domain, to more extensive insertions of elaborate structural elements or entire domains that interact with bound DNA and contribute additional base-specific DNA contacts. Most relevant to this study, two structures (SdaI and NotI) now exist that demonstrate structural elaborations incorporated in addition to the endonuclease core fold that are associated with recognition of long, uninterrupted target sites and rare cutting behavior. The structure of SdaI reveals the presence of a winged helix-turn-helix (wHTH) domain which has been tethered to a PD…(D/E)xK nuclease core domain (Tamulaitiene et al., 2006). The restriction activity of the SdaI enzyme was attributed to the isolated nuclease domain while all DNA recognition activity is predicted to be performed by the wHTH domain, a common DNA-binding motif. Although the crystal structure did not include bound DNA, this prediction is supported by electrostatic and mutation analysis (Tamulaitiene et al., 2006). Therefore, the SdaI endonuclease displays DNA recognition behavior similar to type IIS endonucleases (such as FokI) while also maintaining a homodimeric structure, palindromic DNA target, and cleavage pattern similar to canonical type IIP endonucleases.
NotI employs a separate strategy, completely unique from that employed by SdaI, to also achieve long target site recognition. First, it uses a ~ 65 residue insertion (the “clamp” domain and recognition helix) between strands β9 and β10 of the nuclease core for the purpose of making intimate, specific contacts with bound DNA (Figure 1). Additionally, NotI has acquired an N-terminal 85-residue metal-binding domain. Positioned near to the DNA as well, this domain makes three non-specific DNA-backbone contacts, and also participates in positioning the clamp domain for proper contact with the DNA (see discussion below). The unique aspect of structural elaborations within NotI is that the enzyme structure combines the structural insertions and additional domain into a compact, single-subunit design rather than the modular, multi-domain architecture observed in the SdaI (and FokI) REase structures (Tamulaitiene et al., 2006; Wah et al., 1997).
In addition to these two type IIP 8-basepair cutters, the structure of SfiI (a type IIF tetrameric endonuclease that recognizes a discontinuous 8-basepair target site) has also been determined (Vanamee et al., 2005). Unlike the structures of NotI and SdaI, this enzyme achieves recognition of four basepairs in each DNA half-site strictly through interactions with the core PD…(D/E)xK nuclease domain, without the use of additional protein motifs.
The metal binding domain
Crystals of NotI were analyzed by x-ray fluorescence absorption and emission scans for the presence of both iron and zinc (Supplementary Fig. S4). The metal content of the protein (under expression conditions that were not augmented with either metal) implicates the N-terminal domain in NotI as a Cys4 iron-sulfur center. The Fe-Cys4 iron-binding motif is commonly found in redox active electron transport proteins such as rubredoxin, desulforedoxin, and rubrerythrin (Archer et al., 1995; deMare et al., 1996; Stenkamp et al., 1990). Such iron centers have been shown to be one-electron carriers, and Fe/S clusters are often found in components of electron transport chains such as those found in photosynthesis and aerobic respiration (Johnson et al., 2005). However, the iron atoms of NotI are located far from the enzyme’s active sites (15Å), and there are no sidechains suitable for electron transfer between the irons and the DNA which might indicate or allow for metal participation in phosphoryl bond hydrolysis. Given that all canonical requirements for DNA-bond cleavage are present and accounted for in the identified PD…(D/E)xK nuclease active site (Figure 6), no iron participation in catalysis of phosphoryl hydrolysis would appear necessary. Therefore, the N-terminal iron-bound domain of NotI appears to be playing a structural rather than catalytic role. Mutation of Cys42, Cys55, or Cys81 has been shown to inactivate enzyme activity (X.-Y. Xu, unpublished data).
Given that iron is a highly limited resource under many bacterial growth conditions (Imlay, 2006), it is unusual to see this metal used solely to nucleate and stabilize a structural motif, rather than serve a redox-active catalytic role in an enzyme. However, in certain cases iron-sulfur clusters appear to fill noncatalytic roles in DNA modification enzymes (Flint and Allen, 1996). In the DNA repair glycosylase MutY, DNA-binding appears to promote oxidation of a noncatalytic Cys4 4Fe-4S cluster, which in turn sends an electron through the π-stacked DNA bases (“DNA-mediated charge-transfer”) until it encounters either a second MutY Fe-S cluster (of an adjacent bound enzyme) or a site of DNA damage. This process is therefore thought to provide a method for communication between bound repair enzymes, thus promoting more efficient location of DNA damage (Boal et al., 2005; Lukianova and David, 2005). Homologues of this same Cys4 iron-sulfur cluster are found in a wide range of deglycosylase enzymes, many of which are involved in DNA repair or in removal of methylated cytosine bases (Morales-Tuiz et al., 2006; Roloff et al., 2003). A variety of spectroscopic experiments on the 4Fe-4S cluster of Nth1 base excision enzymes (also known as endonuclease III) have shown that the cluster does not directly interact with bound DNA and does not participate in catalysis (Cunningham et al., 1989). The crystal structure of endonuclease III suggests that the Fe-S cluster is involved in correctly aligning important positively-charged residues for interaction with DNA (Kuo et al., 1992).
It is possible that the ancestor of the iron-bound domain of NotI originated in response to selective conditions experienced by a microbe that evolved in an unusual iron-rich environment, and then arrived in NotI and Nocardia via horizontal transfer of a mobile endonuclease system. A recent study of the archaea Ferroplasma acidiphilum, which is representative of bacterial species that flourish in acidic metal-rich environments, revealed an astoundingly iron-dominated repertoire of proteins (Ferrer et al., 2007). Many of these iron metalloenzymes were found to be metal-free in homologous proteins of related species. It was concluded that over 85% of the entire Ferroplasma proteome may consist of iron-metalloproteins, and that iron had become indispensable components of specialized structural domains in these protein structures, acting as “iron rivets.”
A BLAST (Altschul et al., 1997) search for sequence homologues of NotI, and comparison of its sequence with that of the EagI restriction endonuclease (Roberts et al., 2007) indicate that its N-terminal metal binding domain is found in a large number of putative bacterial endonucleases; many of these sequences display significant (25% to 35%) sequence identity with NotI. Alignment of these reading frames (Figure 1d) using program ClustalW (Thompson et al., 1994) demonstrates that the cysteine ligands involved in metal coordination are strongly conserved (with one position displaying some variability in its position within the primary sequence of the domain). As well, these homologues all display strong conservation of the catalytic residues in the NotI active site, corresponding to a consensus motif of "F/M/V-D…ExQ".
Diversity of restriction endonuclease active sites
Although Type II REases show little homology on the sequence level, crystallographic analyses have allowed for the identification and validation of the PD…(D/E)xK nuclease catalytic motif common to 24 of the 26 REase structures solved to date. The two exceptions that have been visualized are the endonuclease BfiI, which contains a phospholipase D-family nuclease domain linked to a specific DNA-binding domain (Grazulis et al., 2005) and the endonuclease PabI, which contains a novel catalytic fold termed the “half-pipe” (Miyazono et al., 2007). Additional sequence-based bioinformatics studies have predicted that additional endonuclease active site motifs are employed by some restriction endonucleases, including the HNH motif (for example KpnI; 5’-GGTAC/C-3’) (Saravanan et al., 2004b) and the GIY-YIG motif (Eco29kI; 5’-CCGC/GG-3’) (Ibryashkina et al., 2007b).
The exact requirements for residues of the PD…(D/E)xK motif during DNA cleavage vary widely, with related mechanisms defined both by the number of metal ions present in the enzyme active site (corresponding to single metal, two-metal, and three-metal mechanisms) and by the exact identity and necessity of the general base (often a lysine) in the REase mechanism (Pingoud et al., 2005). Within the family of PD…(D/E)xK REases, the sequence position and identity of individual active site residues have been observed to undergo considerable drift while maintaining remarkable structural conservation in the active site: (i) The number of amino acids between the first and second acidic metal-coordinating residues can range from 4 to 34 residues; (ii) the conserved lysine residue and the latter acidic residue of the motif can often be 'swapped' in the active site, requiring the modified nuclease motif of PD-(X21–55)-K-(X12–13)-E (Bochtler et al., 2006; Deibert et al., 2000; Grazulis et al., 2002; Skirgaila et al., 1998; Zhou et al., 2004); (iii) the identify of metal-binding residues can be highly variable, with Glu, Asp, and Asn all capable of substituting for one another (Bochtler et al., 2006; Xu et al., 2004); and (iv) the basic lysine residue found in many REase active sites can be replaced by a either a glutamine (BglII (Lukacs et al., 2000), BstYI (Townson et al., 2004) and now NotI) or by a glutamate residue (BamHI (Xu and Schildkraut, 1991)) (Supplementary Fig. S7).
The active site of NotI represents the third example of a PD…(D/E)xK motif with a glutamine residue in the general base position. NotI active site residues Asp160, Glu182 and Gln184 match up with the conserved residues of the canonical PD…(D/E)xK nuclease motif, and superimpose closely with the same residues from BglII (Figure 6b). However, while BglII is classified as following a one-metal ion mechanism (Lukacs et al., 2000), the active site of NotI appears to accommodate two bound metal ions, as a result of the presence of an additional catalytic acidic residue (Glu 145) that produces an metal-binding geometry in the active site similar to that observed for BamHI (Figure 6c).
Are rare-cutting restriction endonucleases gaining, losing or regaining specificity?
A growing body of evidence indicates that restriction-modification enzyme (R-M) systems have arisen within bacterial genomes via invasive horizontal transfer events. Comparative sequence analyses of R-M genes, and their association with mobile DNA vectors such as plasmids, viruses and transposons indicate extensive and frequent horizontal transfer. At least one study has demonstrated that a restriction endonuclease gene (encoding EcoRI) displays homing-type mobility when placed into the appropriate sequence context (Eddy and Gold, 1992).
Upon residence in a bacterial genome, R-M gene systems act as selfish elements and promote their own survival through two mechanisms: participation in cellular defense (which provides an advantage to the host) and post-segregational host killing (in which loss of the R-M system results in cell death as unmethylated sites in newly replicated DNA are cleaved by residual levels of restriction endonuclease) (Handa et al., 2000; Handa and Kobayashi, 1999). Both mechanisms exert selection pressure on invasive R-M systems to experience specificity decreases, both to ensure frequent cleavage of invading foreign DNA and to compete with alternative R-M genes that recognize overlapping cognate target sites (Chinen et al., 2000).
Given that restriction endonucleases display many of the characteristics of invasive genes and selfish DNA elements, it seems plausible that such gene systems might share distant evolutionary relationships with modern-day microbial invasive elements, such as homing endonucleases, and may even have initially descended from such genes . In such a scenario (Figure 7), an invasive endonuclease (which initially would not be paired with a corresponding protective activity, such as a methyltransferase enzyme) would be required to display sufficiently long site recognition properties, or have its expression and/or activity down-regulated, to avoid undesirable cleavage activity and toxicity in the host. Such an element, if harboring no additional activities of benefit to the host, would be subject to strong selective pressure to eventually be eliminated from the bacterial genome (Edgell et al., 2000). However, if subsequently coupled within the host to a protective genome editing activity, the endonuclease would be free to experience reduction in its target site length and specificity-- generating advantages for both the host and for itself, as described above. Physical linkage of the genes of such a rudimentary R-M system would then promote its mobilization. The final length of the target site recognized by the restriction endonuclease would be dependent on the base composition of the host and the resulting frequency of the target site both in the host genome and in its corresponding phage population.
Figure 7. From invasive endonuclease to restriction endonuclease?
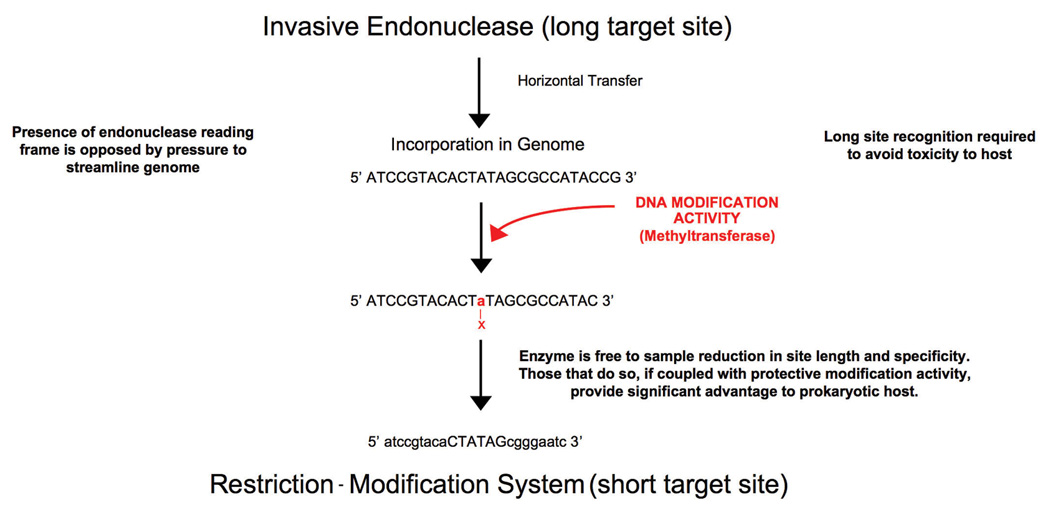
A variety of studies, described in the text, indicate that restriction endonucleases may have originated from ancestral invasive endonucleases (and in fact may still display features of mobility and 'selfishness'. The original invasive mobile endonucleases would be required to display long target site recognition, in order to avoid toxicity to the host; the persistance of such elements would generally be opposed by pressure to minimize and streamline genome size. The acquisition of a beneficial secondary activity would improve the fitness of the host in the presence of the endonuclease gene. One such mechanism for the development of a beneficial function would be the coupling of a DNA modification activity that blocks the cleavage activity of the endonuclease in the host, with subsequent reduction in its cleavage specificity. If invasive endonuclease families (such as homing endonucleases) were related to defensive restriction endonucleases, one would expect to detect the presence of restriction endonucleases with nonoptimal, overly long target sites that are obviously related to more 'advanced' short site cutters. The NotI endonuclease (with its reduced contacts to the basepairs at positions +/−4) and its relationship to the six-basepair cutter EagI appears to fulfill the requirements of an invasive/defensive restriction endonuclease 'intermediate'.
If this hypothesis is true, examples of catalytic protein folds that transcend the boundaries of modern homing and restriction endonuclease families should be observed. This prediction has been fulfilled in multiple systems. Two separate folds typically associated with homing endonucleases have been observed in canonical restriction endonuclease systems (for example, R.KpnI is an HNH endonuclease, and R.Eco29kI is a GIY-YIG endonuclease) (Ibryashkina et al., 2007a; Saravanan et al., 2004a). Similarly, the homing endonuclease I-Ssp6803I ("I-SspI", which is found embedded in persistent group I introns in the fMet-tRNA genes of cyanobacteria) is a tetrameric PD…(D/E)xK endonuclease (Zhao et al., 2007) --the most common catalytic protein fold in restriction endonuclease systems.
An additional prediction arising from this model is that endonucleases that descend from common ancestors, that become resident in host genomes with significantly different base compositions, should recognize DNA target sites with similar sequences but different lengths (so that each recognizes a target that is found at an appropriate frequency in its host and in corresponding phage). The length of the NotI target site (8 basepairs, occuring once per 65kB of random DNA sequence) initially appears inappropriate for restriction. However, the genome base composition of Nocardia bacterial species appear significantly enhanced for G:C content (the Nocardia farcinica genome is 73% G:C (Ishikawa et al., 2004); a variety of sequenced genes from Nocardia otitidis-caviarum are 65 to 70% G:C), which would increase the frequency of the NotI target site. In contrast, the EagI endonuclease from Enterobacter agglomerans (GenBank entry EU371940, Figure 1) (Sznyter and Brooks, 1988) harbors significant sequence identity with NotI, but only recognizes the central six basepairs of the NotI target site. The genome composition of reading frames cloned and sequenced from that bacterial host is much closer to 50% G:C (B. L. Stoddard, personal observation), which would make its shorter target site appropriate within the context of its genomic location.
Therefore, the target site lengths for NotI and EagI appears to have stabilized at eight and six basepairs, respectively, in response to the base content of their bacterial host genomes and the resulting frequency of target site occurence. The longer target site recognized by NotI could either reflect a situation where the enzyme has 'stalled' en route to a shorter site, during its evolution from an invasive ancestor (as a result of the skewed basepair content of its host), or a situation where the enzyme has 'regained' additional target site length from a canonical restriction endonuclease ancestor. In either case, recognition of the most distal basepair in each half site (positions +/− 4) in NotI is unusual (Figure 5). Unlike contacts made to the central six basepairs (in which every potential hydrogen bond donor and acceptor in the major groove is fully engaged by the endonuclease), these outer basepairs are engaged in the major groove through a single contact with a histidine residue, leading to alternate recognition of an A:T basepair (star activity) under nonoptimal buffer conditions (Samuelson et al., 2006). The disparity of this contact pattern, relative to the usual over-saturation of DNA bases by restriction endonucleases, is most easily explained by a model of evolution in which there is less pressure for perfect recognition of the outer-most basepairs. The observations described above for the NotI/EagI endonucleases have also been reported for for the SfiI endonuclease and a corresponding six basepair cutting enzyme, BglI.
Experimental Procedures
Expanded methods are provided in supplementary information. Briefly, the protein was expressed as a C-terminal fusion to the Sce intein and chitin binding domain in E. coli strain ER2744 and purified according to the manufacturer's protocol for the IMPACT system (New England Biolabs). The protein was purified via chitin affinity purification and cation exchange chromatography. Se-Met labeled protein was expressed in a similar manner in T7 Express Crystal strain (NEB #C3022). Prior to crystallization, all protein was dialyzed into a crystallization buffer containing 5% glycerol, 50 mM Tris-HCl pH 7.6, 20 mM NaCl, and 1mM CaCl2. Crystals were grown of the apo enzyme (2.0 M (NH4)2SO4, 100 mM MES pH 6.5, 10 mM CaCl2, and 20 mM NaCl) and the DNA-bound complex (29–39% PEG 4000, 130–240 mM LiSO4, 100mM Tris HCl pH 8.5, in the presence of a 2:1 molar ratio of a DNA duplex containing 5’-CGGAGGCGCGGCCGCGCCGCCG-3’ and 5’- CGGCGGCGCGGCCGCGCCTCCG-3′). The structure of the apo enzyme was determined by a combination of MAD and SIR phasing (the latter using a mercury derivative prepared by soaking with mercury cyanide); the DNA complex was determined by MAD phasing and molecular replacement. The structures were built using COOT and refined with REFMAC. Data and refinement statistics are provided in Table I.
Table I.
Data Collection and Refinement Statistics
| Crystal Form #2 – Apo NotI (Dimer per asymmetric unit) | |||||
|---|---|---|---|---|---|
| Crystal | Native | Semet | HgCN2 | ||
| λ1 – Peak | λ2 – Inflection | λ3 – Remote | |||
| ALS Beamline | 5.0.1 | 5.0.2 | 5.0.2 | 5.0.2 | 5.0.2 |
| Wavelength (Å) | 1.0000 | 0.9804 | 0.9805 | 0.9744 | 1.0048 |
| Space group | P43212 | P43212 | P43212 | P43212 | P43212 |
| a = b, c (Å) | 75.0, 274.8 | 74.6, 273.2 | 74.7, 273.6 | 74.9, 274.1 | 75.0, 273.7 |
| Resolution (Å)* | 50.0 – 2.86 (2.96–2.86) | 50.0 – 2.80 (2.85–2.80) | 50.0 – 2.80 (2.85–2.80) | 50.0 – 2.80 (2.90–2.80) | 50.0 – 3.30 (3.42–3.30) |
| Unique Reflections | 18,707 | 19,948 | 20,050 | 20,201 | 12,564 |
| Redundancy | 6.5 (5.5) | 16.1 (14.3) | 11.0 (9.6) | 26.0 (21.0) | 15.1 (15.4) |
| Completeness (%) | 97.6 (91.0) | 99.5 (98.4) | 99.6 (99.4) | 99.6 (97.1) | 99.7 (100.0) |
| I/σ (I) | 25.1 (15.3) | 29.4 (8.8) | 25.1 (7.9) | 32.0 (7.9) | 21.2 (7.0) |
| Rmerge | 0.063 (0.192) | 0.089 (0.513) | 0.083 (0.424) | 0.109 (0.317) | 0.155 (0.469) |
| Rcryst | 26.2% | ||||
| Rfree (5% of reflections) | 33.5% | ||||
| Protein atoms / Ions | 5150 / 2 SO42− | ||||
| Rms deviation | |||||
| Bond length (Å) | 0.007 | ||||
| Bond angle (°) | 1.103 | ||||
| Crystal Form #1 – NotI–DNA Complex (Dimer plus 2bp DNA per asymmetric unit) | |||||
| Crystal | Native | Semet | |||
| λ1 – Peak | λ2 – Remote | ||||
| ALS Beamline | 8.2.1 | 5.0.2 | 5.0.2 | ||
| Wavelength (Å) | 1.5000 | 0.9794 | 0.9755 | ||
| Space group | P21 | P21 | P21 | ||
| a, b, c (Å) | 73.9, 81.7, 73.6 | 73.4, 81.0, 73.8 | 73.5, 81.1, 73.9 | ||
| β (°) | 99.5 | 98.9 | 98.9 | ||
| Resolution (Å) | 50.0 – 2.50 (2.59–2.50) | 50.0 – 3.00 (3.11–3.00) | 50.0 – 3.00 (3.11–3.00) | ||
| Unique Reflections | 29,473 | 16,881 | 16,852 | ||
| Redundancy | 6.8 (5.0) | 6.8 (4.7) | 7.1 (5.7) | ||
| Completeness (%) | 97.7 (82.2) | 96.6 (78.9) | 96.8 (79.5) | ||
| I/σ (I) | 27.7 (3.9) | 21.5 (3.1) | 25.2 (3.8) | ||
| Rmerge | 0.056 (0.274) | 0.088 (0.374) | 0.062 (0.333) | ||
| Rcryst | 22.3% | ||||
| Rfree (5% of reflections) | 27.7% | ||||
| Protein atoms / DNA / waters | 5532 / 896 / 145 | ||||
| Rms deviation | |||||
| Bond length (Å) | 0.007 | ||||
| Bond angle (°) | 1.094 | ||||
Numbers in parentheses represent values in the highest resolution shell.
Coordinate and Data Deposition
The X-ray structure factor amplitudes and corresponding refined coordinates for the NotI apo enzyme and DNA-bound complex have been deposited in the RCSB database for immediate release (PDB ID codes 3BVQ and 3BVR).
Supplementary Material
Acknowledgements
X-ray data was collected at the Advanced Light Source (ALS) synchrotron facility at the Lawrence Berkeley National Laboratory (University of California) on beamlines 5.0.1, 5.0.2 and 8.2.1 with the assistance of multiple staff. We thank members of the laboratories of Roland Strong and Adrian Ferre-D'Amare for advice and assistance during structure determination. Molecular graphics images were produced using the UCSF Chimera package (Pettersen et al., 2004) from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081). This work was supported by funding from the NIH to BLS (R01 GM49857) and by NIH training grant support to ARL (T32 GM07270).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickle TA, Kruger DH. Biology of DNA restriction. Microbiol Rev. 1993;57:434–450. doi: 10.1128/mr.57.2.434-450.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourniquel AA, Bickle TA. Complex restriction enzymes: NTP-driven molecular motors. Biochimie. 2002;84:1047–1059. doi: 10.1016/s0300-9084(02)00020-2. [DOI] [PubMed] [Google Scholar]
- Chevalier BS, Monnat RJ, Jr, Stoddard BL. The homing endonuclease I-CreI uses three metals, one of which is shared between the two active sites. Nat Struct Biol. 2001;8:312–316. doi: 10.1038/86181. [DOI] [PubMed] [Google Scholar]
- Chinen A, Naito Y, Handa N, Kobayashi I. Evolution of sequence recognition by restriction-modification enzymes: selective pressure for specificity decrease. Mol Biol Evol. 2000;17:1610–1619. doi: 10.1093/oxfordjournals.molbev.a026260. [DOI] [PubMed] [Google Scholar]
- Cunningham RP, Asahara H, Bank JF, Scholes CP, Salerno JC, Surerus K, Munck E, McCracken J, Peisach J, Emptage MH. Endonuclease III is an iron-sulfur protein. Biochemistry. 1989;28:4450–4455. doi: 10.1021/bi00436a049. [DOI] [PubMed] [Google Scholar]
- Eddy SR, Gold L. Artificial mobile DNA element constructed from the EcoRI endonuclease gene. Proc Natl Acad Sci U S A. 1992;89:1544–1547. doi: 10.1073/pnas.89.5.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgell DR, Belfort M, Shub DA. Barriers to intron promiscuity in bacteria. J Bacteriol. 2000;182:5281–5289. doi: 10.1128/jb.182.19.5281-5289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer M, Golyshina OV, Beloqui A, Golyshin PN, Timmis KN. The cellular machinery of Ferroplasma acidiphilum is iron-protein-dominated. Nature. 2007;445:91–94. doi: 10.1038/nature05362. [DOI] [PubMed] [Google Scholar]
- Flint DH, Allen RM. Iron-Sulfur Proteins with Nonredox Functions. Chem Rev. 1996;96:2315–2334. doi: 10.1021/cr950041r. [DOI] [PubMed] [Google Scholar]
- Grazulis S, Manakova E, Roessle M, Bochtler M, Tamulaitiene G, Huber R, Siksnys V. Structure of the metal-independent restriction enzyme BfiI reveals fusion of a specific DNA-binding domain with a nonspecific nuclease. Proc Natl Acad Sci U S A. 2005;102:15797–15802. doi: 10.1073/pnas.0507949102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa N, Ichige A, Kusano K, Kobayashi I. Cellular responses to postsegregational killing by restriction-modification genes. J Bacteriol. 2000;182:2218–2229. doi: 10.1128/jb.182.8.2218-2229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa N, Kobayashi I. Post-segregational killing by restriction modification gene complexes: observations of individual cell deaths. Biochimie. 1999;81:931–938. doi: 10.1016/s0300-9084(99)00201-1. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- Ibryashkina EM, Zakharova MV, Baskunov VB, Bogdanova ES, Nagornykh MO, Den'mukhamedov MM, Melnik BS, Kolinski A, Gront D, Feder M, et al. Type II restriction endonuclease R.Eco29kI is a member of the GIY-YIG nuclease superfamily. BMC Structural Biology. 2007a;7 doi: 10.1186/1472-6807-7-48. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibryashkina EM, Zakharova MV, Baskunov VB, Bogdanova ES, Nagornykh MO, Den'mukhamedov MM, Melnik BS, Kolinski A, Gront D, Feder M, et al. Type II restriction endonuclease R.Eco29kI is a member of the GIY-YIG nuclease superfamily. BMC Struct Biol. 2007b;7:48. doi: 10.1186/1472-6807-7-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay JA. Iron-sulphur clusters and the problem with oxygen. Mol Microbiol. 2006;59:1073–1082. doi: 10.1111/j.1365-2958.2006.05028.x. [DOI] [PubMed] [Google Scholar]
- Ishikawa J, Yamashita A, Mikami Y, Hoshino Y, Kurita H, Hotta K, Shiba T, Hattori M. The complete genomic sequence of Nocardia farcinica IFM 10152. Proc Natl Acad Sci U S A. 2004;101:14925–14930. doi: 10.1073/pnas.0406410101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Dean DR, Smith AD, Johnson MK. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 2005;74:247–281. doi: 10.1146/annurev.biochem.74.082803.133518. [DOI] [PubMed] [Google Scholar]
- Kuo CF, McRee DE, Fisher CL, O'Handley SF, Cunningham RP, Tainer JA. Atomic structure of the DNA repair [4Fe-4S] enzyme endonuclease III. Science. 1992;258:434–440. doi: 10.1126/science.1411536. [DOI] [PubMed] [Google Scholar]
- Lukacs CM, Kucera R, Schildkraut I, Aggarwal AK. Understanding the immutability of restriction enzymes: crystal structure of BglII and its DNA substrate at 1.5 A resolution. Nat Struct Biol. 2000;7:134–140. doi: 10.1038/72405. [DOI] [PubMed] [Google Scholar]
- Miyazono K, Watanabe M, Kosinski J, Ishikawa K, Kamo M, Sawasaki T, Nagata K, Bujnicki JM, Endo Y, Tanokura M, Kobayashi I. Novel protein fold discovered in the PabI family of restriction enzymes. Nucleic Acids Res. 2007;35:1908–1918. doi: 10.1093/nar/gkm091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Tuiz T, Ortega-Galisteo AP, Ponferrada-Marin MI, Martinez-Macias MI, Ariza RR, Roldan-Arjona T. Demeter and repressor of silencing 1 encode 5-methylcytosine DNA clycosylases. Proc Natl Acad Sci U S A. 2006;103:6853–6858. doi: 10.1073/pnas.0601109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pingoud A, Fuxreiter M, Pingoud V, Wende W. Type II restriction endonucleases: structure and mechanism. Cell Mol Life Sci. 2005;62:685–707. doi: 10.1007/s00018-004-4513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang BQ, Schildkraut I. NotI and SfiI: restriction endonucleases with octanucleotide recognition sequences. Methods Enzymol. 1987;155:15–21. doi: 10.1016/0076-6879(87)55005-4. [DOI] [PubMed] [Google Scholar]
- Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE--enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 2007;35:D269–D270. doi: 10.1093/nar/gkl891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roloff TC, Ropers HH, Nuber UA. Comparative study of methyl-CpG-binding domain proteins. BMC Genomics. 2003;4:1. doi: 10.1186/1471-2164-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush LJ, Plass C. Restriction landmark genomic scanning for DNA methylation in cancer: past, present, and future applications. Anal Biochem. 2002;307:191–201. doi: 10.1016/s0003-2697(02)00033-7. [DOI] [PubMed] [Google Scholar]
- Samuelson JC, Morgan RD, Benner JS, Claus TE, Packard SL, Xu SY. Engineering a rare-cutting restriction enzyme: genetic screening and selection of NotI variants. Nucleic Acids Res. 2006;34:796–805. doi: 10.1093/nar/gkj483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravanan M, Bujnicki JM, Cymerman IA, Rao DN, Nagaraja V. Type II restriction endonuclease R.KpnI is a member of the HNH nuclease superfamily. Nucleic Acids Res. 2004a;32:6129–6135. doi: 10.1093/nar/gkh951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saravanan M, Bujnicki JM, Cymerman IA, Rao DN, Nagaraja V. Type II restriction endonuclease R.KpnI is a member of the HNH nuclease superfamily. Nucleic Acids Res. 2004b;32:6129–6135. doi: 10.1093/nar/gkh951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smiraglia DJ, Plass C. The study of aberrant methylation in cancer via restriction landmark genomic scanning. Oncogene. 2002;21:5414–5426. doi: 10.1038/sj.onc.1205608. [DOI] [PubMed] [Google Scholar]
- Sznyter LA, Brooks JE. The characterization and cloning of the EagI restriction-modification system. Gene. 1988;74:53. doi: 10.1016/0378-1119(88)90250-8. [DOI] [PubMed] [Google Scholar]
- Tamulaitiene G, Jakubauskas A, Urbanke C, Huber R, Grazulis S, Siksnys V. The crystal structure of the rare-cutting restriction enzyme SdaI reveals unexpected domain architecture. Structure. 2006;14:1389–1400. doi: 10.1016/j.str.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townson SA, Samuelson JC, Vanamee ES, Edwards TA, Escalante CR, Xu SY, Aggarwal AK. Crystal structure of BstYI at 1.85A resolution: a thermophilic restriction endonuclease with overlapping specificities to BamHI and BglII. J Mol Biol. 2004;338:725–733. doi: 10.1016/j.jmb.2004.02.074. [DOI] [PubMed] [Google Scholar]
- Vanamee ES, Viadiu H, Kucera R, Dorner L, Picone S, Schildkraut I, Aggarwal AK. A view of consecutive binding events from structures of tetrameric endonuclease SfiI bound to DNA. Embo J. 2005;24:4198–4208. doi: 10.1038/sj.emboj.7600880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S-Y, Schildkraut I. Isolation of BamHI variants with reduced cleavage activities. J Biol Chem. 1991;266:4425–4429. [PubMed] [Google Scholar]
- Zhang P, Bao Y, Higgins L, Xu S-Y. rational design of a chimeric endonuclease targeted to NotI recognition site. Protein Engineering Design and Selection. 2007 doi: 10.1093/protein/gzm049. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Bonocora RP, Shub DA, Stoddard BL. The restriction fold turns to the dark side: a bacterial homing endonuclease with a PD-(D/E)-XK motif. Embo J. 2007;26:2432–2442. doi: 10.1038/sj.emboj.7601672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.