

Abstract
Objective
The anti-apoptotic action of HBEGF and its regulation by O2 constitutes a key factor for trophoblast survival. The hypothesis that cytotrophoblast survival is compromised by exposure to hypoxia–reoxygenation (H/R) injury, which may contribute to preeclampsia and some missed abortions, prompted us to investigate HBEGF regulation and its role as a survival factor during H/R in cytotrophoblast cells
Study Design
A transformed human first trimester cytotrophoblast cell line HTR-8/SVneo was exposed to H/R (2% O2 followed by 20% O2) and assessed for HBEGF expression and cell death.
Results
Cellular HBEGF declined significantly within 30 minutes of reoxygenation after culture at 2% O2. H/R significantly reduced proliferation and increased cell death when compared to trophoblast cells cultured continuously at 2% or 20% O2. Restoration of cell survival also was achieved by adding recombinant HBEGF during reoxygenation. HBEGF inhibited apoptosis through its binding to either HER1 or HER4, its cognate receptors.
Conclusion
These results provide evidence that cytotrophoblast exposure to H/R induces apoptosis and decreased cell proliferation. HBEGF accumulation is diminished under these conditions, while restoration of HBEGF signaling improves trophoblast survival.
Keywords: Cytotrophoblast, HBEGF, hypoxia, hypoxia/reoxygenation, apoptosis, EGF receptor, HER4
INTRODUCTION
There is evidence that non-physiological hypoxia/reoxygenation (H/R) occurs in the late first trimester and is responsible for abnormal remodeling of the placenta and its membranes (1). In normal pregnancy the perfusion of maternal blood into the intervillous space is gradual and occurs at the periphery while in missed abortion there is premature onset of perfusion to all regions of the placenta (2). The possible role of H/R in early pregnancy loss has been advanced by several lines of evidence. These include premature onset of the maternal circulation to the placenta, determined by Doppler ultrasonography and correlated to fragmented trophoblastic shell, limited EVT invasion of the placental bed and terminal spiral arteries (1–5). The effect is greatest in pregnancies less than 10 weeks gestation with recent fetal demise suggesting that early gestations are more susceptible to this damage. In pregnancies that survive, H/R has been suggested as the underlying defect that hinders physiological conversion of the spiral arteries, which predisposes to preeclampsia (6–8). Several of the cellular changes observed with H/R exposure of third trimester villous explants are similar to those found in early pregnancy loss, including lipid peroxidation, protein nitrosylation and apoptosis (7, 8). Hypoxia alone does not explain the observed damage by reactive O2 species (ROS) generated during preeclampsia(9). ROS are not efficiently generated by hypoxia in third trimester villous explants, whereas H/R produces ROS within minutes (7).
The capability of some cells, including cytotrophoblast (CT), to survive at low O2 tension requires O2 sensing and multiple early responses, including ion channel changes, glycolytic enzymes, transcription factors and integrins. Heparin-binding EGF-like growth factor (HBEGF) is expressed in the placenta through out pregnancy (10, 11) and is upregulated by low O2 through a post-transcriptional mechanism to inhibit apoptosis (12). Signaling via HER1 or HER4, known HBEGF receptors, is required for both HBEGF upregulation and protection against apoptosis (12). Consistent with its proposed role as a prosurvival factor in response to ROS in first trimester cytotrophoblast, HBEGF expression is expressed robustly in trophoblast, but is reduced about five-fold in preeclamptic placentas (11).
Based on its expression in the human placenta during the first trimester and its induction by oxygen, HBEGF constitutes a key regulator of trophoblast survival. The observed decrease in survival of trophoblast exposed to H/R injury prompted us to investigate the role of HBEGF in trophoblast survival during H/R using an in vitro cell culture model.
MATERIALS AND METHODS
Cell culture and O2 regulation
HTR-8/SVneo cytotrophoblast cells were plated and grown in serum containing medium, which was replaced with serum-free medium 24 hours prior to all experiments, as previously described (13). The cell line was routinely phenotyped to assure cytokeratin-7 expression, hCG secretion, invasion of Matrigel™ basement membrane and HLA-G expression by invading cells. A humidified 5% CO2/95% air incubator was used for ambient cell culture (20% O2). Culture at low O2 was achieved by placing cell cultures into a humidified modular incubator chamber (Billups- Rothenberg, Del Mar, CA) that was then flushed for 5 minutes with a mixture of 2% O2, 5% CO2 and 93% N2 (Wilson Gases, Detroit, MI). The concentration of the gas mixture was confirmed as previously described (12). The exposure to 2% O2 continued for a total of 8 hr and represented the hypoxia exposed cells. Cells exposed to H/R were cultured at 2% O2 for 2 hr and then switched for an additional 6 hrs to medium pre-equilibrated at 20% O2.
Cell treatments
Medium was changed to add vehicle or supplements to growing cell cultures 30 minutes before exposure to ambient or 2% O2, which initiated each experiment. Medium was supplemented with 1 nM recombinant HBEGF (R&D Systems, Minneapolis, MN), 10 µg/ml mouse anti-HER1 (Ab-2) or HER4 (Ab-3) blocking antibodies (LabVision, Fremont, CA), 20 µg/ml mouse non-immune IgG (Jackson ImmunoResearch Laboratories, West Grove, PA), 20 µg/ml of the capase inhibitors, Z-DEVD-FMK, Z-VAD-FMK or 200 ug/ml of the negative control Z-FA-FMK (EMD Biosciences), or 2 mM H2O2 (Sigma).
Immunohistochemistry
Cytotrophoblast cells grown in 150 µl of medium in 96-well tissue culture plates (Becton Dickinson) were processed for immunohistochemistry, as previously described(13)Nuclei expressing Ki-67 were labeled with a monoclonal antibody (Ki-S5; DAKO, Carpinteria, CA). Goat polyclonal antibody (R&D Systems) against human recombinant HBEGF, was used at a concentration (5 ug/ml) in the linear portion of its binding curve. Controls were incubated with 10 µg/ml non-immune IgG (Jackson ImmunoResearch). Cells labeled with goat primary antibodies were incubated 1 hour at 25°C with 0.1 ug/ml rabbit anti-goat IgG (Jackson ImmunoResearch). To visualize and quantify (gray level) antigen, an Envision SystemTM peroxidase anti-mouse/rabbit kit (DAKO) was used in conjunction with image analysis, according to our published procedure (11). Values obtained with IgG substituted for primary antibody were subtracted from each sample.
Cell death and proliferation assays
Cytotrophoblast cells were grown and treated in 150 µl of medium in 96-well plates prior to assay. Cell death was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) using a fluorescein-based kit from Roche Applied Science (Indianapolis, IN) and counterstaining with 5 µg/ml 4,6-diamidino-2-phenylindole, HCl (DAPI; EMD Biosciences) as previously described (12). Fluorescent nuclei were viewed at 20x magnification using a Leica (Wetzlar, Germany) DM IRB epifluorescence microscope and representative images of both DAPI and fluorescein fluorescence were acquired with a Hamamatsu Orca digital camera (Hamamatsu City, Japan). Images were processed using Simple PCI (C-Imaging, Cranberry Township, PA) imaging software, adjusting the threshold to optimize automated counting of fluorescent nuclei. The percentage of TUNEL/DAPI-labeled nuclei (TUNEL index) was determined from triplicate fields in each well. Cells labeled with antibody against Ki-67 were counterstained with DAPI and similarly assessed for the percentage of Ki-67/DAPI-labeled nuclei as an index of cell proliferation. Externalized phosphatidylserine was detected by incubating live cells for 15 minutes at room temperature with biotin-labeled annexin V (1:20; Molecular Probes, Portland, OR) in 10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7. Cells were then fixed and incubated overnight at 4°C with 5 µg/ml UltraAvidin-Texas Red (Leinco Technologies, St. Louis, MO) and 5 mg/ml BSA in PBS. Cell lysis (necrosis) was assessed as previously described by measuring lactate dehydrogenase (LDH) activity released into the culture medium with a DHLTM Cell Cytotoxicity Assay Kit (AnaSpec, San Jose, CA), according to the manufacturer’s instructions (12). Cells grown in black, clear-bottom 96-well tissue culture plates (Corning, Corning, NY) were treated (n=3) as detailed in the Results section in 100 □l of modified BWW medium (Irvine Scientific, Santa Ana, CA). After medium was collected for assay in separate wells, cells were washed three times with BWW medium and lysed for assay of total LDH. The fluorescent reaction product was quantified using a SpectraMax M2 multiplate spectrofluorometer (Molecular Devices, Sunnyvale, CA). To calculate the LDH release index, the value of LDH activity in the medium, multiplied by 100, was divided by the value obtained from the lysed cells.
HBEGF ELISA
HBEGF was quantified using an ELISA kit (R&D Systems), essentially as previously described (12). The calculated inter- and intra-assay coefficients of variation for this human HBEGF immunoassay were 3.45% and 2.26%, respectively. The detection limit of the assay was 12 pg/ml. The HBEGF concentration of each sample was calculated by interpolation from the corresponding standard curve and was expressed as picogram per microgram cellular protein.
Real-time RT-PCR
Cytotrophoblast cells grown and treated in 2 ml of medium in six-well plates were washed three times with sterile PBS (Cambrex Bio Sciences, Verviers, Belgium) and RNA was extracted using RNeasy Mini Kits from Qiagen (Valencia, CA). The primers 5’-TGG TGC TGA AGCTCT TTC TGG-3’ (sense) and 5’GTG GGA ATT AGT CAT GCC CAA- 3’ (antisense) were used to measure HBEGF mRNA copy number by real time PCR as previously described (12).
Statistics
Assays were conducted using replicate samples and all experiments were repeated at least three times. The program SPSS version 12.0 (SPSS, Chicago, IL) was used to determine statistical significance. For immunohistochemical quantification, the level of each growth factor at 20% and 2% O2 was compared with a two-tailed Student’s independent t-test. Comparisons were made to vehicle-treated controls for Ki-67, TUNEL and LDH data. TUNEL data that did not meet the assumption of equal variances among groups were log transformed before analysis. All ELISA data were analyzed by the Kruskal-Wallis non-parametric ANOVA with the Mann-Whitney posthoc test, using the Holm modification to the Bonferonni correction. All graphed data are presented as mean ± s.e.m.
RESULTS
We previously reported that HBEGF protein levels increase at least 100-fold in HTR-8/SVneo cytotrophoblast cells when the O2 concentration is reduced to 2% (12). HBEGF accumulation in human cytotrophoblast cells, determined by immunohistochemistry and image analysis, was reduced significantly after reoxygenation as compared to 2% O2 treatment (Fig. 1A). Down regulation of HBEGF occurred rapidly (Fig. 1B), reaching significance within 30 min according to a quantitative ELISA. HBEGF mRNA levels do not vary between ambient and 2% O2 (12), nor did reoxygenation alter transcript expression when determined by real time RT-PCR (data not shown). It appeared that O2 regulates HBEGF through a post-transcriptional mechanism and that its cytoprotective activity becomes unavailable soon after O2 levels rise in the course of an H/R paradigm.
Figure 1. HBEGF protein levels during hypoxia and reoxygenation.
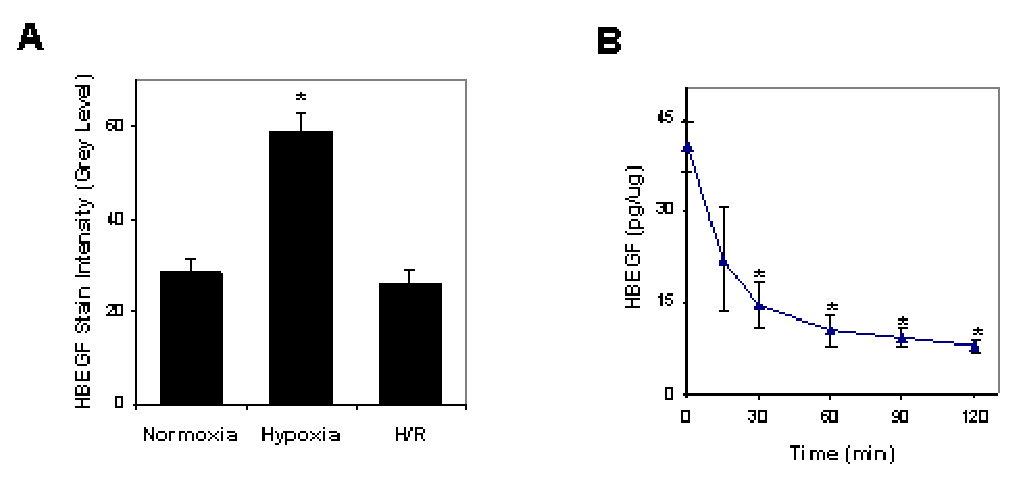
A. Cytotrophoblast cells were cultured at 20% O2 for 8 hr (normoxia), 2% O2 for 8 hr (hypoxia) or 2% O2 for 2 hr followed by 20% O2 for 6 hr (H/R). HBEGF was assessed by quantitative immunohistochemistry and image analysis, as described in the Materials & Methods. B. HBEGF was quantified by ELISA in cell extracts during reoxygenation after 5 hr of culture at 2% O2. *, p<0.05 compared to normoxia (A) or 0 time at normoxia (B).
Cell proliferation, assessed by nuclear Ki67 expression, was increased by culture at 2% compared to 20% O2 (Fig. 2A), but was reduced below the normoxia value upon reoxygenation. As a further measure of cellular distress, cell death was gauged by TUNEL assay. Cells maintained at 20% or 2% O2 showed no difference in apoptosis (Fig. 2B). However, 6 h after reoxygenation of cells maintained at 2% O2, TUNEL increased 2-fold (Fig. 2B). The induction of cell death by H/R was reversed by pretreatment with inhibitor caspase 3 or a pan caspase inhibitor (Fig. 2C), providing evidence of apoptotic pathway activation (14). Moreover, elevated phosphatidylserine exposure on the outer surface of cytotrophoblast cells exposed to H/R was detected by annexin V binding (Fig. 3), a hallmark of apoptotic pathway activity (14). H/R did not cause necrosis of cytotrophoblast cells, according to the lack of LDH release to the medium (Fig. 2D), which monitors cell lysis occurring as a result of necrosis (14). As a positive control for necrosis, exposure to peroxide treatment caused LDH accumulation in the medium.
Figure 2. Cytotrophoblast death is due to apoptosis after H/R.
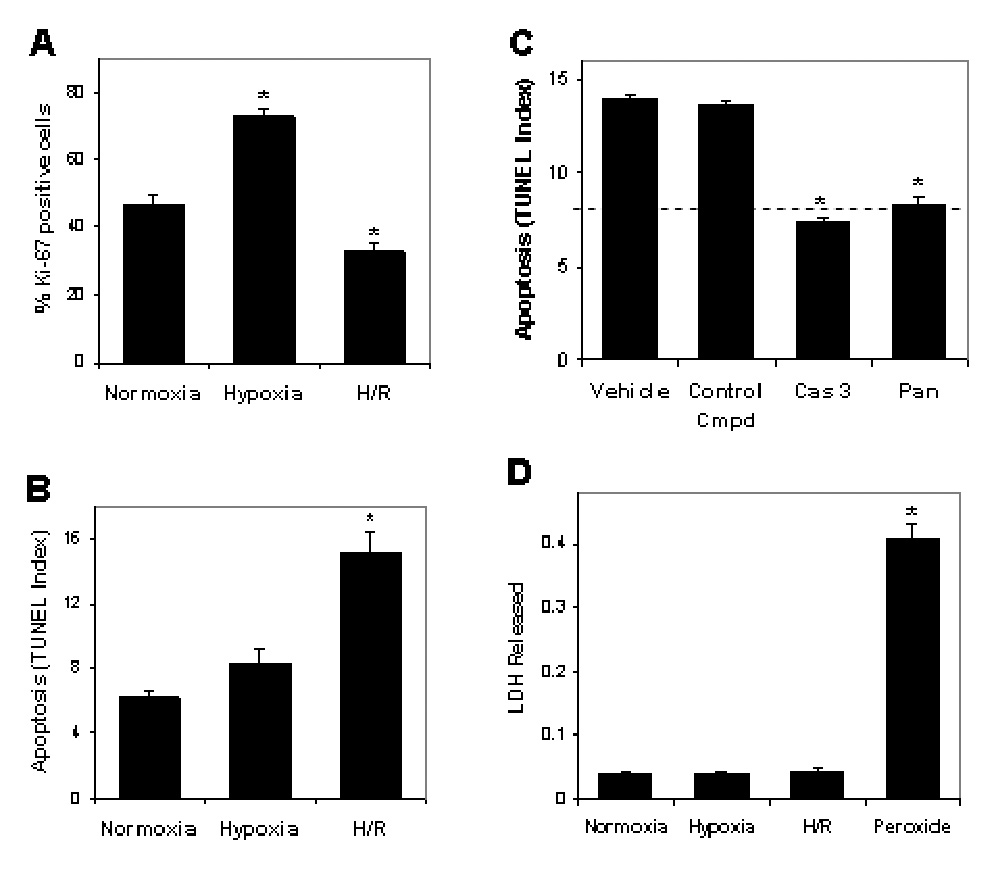
A. Proliferation of cytotrophoblast cells cultured as in Fig. 1A was assessed by immunofluorescence using anti-Ki-67 antibody and DAPI counterstain to derive the percentage of Ki67-positive cells. B. Cell death was quantified by TUNEL assay of cytotrophoblast cells cultured as in Fig 1A. C. Cytotrophoblast were exposed to H/R in the absence (vehicle) or presence of 20 µM of caspase 3 inhibitor (Cas 3), an inhibitor of all caspases (Pan) or an inactive peptide analogue (Control Cmpd). The dashed line indicates the level of TUNEL under normoxic conditions. D. LDH released from cytotrophoblast cells cultured as in Fig. 1A was measured as described in the Materials & Methods. As positive control for necrosis, cells were treated for 30 min with 10 nM H2O2 (Peroxide). *, p<0.05 compared to normoxia.
Figure 3. H/R increases annexin V binding to cytotrophoblast cells.
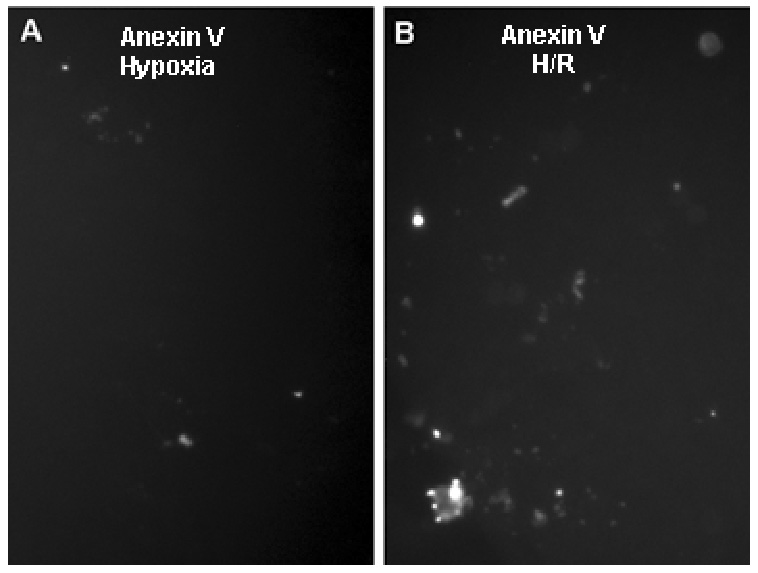
Cells were cultured for 8 hr at 2% O2 (A) or at 2% O2 for 2 hr followed by 20% O2 for 6 hr (B) to generate H/R. Cells were then labeled with annexin V, as described in the Materials & Methods. Immunoflurorescence is shown for fields of equivalent cell density. Magnification, 20×.
The foregoing experiments reveal an inverse relationship between HBEGF expression and survival of cytotrophoblast cells during oxidative stress. To rigorously test this hypothesis, we supplemented cells with human recombinant HBEGF during reoxygenation to determine if survival could be improved. As shown in Fig. 4, 1 nM HBEGF attenuated apoptosis during H/R to levels comparable to normoxic controls. The interaction of HBEGF with its receptors was examined using function-blocking antibodies against HER1 or HER4. Neither antibody alone prevented HBEGF rescue; however, the combination of both antibodies proved effective in returning TUNEL to significant levels above the control (Fig. 4). These results indicate that HBEGF prevents apoptosis through interaction with either of its two receptors and directly establishes that HER downstream signaling can regulate survival of cytotrophoblast cells exposed to oxidative stress caused by H/R.
Figure 4. Cytotrophoblast apoptosis after H/R is rescued by recombinant HBEGF signaling through its cognate receptors, HER1 and HER4.
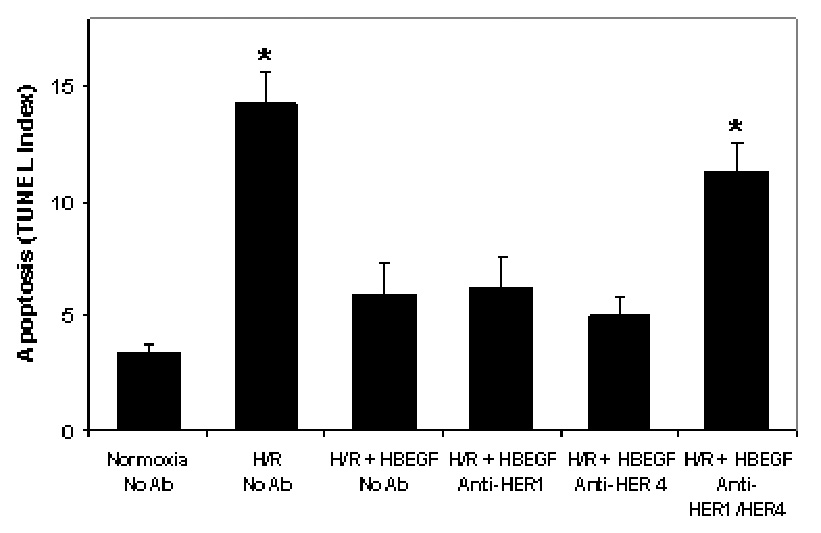
TUNEL was determined as in Fig. 2B for cytotrophoblast cells cultured under the indicated O2 treatment regimes with or without addition of 1 nM recombinant HBEGF, or 10 µM of blocking antibodies against HER1 and HER4, as shown on the × axis. *, p< 0.05 compared to control (Normoxia no Ab).
COMMENT
The regulation of HBEGF expression by H/R was examined in the immortalized first trimester cytotrophoblast cell line, HTR-8/SVneo. This cell line displays several characteristic features of first trimester cytotrophoblast, including expression of HLA-G, integrin switching from α6β4 to α1β1 during basement membrane invasion, and increased proliferation under physiologic hypoxic conditions (13). We previously demonstrated that HBEGF upregulation is critical for the survival of human cytotrophoblast cells during exposure to low concentrations of O2 (12), such as those encountered in utero during the first trimester of gestation (15). Disruption of the relatively hypoxic environment of the early placenta by reoxygenation exposes embryonic tissues to ROS and could potentially result in cell death, as observed in an in vitro perfusion model with term placenta (6–8). Our findings, using a first trimester cytotrophoblast cell line, reveal that the protective activity of endogenous HBEGF is lost when O2 levels are abruptly increased. The resulting cell death requires caspase activity and is accompanied by phosphatidylserine randomization, while cells remain intact and retain cytoplasmic LDH activity; all indicative of apoptosis(14) It has been suggested that excessive oxidative stress in the first trimester can compromise trophoblast function or survival (1–5, 9). Trophoblast survival and extravillous differentiation required for remodeling of the uterine spiral arteries are deficient in both missed abortion and preeclampsia (1), perhaps due to elevated levels of oxidative stress. The data presented in this report indicate that signaling downstream of the HBEGF receptors HER1 and HER4 is capable of stemming progression toward apoptosis. Ethanol-induced apoptosis is similarly blocked by the EGF signaling system in HTR-8/SVneo cells (16) suggesting a broad survival function for this pathway.
A role for H/R injury in early pregnancy loss has been advanced by several lines of molecular evidence, including morphological evidence of increased apoptosis and markers of cellular stress, expression of heat shock protein 70, protein nitrosylation and lipid peroxidation in tissues obtained from missed abortions with early onset placental perfusion (1–5). Xanthine dehydrogenase, which normally utilizes NAD as an electron acceptor, is converted under the conditions of ischemia/reperfusion into xanthine oxidase. During ischemia, excessive ATP consumption leads to the accumulation of the purine catabolites, hypoxanthine and xanthine. With subsequent reperfusion of oxygen, they are metabolized by xanthine oxidase to yield massive amounts of superoxide (17). In experimental systems, H/R is associated with induction of the redox-responsive trancription factors NF-κB and AP-1, as well as the MAPKs c-jun NH2-terminal kinase (JNK), p38 and the extracellular signal-regulated kinases ERK1 and ERK2 (18, 19). This activation could account for inflammatory responses and apoptotic cell death in the affected tissue. In proximal renal tubule cells, reoxygenation following hypoxia results in activation of MEK/ERK and PI3K/Akt/XIAP survival signaling pathways, which are mediated through HER1 (20). The anti-apoptotic action of neuregulin 1-β, another member of the EGF-like family, requires phosphorylation of HER-4 in rat hearts exposed to ischemia-reperfusion injury (21).
We have extended the above findings to first trimester cytotrophoblast by demonstrating that re-exposure to 20% O2 for as little as 2 hours decreases cellular HBEGF significantly, with a concomitant increase in apoptosis. Cleaved, secreted HBEGF and subsequent downstream signaling are required for its prosurvival action at 2% O2 (12). The rapidity of the decline in HBEGF during reoxygenation was facilitated through a post-transcriptional mechanism, suggested by the lack of change in HBEGF mRNA expression with fluctuating O2 concentrations. These findings contrast with HBEGF regulation in response to oxidative stress in other cell contexts, including intestinal and bladder epithelial cells, which occurs at the mRNA level (22–25).
HBEGF accumulation has been examined in other cell types and organ systems during H/R. HBEGF mRNA and protein levels rose after reoxygenation after rat intestinal and renal epithelial cells were exposed to anoxia in vitro (26, 27). The apparent discrepancy in HBEGF response between these studies and our findings in cytotrophoblast may reflect the lower O2 levels (0%) present in the rat studies, duration of anoxic exposure prior to reoxygenation or differences in cell context. HBEGF expression after ischemia and reperfusion injury in vivo has been studied in several organ systems. Rat kidneys subjected to H/R increased HBEGF mRNA and protein accumulation in the distal tubule, suggesting its role as a prosurvival factor in this setting (28). HBEGF expression is increased in rat intestinal epithelium after H/R and when injected into the intestinal lumen, epithelial and crypt cells survive by down-regulating inducible nitric oxide synthase and nitric oxide production (29).
HTR-8/SVneo cell proliferation increases at 2% O2 (12, 13) as in primary cultured cytotrophoblast and villous explants under similar O2 conditions (30, 31). The inhibition of HBEGF secretion or its binding to HER 1 and HER 4 during exposure to 2% O2 did not prevent the increase in cell proliferation (12). During reoxygenation, Ki67 expression declined significantly below the basal level obtained from cytotrophoblast cells cultured at 20% O2. Therefore, proliferation was negatively impacted by H/R. It is not known whether the decline in HBEGF during reoxygenation is a factor in the consequential reduction in proliferation.
Premature exposure of the trophoblast to elevated O2 levels during early gestation could diminish cytotrophoblast survival or functions dependent on HBEGF and other hypoxia-induced molecules. These effects could be catastrophic for the conceptus, while sub-lethal oxidative stress may alter invasion and apoptosis in the first trimester to manifest as abnormal placentation that leads to pathology later in pregnancy. The present study establishes that H/R causes a rapid and significant reduction of HBEGF accumulation in an immortalized human first trimester cytotrophoblast cell line. H/R may decrease HBEGF availability through several possible mechanisms that include decreased HBEGF mRNA translation, increased HBEGF protein turnover, or reduced metalloproteinase-mediated shedding of HBEGF from the plasma membrane. Further investigation of these mechanisms will provide an understanding of the relationship between reoxygenation injury and HBEGF availability in the developing trophoblast.
ACKNOWLEDGEMENTS
We thank Dr Charles Graham, Queen’s University, Kingston, Ontario, Canada, for his generous gift of HTR-8/SVneo cells used in this study. We are grateful for the technical assistance of Dr. Zophia Duniec-Dmuchowski and Po Jen Chiang and statistical analysis conducted by Michel Kruger.
This work was supported by grants from the National Institutes of Health to R.E.L. (HD37500) and D.R.A. (AA12057) and by the Intramural Research Program of the National Institute of Child Health and Human Development, NIH, DHHS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain
Human cytotrophoblast cells exposed to hypoxia-reoxygenation downregulate HBEGF and begin to undergo apoptosis. Supplementation with HBEGF to activate its cognate HER receptors prevents cell death under these conditions, revealing an endogenous survival mechanism.
REFERENCES
- 1.Burton GJ, Jauniaux E. Placental oxidative stress: from miscarriage to preeclampsia. J Soc Gynecol Investig. 2004;11:342–352. doi: 10.1016/j.jsgi.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 2.Burton GJ, Jauniaux E, Watson AL. Maternal arterial connections to the placental intervillous space during the first trimester of human pregnancy: the Boyd collection revisited. Am J Obstet Gynecol. 1999;181:718–724. doi: 10.1016/s0002-9378(99)70518-1. [DOI] [PubMed] [Google Scholar]
- 3.Burton GJ, Hempstock J, Jauniaux E. Oxygen, early embryonic metabolism and free radical-mediated embryopathies. Reprod Biomed Online. 2003;6:84–96. doi: 10.1016/s1472-6483(10)62060-3. [DOI] [PubMed] [Google Scholar]
- 4.Hempstock J, Jauniaux E, Greenwold N, Burton GJ. The contribution of placental oxidative stress to early pregnancy failure. Hum Pathol. 2003;34:1265–1275. doi: 10.1016/j.humpath.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Jauniaux E, Watson AL, Hempstock J, Bao YP, Skepper JN, Burton GJ. Onset of maternal arterial blood flow and placental oxidative stress. A possible factor in human early pregnancy failure. Am J Pathol. 2000;157:2111–2122. doi: 10.1016/S0002-9440(10)64849-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hung TH, Burton GJ. Hypoxia and reoxygenation: a possible mechanism for placental oxidative stress in preeclampsia. Taiwan J Obstet Gynecol. 2006;45:189–200. doi: 10.1016/S1028-4559(09)60224-2. [DOI] [PubMed] [Google Scholar]
- 7.Hung TH, Skepper JN, Burton GJ. In vitro ischemia-reperfusion injury in term human placenta as a model for oxidative stress in pathological pregnancies. Am J Pathol. 2001;159:1031–1043. doi: 10.1016/S0002-9440(10)61778-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hung TH, Skepper JN, Charnock-Jones DS, Burton GJ. Hypoxia-reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res. 2002;90:1274–1281. doi: 10.1161/01.res.0000024411.22110.aa. [DOI] [PubMed] [Google Scholar]
- 9.Many A, Hubel CA, Fisher SJ, Roberts JM, Zhou Y. Invasive cytotrophoblasts manifest evidence of oxidative stress in preeclampsia. Am J Pathol. 2000;156:321–331. doi: 10.1016/S0002-9440(10)64733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leach RE, Khalifa R, Ramirez ND, Das SK, Wang J, Dey SK, Romero R, Armant DR. Multiple roles for heparin-binding epidermal growth factor-like growth factor are suggested by its cell-specific expression during the human endometrial cycle and early placentation. J Clin Endocrinol Metab. 1999;84:3355–3363. doi: 10.1210/jcem.84.9.5980. [DOI] [PubMed] [Google Scholar]
- 11.Leach RE, Romero R, Kim YM, Chaiworapongsa T, Kilburn B, Das SK, Dey SK, Johnson A, Qureshi F, Jacques S, Armant DR. Pre-eclampsia and expression of heparin-binding EGF-like growth factor. Lancet. 2002;360:1215–1219. doi: 10.1016/S0140-6736(02)11283-9. [DOI] [PubMed] [Google Scholar]
- 12.Armant DR, Kilburn BA, Petkova A, Edwin SS, Duniec-Dmuchowski ZM, Edwards HJ, Romero R, Leach RE. Human trophoblast survival at low oxygen concentrations requires metalloproteinase-mediated shedding of heparin-binding EGF-like growth factor. Development. 2006;133:751–759. doi: 10.1242/dev.02237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kilburn BA, Wang J, Duniec-Dmuchowski ZM, Leach RE, Romero R, Armant DR. Extracellular matrix composition and hypoxia regulate the expression of HLA-G and integrins in a human trophoblast cell line. Biol Reprod. 2000;62:739–747. doi: 10.1095/biolreprod62.3.739. [DOI] [PubMed] [Google Scholar]
- 14.Allen RT, Hunter WJ, 3rd, Agrawal DK. Morphological and biochemical characterization and analysis of apoptosis. J Pharmacol Toxicol Methods. 1997;37:215–228. doi: 10.1016/s1056-8719(97)00033-6. [DOI] [PubMed] [Google Scholar]
- 15.Jauniaux E, Watson A, Burton G. Evaluation of respiratory gases and acid-base gradients in human fetal fluids and uteroplacental tissue between 7 and 16 weeks' gestation. Am J Obstet Gynecol. 2001;184:998–1003. doi: 10.1067/mob.2001.111935. [DOI] [PubMed] [Google Scholar]
- 16.Kilburn BA, Chiang PJ, Wang J, Flentke GR, Smith SM, Armant DR. Rapid induction of apoptosis in gastrulating mouse embryos by ethanol and its prevention by HB-EGF. Alcohol Clin Exp Res. 2006;30:127–134. doi: 10.1111/j.1530-0277.2006.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Granger DN. Role of xanthine oxidase and granulocytes in ischemia-reperfusion injury. Am J Physiol. 1988;255:H1269–H1275. doi: 10.1152/ajpheart.1988.255.6.H1269. [DOI] [PubMed] [Google Scholar]
- 18.Clerk A, Fuller SJ, Michael A, Sugden PH. Stimulation of "stress-regulated" mitogen-activated protein kinases (stress-activated protein kinases/c-Jun N-terminal kinases and p38-mitogen-activated protein kinases) in perfused rat hearts by oxidative and other stresses. J Biol Chem. 1998;273:7228–7234. doi: 10.1074/jbc.273.13.7228. [DOI] [PubMed] [Google Scholar]
- 19.Lazou A, Bogoyevitch MA, Clerk A, Fuller SJ, Marshall CJ, Sugden PH. Regulation of mitogen-activated protein kinase cascade in adult rat heart preparations in vitro. Circ Res. 1994;75:932–941. doi: 10.1161/01.res.75.5.932. [DOI] [PubMed] [Google Scholar]
- 20.Kwon DS, Kwon CH, Kim JH, Woo JS, Jung JS, Kim YK. Signal transduction of MEK/ERK and PI3K/Akt activation by hypoxia/reoxygenation in renal epithelial cells. Eur J Cell Biol. 2006;85:1189–1199. doi: 10.1016/j.ejcb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Kuramochi Y, Cote GM, Guo X, Lebrasseur NK, Cui L, Liao R, Sawyer DB. Cardiac endothelial cells regulate reactive oxygen species-induced cardiomyocyte apoptosis through neuregulin-1beta/erbB4 signaling. J Biol Chem. 2004;279:51141–51147. doi: 10.1074/jbc.M408662200. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Adam RM, Freeman MR. Trafficking of nuclear heparin-binding epidermal growth factor-like growth factor into an epidermal growth factor receptor-dependent autocrine loop in response to oxidative stress. Cancer Res. 2005;65:8242–8249. doi: 10.1158/0008-5472.CAN-05-0942. [DOI] [PubMed] [Google Scholar]
- 23.Kim J, Lin J, Adam RM, Lamb C, Shively SB, Freeman MR. An oxidative stress mechanism mediates chelerythrine-induced heparin-binding EGF-like growth factor ectodomain shedding. J Cell Biochem. 2005;94:39–49. doi: 10.1002/jcb.20276. [DOI] [PubMed] [Google Scholar]
- 24.Michalsky MP, Kuhn A, Mehta V, Besner GE. Heparin-binding EGF-like growth factor decreases apoptosis in intestinal epithelial cells in vitro. J Pediatr Surg. 2001;36:1130–1135. doi: 10.1053/jpsu.2001.25730. [DOI] [PubMed] [Google Scholar]
- 25.Pillai SB, Turman MA, Besner GE. Heparin-binding EGF-like growth factor is cytoprotective for intestinal epithelial cells exposed to hypoxia. J Pediatr Surg. 1998;33:973–978. doi: 10.1016/s0022-3468(98)90517-6. discussion 978–979. [DOI] [PubMed] [Google Scholar]
- 26.Sakai M, Tsukada T, Harris RC. Oxidant stress activates AP-1 and heparin-binding epidermal growth factor-like growth factor transcription in renal epithelial cells. Exp Nephrol. 2001;9:28–39. doi: 10.1159/000020705. [DOI] [PubMed] [Google Scholar]
- 27.Xia G, Rachfal AW, Martin AE, Besner GE. Upregulation of endogenous heparin-binding EGF-like growth factor (HB-EGF) expression after intestinal ischemia/reperfusion injury. J Invest Surg. 2003;16:57–63. [PubMed] [Google Scholar]
- 28.Takemura T, Kondo S, Homma T, Sakai M, Harris RC. The membrane-bound form of heparin-binding epidermal growth factor-like growth factor promotes survival of cultured renal epithelial cells. J Biol Chem. 1997;272:31036–31042. doi: 10.1074/jbc.272.49.31036. [DOI] [PubMed] [Google Scholar]
- 29.Xia G, Lara-Marquez M, Luquette MH, Glenn S, Haque A, Besner GE. Heparin-binding EGF-like growth factor decreases inducible nitric oxide synthase and nitric oxide production after intestinal ischemia/reperfusion injury. Antioxid Redox Signal. 2001;3:919–930. doi: 10.1089/15230860152665073. [DOI] [PubMed] [Google Scholar]
- 30.Genbacev O, Joslin R, Damsky CH, Polliotti BM, Fisher SJ. Hypoxia alters early gestation human cytotrophoblast differentiation/invasion in vitro and models the placental defects that occur in preeclampsia. J Clin Invest. 1996;97:540–550. doi: 10.1172/JCI118447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jurisicova A, Detmar J, Caniggia I. Molecular mechanisms of trophoblast survival: from implantation to birth. Birth Defects Res C Embryo Today. 2005;75:262–280. doi: 10.1002/bdrc.20053. [DOI] [PubMed] [Google Scholar]