

Summary
Canonical Wnt signaling via β-catenin-dependent transcription critically regulates cell fate and proliferation in development and disease. However, mechanisms governing β-catenin nuclear localization are not well understood. Here we demonstrate that nuclear accumulation of β-catenin and gene activation in response to Wnt requires activation of Rac1, a Rho-family small GTPase, via Gαq/11βγ signaling involving phosphatidylinositol-3 kinase (PI-3K). Moreover, the role of Rac1 depends on phosphorylation of β-catenin at Ser191 and Ser605, an event chiefly mediated by c-Jun NH2-terminal kinase 2 (JNK2) in ST2 cells; mutations of these residues significantly affect β-catenin nuclear accumulation in response to Wnt. Genetic ablation of Rac1 in the mouse embryonic limb bud ectoderm disrupts canonical Wnt signaling, and phenocopies deletion of β-catenin in causing severe truncations of the limb. Finally, Rac1 interacts genetically with β-catenin and Dickkopf 1 (Dkk1) in controlling limb outgrowth. Together these results uncover Rac1 activation and subsequent β-catenin phosphorylation as a hitherto uncharacterized mechanism controlling canonical Wnt signaling, and may provide additional targets for therapeutic intervention of this important pathway.
Introduction
Wnt signaling is critical for normal development of multicellular organisms via regulation of cell fate, proliferation and behavior. Aberrant Wnt signaling has been implicated in human diseases (Clevers, 2006). In the canonical Wnt pathway, Wnt binding to Frizzled receptors and the low-density lipoprotein receptor-related protein 5 or 6 (LRP5/6), activates the cytoplasmic signaling protein Dishevelled (Dvl) to stabilize cytosolic β-catenin; β-catenin, upon entering the nucleus, in turn activates transcription of downstream target genes via lymphoid enhancer-binding factor-1 (Lef-1) and T cell factors (Tcf1, 3, 4) (Huelsken and Birchmeier, 2001; Wodarz and Nusse, 1998). The amplitude of signaling is fine tuned in part via negative feedback mechanisms that include the secreted molecule Dickkopf 1 (Dkk1) (Glinka et al., 1998), itself a direct transcriptional target of β-catenin (Chamorro et al., 2005; Niida et al., 2004). Dkk1 antagonizes the pathway by interfering with LRP5/6-Wnt interactions (Bafico et al., 2001; Mao et al., 2001; Semenov et al., 2001), and was recently shown to also activate the Wnt/planar cell polarity (PCP) pathway through interaction with the glypican 4/6 homolog Knypek in Xenopus (Caneparo et al., 2007).
Although nuclear localization of β-catenin in response to Wnt is essential for canonical signaling, mechanisms controlling this process are not well understood. Although previous reports suggested that BCL9 (Townsley et al., 2004) may actively import β-catenin to the nucleus whereas APC (Henderson, 2000; Neufeld et al., 2000) and Axin (Cong and Varmus, 2004) may export it to the cytoplasm, a recent study using fluorescence recovery after photobleaching (FRAP) in living cells expressing fluorescence-tagged β-catenin indicated that these molecules function mainly by retaining β-catenin in either the nucleus or the cytoplasm (Krieghoff et al., 2006).
The Rho family of small GTPases regulates cytoskeleton and transcription by virtue of cycling between inactive GDP-bound and active GTP-bound forms (Hall, 1998). Members of the family, including RhoA, Rac1 and Cdc42 have been shown to participate in noncanonical Wnt signaling pathways that control planar cell polarity (PCP) in Drosophila (Eaton et al., 1996; Fanto et al., 2000; Strutt et al., 1997) or convergent extension (CE) in Xenopus (Choi and Han, 2002; Habas et al., 2003; Habas et al., 2001; Penzo-Mendez et al., 2003). Moreover, Rac1 may function in part by activating c-Jun NH2-terminal kinase (JNK) (Habas et al., 2003), itself important for both Drosophila PCP (Boutros et al., 1998) and Xenopus CE (Yamanaka et al., 2002). JNK was also shown to be activated by overexpressed Dvl in mammalian cell cultures (Li et al., 1999; Moriguchi et al., 1999). The signaling cascade leading to Rac1 activation in response to Wnt is not understood, but heterotrimeric G protein signaling in neutrophils was shown to activate Rac through Gβγ subunits and PtdIns(3,4,5)P3 produced by PI-3K, both of which directly bind and activate a guanine-nucleotide exchange factor P-Rex1 (Dong et al., 2005; Welch et al., 2002; Welch et al., 2005).
Here we report that Rac1 activation is a critical component of canonical Wnt signaling. Specifically, in ST2 cells we show that Rac1 activates JNK2 that in turn phosphorylates β-catenin on critical residues and controls its nuclear translocation.
Results
Rac1 activation by Wnt3a via Gαq/11βγ and PI-3K is required for β-catenin signaling
We have studied the potential role of Rho small GTPases in Wnt signaling during osteoblast differentiation. The murine bone marrow-derived stromal cell line ST2 undergoes robust osteoblastogenesis in response to Wnt (Tu et al., 2007). We used an established binding assay to determine whether the GTP-bound (active) forms of Rho GTPases were increased upon Wnt signaling (see Methods). Wnt3a consistently activated Rac1 by 2-3 fold over the control at 30 and 60 minutes after stimulation (average fold change at 60 minutes: 2.8±0.7, n=7) (Fig. 1A). Wnt3a activated Cdc42 to a similar extend but did not significantly affect RhoA (Fig. 1B-C). We confirmed the activation of Rac1 with purified recombinant Wnt3a protein (Fig. 1D). To examine whether Rac1 or Cdc42 participate in canonical Wnt signaling, ST2 cells were infected with retroviruses expressing a dominant negative form of each molecule (N17Rac1 or N17Cdc42), and assayed for their response to Wnt3a in up-regulating expression of a Lef1-luciferase reporter. The Rac1 mutant (dnRac1) completely abolished the induction by Wnt3a, whereas dnCdc42 did not have a significant effect (Fig. 1E). The specificity of dnRac1 was confirmed by Rac1 siRNA, which reduced Rac1 protein to an undetectable level and significantly diminished Lef1-luciferase induction by Wnt3a, whereas the scrambled control RNA did not have any effect (Fig. 1F-G). To confirm the biological relevance of Rac1 activity in Wnt signaling, ST2 cells either transfected with Rac1 siRNA or expressing dnRac1 were examined for their ability to undergo osteoblast differentiation in response to Wnt3a. Disruption of Rac1 activity by either means reduced approximately 70% of Wnt3a-induced expression of alkaline phosphatase (AP), a common osteoblast marker (Fig. 1H-I). The remaining AP expression was likely due to differentiation induced by noncanonical Wnt signaling also activated by Wnt3a in these cells (Tu et al., 2007). Thus, Wnt3a activates Rac1, and Rac1 activity is required for canonical Wnt signaling in ST2 cells.
Figure 1.
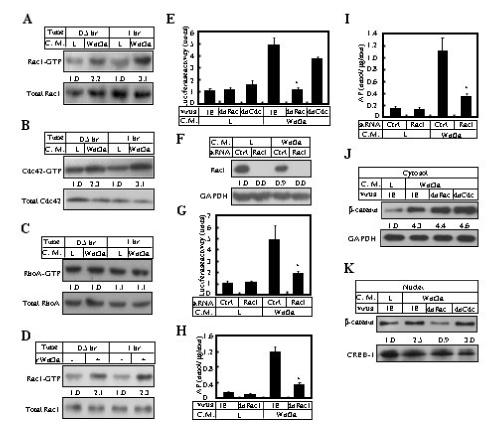
Rac1 activation is required for canonical Wnt signaling. (A-C) Western analyses to detect GTP-bound forms and total amounts of Rac1, Cdc42 and RhoA, in ST2 cells cultured in Wnt3a versus L conditioned medium (C. M.) for 0.5 or 1 hr. The relative amount of the GTP-bound form normalized to the total amount, in L medium, is designated 1.0. (D) Rac1 activation by purified recombinant Wnt3a protein (rWnt3a) at 50 ng/ml. (E) Expression of Lef1-Luciferase in cells infected with a control virus (IE, expressing GFP) or viruses expressing dnRac1 or dnCdc42. The cells were first infected with the viruses, then transiently transfected and cultured in regular growth medium for 47 hrs, and finally incubated in Wnt3a or L conditioned medium for 1 hr before being harvested. (F) Western analyses of Rac1 in cells at ∼96 hrs after transfection with control (Ctrl) or Rac1 siRNA. (G) Expression of Lef1-Luciferase following siRNA transfections. (H-I) Expression of AP following viral infection (H) or siRNA transfection (I). (J-K) Western analyses of β-catenin in cytosolic (J) or nuclear (K) fractions of cells cultured in L or Wnt3a medium for 1 hr following viral infections. Cytosolic and nuclear signals were normalized to GAPDH and CREB-1, respectively. *: p<0.05, n=3.
To explore the mechanism underlying the role of Rac1 in canonical Wnt signaling, Western analyses were performed for β-catenin in cytosolic versus nuclear fractions of ST2 cells expressing dnRac1, with or without Wnt stimulation. Expression of dnRac1 did not affect β-catenin stabilization in the cytosol, but completely abolished β-catenin accumulation in the nucleus in response to Wnt3a; the nuclear effect was not observed with dnCdc42 (Fig. 1, J-K). It should be noted that although dnRac1 markedly decreased β-catenin levels in the nucleus, it did not cause an obvious increase in the cytoplasm. This can be explained by the fact that the relative amount of nuclear versus cytoplasmic β-catenin is small in these cells, approximately 2% with or without Wnt3a stimulation, as per our estimation by Western analyses. Finally, immunofluorescence confocal microscopy confirmed that dnRac1 indeed prevented nuclear accumulation of endogenous β-catenin in response to Wnt3a (Fig. 5, compare B’ and C’). Thus, Rac1 activity is required for β-catenin nuclear localization in response to Wnt signaling.
Figure 5.
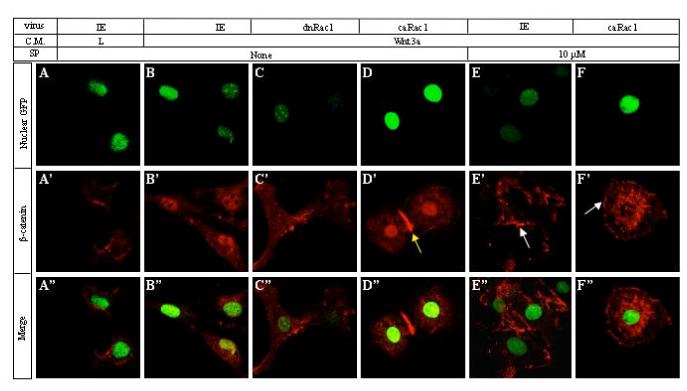
Rac1 and JNK activity are required for β-catenin nuclear localization in response to Wnt. ST2 cells were infected with indicated viruses and cultured in L or Wnt3a medium for 1 hr, with or without SP600125 (10μM), before being subjected to immunofluorescence confocal microscopy. (A-F) Nuclei as revealed by NLS-EGFP expressed by each virus via IRES. (A’-F’) Endogenous β-catenin detected by immunostaining. Yellow arrow in D’: enrichment of β-catenin at a cell-cell junction. White arrow in E’ and F’: enrichment of β-catenin in certain areas of the cell periphery. Note characteristic “fried-egg” cellular morphology in D’ and F’, caused by expression of caRac1. (A“-F”) Merged pictures of GFP and β-catenin signals.
We next investigated the molecular mechanism underlying Rac1 activation by Wnt3a. To assess the potential involvement of Dishevelled (Dvl) proteins, we first tested ST2 cells expressing Dvl2 variants individually missing each of the three conserved regions (ΔDIX, ΔDEP and ΔPDZ) for their ability to activate Rac1 in response to Wnt3a. All three variants were previously shown to inhibit both β-catenin stabilization and PKCδ activation by Wnt3a (Tu et al., 2007), and were expressed at similar levels in these experiments (data not shown). Whereas ΔDEP had little effect, ΔDIX and ΔPDZ either abolished or significantly diminished Rac1 activation (Fig. 2A).
Figure 2.
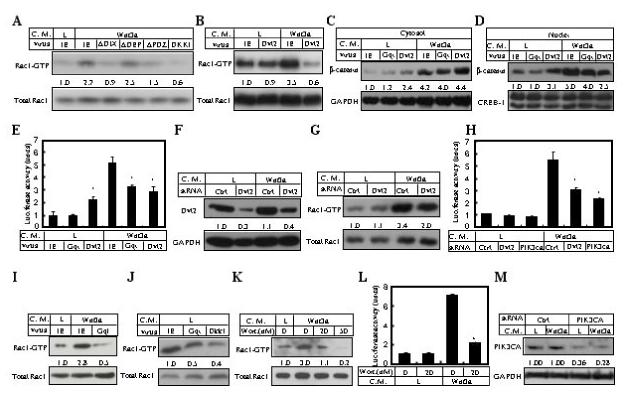
Mechanisms controlling Rac1 activation and canonical Wnt signaling. (A-B) Rac1 activation assays in cells cultured in L or Wnt3a medium for 1 hr following infection of indicated viruses. (C-D) Western analyses of cytosolic (C) versus nuclear β-catenin (D) in cells cultured in L or Wnt3a medium for 1 hr following infection of indicated viruses. Cytosolic and nuclear signals were normalized to GAPDH and CREB-1, respectively. (E) Lef1-luciferase expression by cells infected with indicated viruses and culture in L versus Wnt3a medium. (F) Western analyses of Dvl2 in ST2 cells transfected with Dvl2-specific or control siRNA. (G) Rac1 activation assays following Dvl2 knockdown. (H) Lef1-luciferase expression by cells transfected with gene-specific or control siRNA and culture in L versus Wnt3a medium. (I-J) Rac1 activation assays in cells infected with indicated viruses and cultured in L versus Wnt3a medium. (K) Effect of wortmannin on Rac1 activation at 1hr following Wnt3a stimulation in ST2 cells. (L) Effect of wortmannin on Lef1-Luciferase expression in ST2 cells. Transiently transfected cells were cultured in regular growth medium for 47 hrs, and then incubated in either L or Wnt3a conditioned medium (with or without 20 nM wortmannin) for 1 hr before being harvested. In all cases, wortmannin was added 3 hrs before and during the culture in conditioned media. (M) Western analyses of PIK3CA in cells transfected with gene-specific or control siRNA. *: p<0.05, n=3.
Interestingly, overexpression of the full-length Dvl2 also inhibited Rac1 activation by Wnt3a (Fig. 2B). This result is unexpected because previous studies have shown that overexpression of Dvl proteins activate canonical signaling in the absence of exogenous Wnt ligands. We suspected that Dvl2 overexpression might have a different effect on β-catenin signaling when Wnt ligands were present. To test this possibility, we examined β-catenin signaling in ST2 cells overexpressing Dvl2 with or without Wnt3a stimulation. Consistent with previous reports, at basal conditions Dvl2 overexpression increased both cytoplasmic and nuclear β-catenin levels, and activated Lef1-luciferase expression, but the activation level as judged by all three parameters was significantly lower than that induced by Wnt3a (Fig. 2C-E). Importantly, in the presence of Wnt3a, both nuclear β-catenin levels and Lef1-luciferase expression were reduced by 50% in the Dvl2-overexpressing cells when compared to the control cells expressing IE virus (Fig. 2D-E), although the level of β-catenin in the cytoplasm was not decreased (Fig. 2C). The mechanism through which Dvl2 overexpression activates β-catenin signaling under basal conditions may be different from that employed by Wnt ligands, as Dvl2 overexpression did not activate Rac1 (Fig. 2B). Taken together, the data so far indicate that Dvl2 overexpression inhibits both β-catenin signaling and Rac1 activation in response to Wnt, and that the DEP domain appears to mediate the inhibition. To test the role of endogenous Dvl2, we knocked down Dvl2 using siRNA. Reduction of Dvl2 protein level by 60-70% (Fig. 2F) correlated with a significant decrease in both Rac1 activation and Lef1-luciferase expression in response to Wnt3a (Fig. 2G-H). The remaining activity is likely due to the residual Dvl2 and/or Dvl1 and Dvl3 that are also expressed in these cells (Tu et al., 2007). Overall, the data indicate that Dvl proteins play an important role in mediating Wnt-induced Rac1 activation.
Additional molecules were evaluated for their role in Wnt-induced Rac1 activation. To assess the potential involvement of LRP5/6 signaling, cells overexpressing Dkk1 were assayed; Rac1 activation by Wnt3a was completely abolished by Dkk1 (Fig. 2A). Because overexpression of GqI, a dominant negative reagent for Gαq/11βγ signaling, impaired PKCδ activation but not β-catenin stabilization in response to Wnt (Tu et al., 2007), we examined whether it would affect Wnt-induced Rac1 activation. GqI not only negated Rac1 activation by Wnt3a, but also reduced the basal level of Rac1-GTP (Fig. 2I-J). Because Dkk1 also similarly inhibited the basal level of Rac1-GTP (Fig. 2J), it appears that the basal level Rac1 activation is in part due to endogenous Wnt signaling. GqI also significantly reduced Lef1-luciferase expression in response to Wnt3a (Fig. 2E), consistent with a decrease in nuclear β-catenin levels even though the cytoplasmic amount was not significantly affected (Fig. 2C-D). Since PI-3K is known to mediate Rac1 activation by heterotrimeric G protein signaling, we next examined whether it is required for Rac1 activation by Wnt. Wortmannin, a potent inhibitor for PI-3K, dose-dependently inhibited Rac1 activation by Wnt3a (Fig. 2K), and markedly reduced Wnt-induced Lef1-luciferase expression (Fig. 2L). On the other hand, wortmannin under these conditions had no significant effect on cell survival or general gene expression (Supplementary data, Fig. S3). Finally, siRNA knockdown of the PI-3K p110 catalytic subunit alpha (PIK3CA) (Fig. 2M) markedly reduced Lef1-luciferase expression in response to Wnt3a (Fig. 2H). Taken together, these data indicate that Wnt3a activates Rac1 through a signaling cascade involving LRP5/6, Dvl, Gαq/11βγ and PI-3K.
Constitutively active Rac1 enables canonical signaling by an otherwise inactive Wnt
To further substantiate the role of Rac1 in the canonical Wnt pathway, we studied Wnt7b signaling in ST2 cells, wherein it did not activate Lef1-luciferase expression (Tu et al., 2007). Interestingly, cells overexpressing Wnt7b accumulated >5 fold (versus ∼4 fold induced by Wnt3a, see Fig. 1I) more β-catenin over the control in the cytoplasm (Fig. 3A), but showed little increase in the nucleus (Fig. 3B). Similarly, immunofluorescence confocal microscopy confirmed that no significant levels of endogenous β-catenin were detected in the nucleus following Wnt7b expression (Fig. 3F, compare with Fig. 3E). Importantly, Wnt7b did not activate Rac1 in these cells (Fig. 3C). Thus, the inability for Wnt7b to activate canonical signaling in ST2 cells is not due to deficiency in stabilizing β-catenin, but instead correlates with the lack of Rac1 activation and β-catenin nuclear localization.
Figure 3.
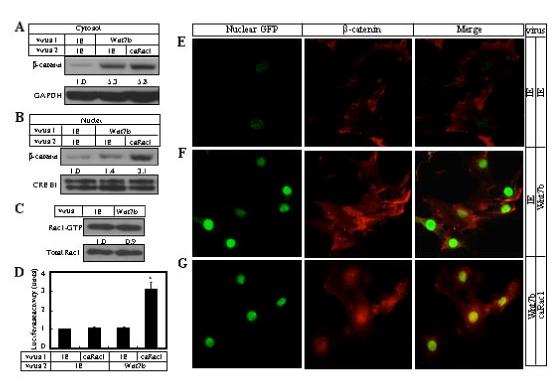
Rac1 activation enables Wnt7b to activate canonical Wnt signaling in ST2 cells. (A-B) Western analyses of β-catenin in cytosolic (A) or nuclear (B) fractions of ST2 cells following co-infection of viruses. Total viral titers were similar in each condition. (C) Rac1 activation assay in cells infected with control IE or Wnt7b virus. (D) Expression of Lef1-luciferase following co-infection. (E-G) Immunofluorescence confocal microscopy of cells following infection by the control IE virus alone (E), both control and Wnt7b viruses (F), or both caRac1 and Wnt7b viruses (G). The cells were infected at a similar titer. Nuclei were labeled green due to expression of nuclear GFP via IRES by all viruses used in the present study. β-catenin signal is in red. *: p<0.05, n=3.
To test whether Rac1 activation is sufficient to enable Wnt7b in activating the canonical pathway, we overexpressed a constitutively active form of Rac1 (V12Rac1, termed caRac1 hereafter) that lacks GTPase activity. Cells expressing caRac1 activated Lef1-luciferase expression in response to Wnt7b, whereas caRac1 alone did not have any effect (Fig. 3D). Consistent with this result, caRac1 increased the nuclear amount of β-catenin by more than 2 fold (Fig. 3B), even though it had little effect on Wnt7b-induced β-catenin accumulation in the cytoplasm (Fig. 3A). When examined by immunofluorescence confocal microscopy, cells expressing caRac1 alone exhibited the typical “fried egg” morphology due to exaggerated lamellipodia as previously described (Welch et al., 2002), but did not show an increase in nuclear β-catenin (data not shown). However, when Wnt7b and caRac1 were co-expressed in the culture, the cells exhibited both the ”fried egg” morphology, and prominent nuclear accumulation of β-catenin (Fig. 3G). Thus, constitutively active Rac1 is sufficient to localize Wnt7b-stabilized cytoplasmic β-catenin to the nucleus and enable canonical signaling in ST2 cells.
Rac1-mediated JNK2 activation by Wnt3a controls β-catenin signaling
To explore downstream events mediating the role of Rac1 in β-catenin nuclear localization, we investigated the potential relevance of JNK. Western analyses using an antibody specific for dual phosphorylation of JNK1 or JNK2 at Thr183 and Tyr185, a prerequisite for JNK activation, revealed that JNK2 was specifically activated by approximately 4 fold at 30 or 60 minutes after Wnt3a stimulation (Fig. 4A), and that the activation was essentially abolished by expression of dnRac1 (Fig. 4B). Inhibition of JNK activity by SP600125 dose-dependently diminished activation of Lef1-luciferase expression by Wnt3a (Fig. 4C). Likewise, SP600125 decreased Wnt3a-activated AP expression by as much as 70% (Fig. 4D), a reduction equivalent to that caused by disruption of Rac1 activity (Fig. 1G-H). Also similar to Rac1 disruption, SP600125 dose-dependently inhibited nuclear accumulation of β-catenin (Fig. 4F), without significantly affecting its stabilization in the cytoplasm (Fig. 4E). We confirmed the importance of JNK2 by siRNA experiments, which showed that JNK2 siRNA reduced the protein level by 60% and diminished Wnt3a-induced Lef1-luciferase induction by ∼50%, whereas the non-targeting control siRNA did not have any effect (Fig. 4G). Knockdown of JNK1 by 80% modestly reduced Wnt3a-induced Lef1-luciferase expression by ∼25%, whereas double knockdown of JNK1 and JNK2 had an effect similar to the JNK2 single knockdown (Fig. 4G). Further supporting the predominant role of JNK2, immunofluorescence confocal microscopy showed that JNK2 siRNA significantly reduced the nucleus β-catenin level in the presence of Wnt3a, whereas JNK1 siRNA had only a modest effect (Fig. 4H). Similarly, SP600125 abolished Wnt-induced nuclear localization of β-catenin, but appeared to enrich β-catenin in certain areas associated with the cell membrane (Fig. 5, compare E’ with B’). Thus, Wnt3a activates JNK2 via Rac1, and JNK activity appears to be required for nuclear localization of β-catenin.
Figure 4.

JNK2 activation is required for canonical Wnt signaling. (A) Western analyses of phospho-JNK and total JNK in ST2 cells cultured in L versus Wnt3a medium for 0.5 or 1 hr. Phospho-JNK2 level was normalized to total JNK2. (B) Western analyses of phospho-JNK and total JNK in ST2 cells cultured in L versus Wnt3a medium for 1 hr following viral infections. (C) Effect of SP600125 on expression of Lef1-luciferase in ST2 cells. (D) Effect of SP600125 on expression of AP in ST2 cells. (E-F) Western analyses of β-catenin in cytolosic (E) or nuclear (F) fractions of ST2 cells cultured in L versus Wnt3a medium for 1 hr with varying concentrations of SP600125. SP600125 was added to the cells 3 hrs before and during the culture in conditioned media. (G) Effects of JNK1/2 knockdown on Lef1-Luciferase expression. Western analyses of JNK performed at ∼96 hrs after siRNA transfection. (H) Effects of JNK1/2 knockdown on nuclear localization of endogenous β-catenin as per immunofluorescence confocal microscopy. (I-K) Co-immunoprecipitation of endogenous β-catenin, JNK1/2 and Rac1 in cytosolic (I) versus nuclear (J) fractions of cells cultured in L versus Wnt3a medium for 1 hr (I-J) or infected with control (IE) or Wnt7b-expressing retrovirus. Purified IgG1 was used as control antibody for precipitation. *: p<0.05, n=3.
To assess potential physical interactions among endogenous β-catenin, Rac1 and JNK, co-immunoprecipitation experiments were performed using cytosolic versus nuclear fractions from ST2 cells incubated with either L or Wnt3a conditioned medium. In the cytosolic fraction, with or without Wnt3a treatment, protein complexes precipitated with a β-catenin antibody, contained total Rac1, total JNK1 and JNK2, in addition to β-catenin as expected (Fig. 4I). Importantly, phospho-JNK2, but not JNK1, was detected in the β-catenin complex only under Wnt3a stimulation. In addition, although the amount of β-catenin detected in the precipitate was markedly increased in response to Wnt3a, the amount of Rac1 was not significantly changed, indicating that Rac1 may be the rate-limiting partner in the complex, and that β-catenin stabilization does not significantly enhance the interaction between the two proteins. Consistent with this notion, the converse experiment using a Rac1 antibody for immunoprecipitation showed that a similar level of β-catenin was detected in the precipitates with or without Wnt3a (Fig. 4I). The Rac1 immunoprecipitates also contained total JNK1 and JNK2 regardless of Wnt3a, but contained a significantly higher level of phospho-JNK2 in the presence of Wnt3a. On the other hand, the same procedures did not detect any co-precipitation among β-catenin, JNK and Rac1 in the nuclear fractions, regardless of Wnt3a (Fig. 4J), even though TCF-4, a member of the LEF/TCF family known to interact with β-catenin, co-precipitated with nuclear β-catenin as expected (data not shown). In all cases, the control experiments using purified IgG1 did not precipitate any of the proteins. Thus, the endogenous β-catenin, Rac1, JNK1 and JNK2 constitutively interact with each other in the cytoplasm of ST2 cell, and Wnt3a signaling specifically activates JNK2 in this context.
To determine whether JNK2 activation within the Rac1-JNK1/2-β-catenin complexes is specific to canonical Wnt signaling, we examined the effect of Wnt7b, which does not activate canonical Wnt signaling in ST2 cells. These experiments detected no phospho-JNK2 in the cytosolic β-catenin immunoprecipitates with or without Wnt7b stimulation (Fig. 4K). This result is consistent with the lack of Rac1 activation by Wnt7b, and indicates that Rac1-mediated JNK2 activation within Rac1-JNK1/2-β-catenin complexes is likely specific to canonical Wnt signaling.
To test further the functional relationship between JNK and Rac1, we examined the effect of SP600125 on caRac1-enhanced β-catenin nuclear localization in response to Wnt3a (Fig. 5). Overexpression of caRac1 accentuated β-catenin in the nucleus in response to Wnt3a (Fig. 5D’). However, SP600125 markedly reduced nuclear β-catenin in caRac1-expressing cells stimulated with Wnt3a, and instead caused prominent accumulation of β-catenin in the perinuclear region (Fig. 5F’). Interestingly, the cells maintained the characteristic “fried-egg” morphology caused by caRac1, but now also exhibited β-catenin enrichment in certain areas of the plasma membrane, a feature common to cells treated with SP600125 (Fig. 5 E’, F’, white arrows). Overall, JNK appears to function downstream of Rac1 in regulating nuclear localization of β-catenin during canonical Wnt signaling.
To investigate the specificity of JNK in mediating canonical Wnt signaling, we examined the other members of the mitogen-activated protein kinase (MAPK) family. Although Wnt3a failed to activate p38 (supplementary data, Fig. S1A), it activated extracellular-signal related kinase 2 (ERK2) by 2-3 fold at 30 or 60 minutes post stimulation (Fig. S1B). However, inhibition of ERK activation by PD98059 or U0126 did not affect Wnt3a-induced Lef1-luciferase expression (Fig. S1D), even though the drugs potently inhibited ERK2 activation (Fig. S1C). Thus, within the MAPK family, JNK appears to be specifically required for canonical Wnt signaling in ST2 cells.
To determine whether the PI3K-Rac1-JNK mechanism is unique to ST2 cells, we examined several other cell lines. In HEK293 cells, Wnt3a treatment for one hour activated Rac1 by approximately 2 fold, and the induction was abolished by wortmaninn (supplementary data, Fig. S2A). Importantly, wortmannin or SP600125 completely inhibited Wnt3a-induced Lef1-luciferase expression (Fig. S2B, C). Similarly, in NIH3T3 cells, Wnt3a induced Rac1 activation by ∼4 fold and Lef1-luciferase expression by ∼3 fold; wortmannin again significantly inhibited both responses, although to a lesser degree than in the ST2 and HEK293 cells (Fig. S2D, E). SP600125 also abolished the induction of Lef1-luciferase expression by Wnt3a in NIH3T3 cells (Fig. S2F). Thus, the role of PI-3K/Rac1/JNK is likely a common feature of canonical Wnt signaling.
Ser191 and Ser605 are critical for β-catenin nuclear localization
Both the role of JNK2 in β-catenin nuclear localization and the physical association between the two proteins prompted us to examine whether JNK directly phosphorylates β-catenin. A search of the β-catenin protein sequence revealed three potential JNK phosphorylation sites, i.e. Ser191, Ser246 and Ser605, with the first two conserved among Drosophila, Xenopus, mouse and human, and the third one common between mouse and Xenopus. To assess their potential importance, we expressed β-catenin variants that harbor mutations at the consensus serine residues (Ser to Ala) individually (S191A, S246A, S605A) or in combination (triple), and evaluated their capacity to mediate Lef1-luciferase expression in ST2 cells in response to Wnt3a. Overexpression of wild type β-catenin increased the expression level by approximately 6 fold over the control cells in response to Wnt3a. Whereas the S246A variant behaved essentially the same as wild type β-catenin, the S191A and S605A variants each caused only a 2-fold increase over the control, and importantly, the triple mutant resulted in no significant increase (Fig. 6A, left top). The differential effects of the variants did not reflect differences in their expression levels, as S605A and the triple mutant were expressed at comparable levels as the wild type, and S191A, S246A were at 85% and 47% of the wild type respectively (Fig. 6A, left bottom). Moreover, endogenous β-catenin levels were similar among the cells expressing the different β-catenin variants. To evaluate the potential importance of phosphorylation at Ser191 and Ser605, we mutated the serine residues to the phosphomimetic aspartate (D). Whereas either S191D or S605D only minimally affected Lef1-luciferase expression under basal conditions (∼10% increase over the wild type construct), S191D showed significant synergy with Wnt3a in further upregulating Lef1-luciferase expression, whereas S605D had a similar but more modest effect (Fig. 6A, right top). Western blotting showed that S191D and the wild type β-catenin construct were expressed at the same level but S605D was ∼80% higher, and that the endogenous β-catenin levels were similar among the cells expressing different constructs (Fig. 6A, rightbottom). Thus Ser191, and to a lesser degree Ser605, play an important role in canonical Wnt signaling.
Figure 6.

Phosphorylation of Ser191 and Ser605 is critical for Wnt-induced nuclear localization of β-catenin. (A) Expression of Lef1-luciferase following transient transfection of β-catenin variants, and Western analyses of Myc-tagged β-catenin variants and endogenous β-catenin in the same lysates used for luciferase assays. (B) In vitro phosphorylation of Myc-tagged β-catenin variants by JNK2α2. Autoradiography signals were normalized to levels of Myc-β-catenin variants as determined by Western analyses using a Myc antibody. (C) In vivo phosphorylation of Myc-β-catenin variants in intact cells. Autoradiography signals were normalized to levels of Myc-β-catenin variants. (D) In vivo phosphorylation of Myc-β-catenin (wild type construct) in intact cells transfected with siRNA. Autoradiography signals were normalized to Myc-β-catenin levels. (E-L, E’-L’, E”-L”) Confocal images for nuclear GFP (E-L), exogenous myc-β-catenin variants (E’-L’) and the merge (E”-L”) in cells cultured with or without recombinant Wnt3a at 50 ng/ml for 1 hr, following infection with IE virus (expressing nuclear GFP) and transient transfection with plasmids expressing Myc-β-catenin variants.*: p<0.05, n=3.
We next determined whether Ser191, Ser246 or Ser605 could indeed be phosphorylated by JNK. Here, overexpressed Myc-tagged β-catenin variants were immunoprecipitated from ST2 cells with a Myc antibody, and subsequently subjected to phosphorylation assays in vitro using purified active JNK2α2, followed by SDS-PAGE and autoradiography. The wild type β-catenin incorporated robust levels of 32P and S246A showed a 20% reduction, but S191A virtually eliminated phosphorylation and S605A reduced it by 75% (Fig. 6B, top). As expected, the triple mutant β-catenin incorporated no 32P. Subsequent Western blotting using the Myc antibody confirmed that the 32P-labeled bands corresponded to the β-catenin variants, and that similar levels of Myc-tagged β-catenin variants were present among the various immunoprecipitates (Fig. 6B, bottom). Thus, consistent with their importance for canonical Wnt signaling, S191 and, to a lesser extent, S605 appear to be the predominant phosphorylation sites for JNK2 in vitro.
To confirm that S191 and S605 are phosphorylated in intact cells responding to Wnt, we performed in vivo phosphorylation assays. Cells expressing the Myc-tagged β-catenin variants were incubated with 32P orthophosphate with or without Wnt3a stimulation, and their lysates were immunoprecipitated with the Myc antibody before being resolved on SDS-PAGE and subjected to autoradiography. Under basal conditions, no significant phosphorylation was detected for any of the β-catenin variants. However, upon Wnt3a stimulation, the wild type β-catenin became highly phosphorylated (Fig. 6C, top). Whereas S246A had little effect (7% reduction), S605A and S191A reduced phosphorylation by 32% and 82% respectively; the triple mutation completely abolished phosphorylation in response to Wnt3a. Western blotting using the Myc antibody revealed similar low levels of Myc-β-catenin variants among the immunoprecipitates under the basal conditions, and similar high levels upon Wnt3a stimulation (Fig. 6C, bottom), indicating that the different variants were expressed at similar levels and that the mutations had no obvious effect on the stabilization of Myc-β-catenin by Wnt3a. Normalization of the 32P signal to the Myc-β-catenin protein level revealed that Wnt3a induced a ∼3 fold increase in phosphorylation on per molecule basis. Thus, β-catenin is phosphorylated in intact cells upon Wnt signaling, and Ser191 and Ser605 are the predominant phosphorylation sites.
To examine directly the role of JNK1/2 in Myc-β-catenin phosphorylation in intact cells in response to Wnt3a, we performed in vivo phosphorylation assays in ST2 cells transfected with JNK1/2 siRNA. JNK2 knockdown notably reduced 32P levels of Myc-β-catenin, whereas JNK1 knockdown had a smaller effect, and the double knockdown exhibited an additive effect (Fig. 6D). Thus, JNK2 appears to be mainly responsible for Wnt3a-induced β-catenin phosphorylation in ST2 cells.
To confirm the effect of specific phosphorylation on nuclear localization of β-catenin, we examined the subcellular localization of the Myc-tagged β-catenin variants in response to Wnt3a by immunofluorescence confocal microscopy. Detection of the Myc tag revealed that the exogenous wild type β-catenin was present in the cytoplasm at a relatively low level, and with clear exclusion from the nucleus under basal conditions (Fig. 6E-E”), but was detected at high levels in both compartments in response to Wnt3a (Fig. 6F-F”). Note that the overexpressed myc-β-catenin did not exhibit the same prominent nuclear localization as the endogenous β-catenin in response to Wnt3a (see Fig. 4H and 5B’). Importantly, the variants harboring either S191A (Fig. 6G-G”), S605A (Fig. 6I-I”), or the triple mutation (Fig. 6J-J”) failed to accumulate in the nucleus when the cells were stimulated with Wnt3a, whereas the S246A form, like the wild type form, was detected in the nucleus in response to Wnt3a (Fig. 6H-H”, compare with 6F-F”). Conversely, the S191D form exhibited more prominent nuclear localization than the wild type construct (Fig. 6K-K”, compare with F-F”), whereas S605D also slightly increased the nuclear signal (Fig. 6L-L”). These results therefore support the notion that both Ser191 and Ser605 participate in the nuclear localization of β-catenin in response to Wnt signaling.
Genetic removal of Rac1 in the mouse limb bud ectoderm phenocopies β-catenin deletion
To determine the physiological relevance of Rac1 in canonical Wnt signaling, we genetically ablated Rac1 from the apical ectodermal ridge (AER) of the mouse embryonic limb bud, where Wnt signaling through β-catenin is critical for limb outgrowth (Barrow et al., 2003). Specifically, we generated embryos of Msx2-Cre;Rac1n/c (Rac1-CKO) by crossing males of Rac1c/c with females of Msx2-Cre;Rac1c/+, taking advantage of the fact that Msx2-Cre is expressed both in the female germline, and in the limb bud ectoderm that gave rise to the AER (Sun et al., 2000). Rac1-CKO embryos at E16.5 lacked all hindlimb structures, and exhibited truncations at various levels in the forelimb (Fig. 7, A-D, A’-D’). These defects are identical to those previously characterized by others in embryos of Msx2-Cre;β-cateninn/c (Barrow et al., 2003). The discrepancy between the fore- and hindlimb, as well as the phenotypic variation among forelimbs were also noted in the β-catenin mutants, and most likely reflect the earlier onset of Msx2-Cre in the hindlimb, and the potential temporal variation in Cre expression among the forelimbs. Overall, genetic removal of either Rac1 or β-catenin from the limb bud ectoderm results in identical limb truncation phenotypes in the mouse.
Figure 7.
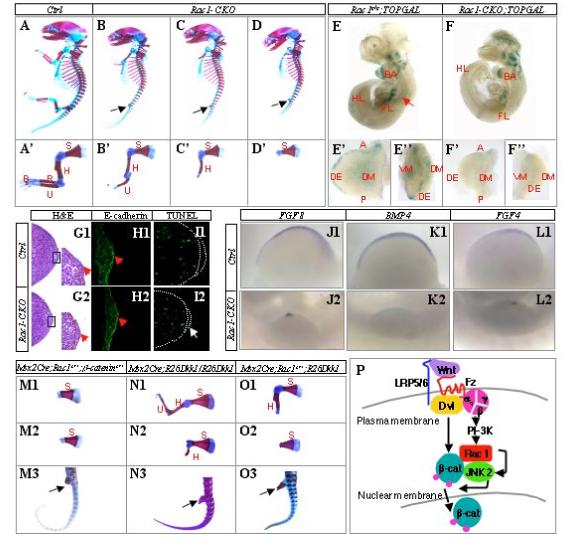
Rac1 interacts genetically with β-catenin and Dkk1 in the limb bud ectoderm of mouse embryos. (A-D) Whole-mount skeletal preparations of E16.5 embryos. Black arrows denote complete absence of hindlimb structures in all Rac1-CKO embryos. The genotype for the control embryo is Msx2-Cre; Rac1c/+. (A’-D’) Forelimb skeletons dissected from embryos above shown at a higher magnification. S: scapula; H: humerus; R: radius; U: ulna; P: phalanges. (E-F) Whole-mount Lac Z staining of E10 littermate embryos. Note that the control embryo (E) was partially squashed during staining so that the dorsal neural tube (red arrow) is now facing the reader instead of to the right. BA: 1st branchial arch; FL: forelimb; HL: hindlimb. (E’, E”, F’,F”) Forelimb buds dissected from the embryos above shown at a higher magnification with either the dorsal (E’, F’) or the distal view (E”, F”). A: anterior; P: posterior; DE: distal ectoderm; DM: dorsal mesenchyme; VM: ventral mesenchyme. (G1-G2) H&E staining of forelimb sections through the AER region at E10.5. Boxed areas shown at a higher magnification. Red dotted line separates mesenchyme from ectoderm. Red arrow denotes AER. (H1-H2) Immunostaining of E-cadherin in the AER region of the forelimb at E10.5. Note thinner AER (red arrow) in the mutant. (I1-I2) TUNEL staining of forelimb sections at E10.5. Dotted white line demarcates ectoderm and AER. White arrow denotes TUNEL signal in the mutant AER. (J1-J2, K1-K2, L1-L2) Whole mount in situ hybridization of the forelimb at E10.5. Ventral view is shown for all limb buds, anterior to the left and posterior to the right. (M1-M3) Representative forelimbs (M1-M2) and hindlimb (M3, black arrow) of Msx1-Cre;Rac1c/+;β-cateninc/+ embryos at E16. (N1-N3) Representative forelimbs (N1-N2) and hindlimb (N3, black arrow) of Msx1-Cre;R26-Dkk1/R26-Dkk1 mice at birth. (O1-O3) Representative forelimbs (O1-O2) and hindlimb (O3, black arrow) of Msx1-Cre;Rac1c/+;R26-Dkk1 embryos at E16.5. (P) A model for canonical Wnt signaling integrating β-catenin stabilization and Rac1-JNK2-mediated phosphorylation at Ser191 and Ser605 (small purple circles) necessary for β-catenin nuclear localization.
To directly monitor the effect of Rac1 removal on canonical Wnt signaling in the limb bud ectoderm, we took advantage of the TOPGAL reporter mouse strain engineered to reflect β-catenin signaling in vivo (DasGupta and Fuchs, 1999). Specifically, we generated E10 embryos with the genotype of Msx2-Cre;Rac1n/c;TOPGAL (Rac1-CKO-TOPGAL), and control littermates also carrying TOPGAL. The control TOPGAL embryos (e.g. Rac1c/c;TOPGAL) exhibited robust activities in multiple tissues including the limb buds (Fig. 7E). Within the forelimb bud, both the distal ectoderm (DE) and the proximal mesenchyme (DM and VM) showed strong signals (Fig. 7E’-E”). In contrast, the forelimb of Rac1-CKO-TOPGAL embryo had little if any LacZ activity, even though the activities in other parts of the embryo appeared normal (Fig. 7F, F’, F”). Moreover, consistent with the limb truncation phenotype, the size of both fore- and hindlimb buds was significantly smaller in the Rac1-CKO-TOPGAL embryo (Fig. 7, E’ vs. F’, and data not shown). Importantly, the limb bud of Rac1-CKO embryos maintained an intact epithelium and a morphologically identifiable AER at E10.5 (Fig. 7G1-G2, red arrows). Immunostaining of E-cadherin revealed normal adherens junctions among epithelial cells and confirmed a thinner AER in the mutant embryo (Fig. 7H1-H2). These results argue against a nonspecific effect of Rac1 removal on limb bud ectoderm, but instead support a specific role for Rac1 in regulating canonical Wnt signaling.
The striking morphological similarity between the Rac1 and the β-catenin conditional knockout embryos prompted us to explore their potential resemblance at the molecular level. Barrow et al demonstrated that the forelimbs of Msx2-Cre;β-cateninn/c embryos failed to maintain FGF8 expression in the AER, and failed to express BMP4 in the distal ventral ectoderm at 34-somite stage (Barrow et al., 2003). Similarly, with the Rac1-CKO embryos we detected only residual FGF8 in the forelimb AER and no BMP4 expression in the ventral ectoderm at E10.5 (34-35 somites) (Fig. 7J1-J2 and K1-K2, respectively). Moreover, AER expression of FGF4 was markedly reduced in Rac1-CKO embryos (Fig. 7L1-L2). Also similar to the β-catenin mutant embryos, none of the molecules were detected in the hindlimb of the Rac1 mutant at E10.5 (data not shown). Finally, removal of β-catenin caused apoptosis in both mesenchyme and the ectoderm (Barrow et al., 2003); we observed an increase in TUNEL staining in both compartments in the forelimb of Rac1-CKO embryos at E10.5 (Fig. 7I1-I2). Thus, both morphologically and molecularly, the Rac1 conditional mutant embryos resemble the β-catenin conditional mutants.
Rac1 interacts genetically with β-catenin and Dkk1 in the AER
To address further the specificity of the role of Rac1 in β-catenin signaling, we examined whether the two molecules genetically interact in the AER. Specifically, we generated mouse embryos lacking one copy each of Rac1 and β-catenin in the limb bud ectoderm (Msx2-Cre;Rac1c/+;β-cateninc/+), by crossing Msx2-Cre;Rac1c/+ males with β-cateninc/c females. Remarkably, all double heterozygous embryos (5/5) developed no hindlimb, and nearly all forelimbs (9/10 from 5 embryos) lacked structures distal to the scapula (Fig. 7, M1-M3), resembling the limb phenotypes of the Msx2-Cre;Rac1n/c or the Msx2-Cre;β-cateninn/c embryos. The one exception forelimb lacked digits 4, 5, and the deltoid tuberosity of the humerus (data not shown). The single heterozygous mice for either Rac1 or β-catenin showed no phenotype (data not shown). Thus, Rac1 in AER controls limb outgrowth likely through regulation of β-catenin.
To further substantiate that the limb phenotype caused by the removal of Rac1 or β-catenin is indeed due to disruption of canonical Wnt signaling, we genetically manipulated the expression levels of Dkk1. Specifically, we generated a mouse strain (R26-Dkk1) with the full-length Dkk1 cDNA knocked into the Rosa26 locus; the cDNA is proceeded by a transcriptional stop signal flanked by a pair of loxP sites, and therefore can only be transcribed when the loxP sites are recombined by Cre (Soriano, 1999; Srinivas et al., 2001). We then crossed the R26-Dkk1 and the Msx2-Cre mice to produce embryos carrying both alleles and hence expressing exogenous Dkk1 in the AER. Embryos with Msx2-Cre and one copy of the R26-Dkk1 allele (Msx2-Cre;R26-Dkk1) did not have any obvious phenotype (data not shown). However, all embryos carrying Msx2-Cre and two copies of R26-Dkk1 (Msx2-Cre;R26-Dkk1/R26-Dkk1) formed no hindlimb, and exhibited a varying degree of truncations in the forelimb (Fig. 7, N1-N3). Thus, overexpression of Dkk1 in the AER results in the same phenotype as removing either β-catenin or Rac1 in the same compartment. Moreover, the dosage effect of Dkk1 underscores both the robustness of the canonical Wnt signaling system, and the sensitivity of the AER to a threshold level of Wnt signaling.
The findings above prompted us to hypothesize that a partial reduction of Rac1 on top of moderate Dkk1 overexpression may be sufficient to diminish Wnt signaling to below the critical threshold level in the AER. To test this notion, we generated embryos carrying Msx2-Cre and one copy each of Rac1c and R26-Dkk1 (Msx2-Cre;Rac1c/+;R26-Dkk1). Remarkably, these embryos developed no hindlimb and had severe truncations in the forelimb (Fig. 7, O1-O3). Thus, Rac1 interacts genetically with Dkk1 and β-catenin in the control of limb outgrowth. Overall, the genetic evidence, together with the in vitro data, argues for a critical role of Rac1 in canonical Wnt signaling.
Discussion
By using both biochemical and genetic approaches, we have uncovered a signaling cascade that operates in conjunction with β-catenin stabilization to activate canonical Wnt signaling. Studies in ST2 cells support a model that Wnt signals through LRP5/6, Dvl and most likely Frizzled receptors to activate a signaling module composed of Gαq/11βγ-PI3K-Rac1-JNK2 (Fig. 7T). As a result, stabilized β-catenin is phosphorylated at Ser191 and Ser605 and thereby localized to the nucleus. Our results are not only consistent with previous findings that abnormally high levels of Rac1 activity promotes β-catenin nuclear localization and TCF/LEF-mediated transcription in cancer cell lines (Esufali and Bapat, 2004), but also identify Rac1 activation as an integral component of canonical Wnt signaling in a normal cellular context.
Both this and a previous study of ours have identified Gαq/11βγ as an important component of Wnt signaling. In the previous study, we provided evidence that Gαq/11βγ mediates a noncanonical Wnt pathway involving PLCβ-PKCΔ (Tu et al., 2007). Here we show that Gαq/11βγ activates a PI3K-Rac1 cascade that participates in canonical Wnt signaling. It is known that βγ subunits can activate PI3K, whereas both Gαq/11 and βγ subunits can activate PLCβ (Malbon, 2005). Our data from both studies indicate that Wnt3a activates at least three distinct signaling cascades in ST2 cells: 1) stabilization of β-catenin, which does not require Gαq/11βγ but is inhibited by Dkk1; 2) activation of PLCβ-PKCδ, which requires Gαq/11βγ signaling but is not inhibited by Dkk1; 3) activation of PI3K-Rac1-JNK2, which both requires Gαq/11βγ signaling and is inhibited by Dkk1. Because of the diverse composition within the Gαq/11βγ family, it is not known whether the same heterotrimeric G proteins mediate both the PLCβ-PKCδ and the PI3K-Rac1 pathway. Moreover, it is not known whether Wnt3a induces formation of three distinct signaling complexes or a single complex with all three signaling properties. The specificity of signaling may be determined by the diverse Wnt receptors at the cell surface.
Rac1 has also been implicated in nuclear transport of other proteins. It was shown to regulate nuclear accumulation of an armadillo protein SmgGDS, through a mechanism dependent upon both the C-terminal polybasic region (PBR) and the activation of Rac1 (Lanning et al., 2003). More recently, Rac1 was reported to control nuclear translocation of STAT transcription factors through interactions with a GTPase-activating protein MgcRacGAP and tyrosine-phosphorylated STAT3 or STAT5A (Kawashima et al., 2006). The present study has identified a mechanism in which Rac1 activation controls nuclear localization through JNK-dependent phosphorylation, but the molecular details for this mechanism remain to be elucidated.
In addition to the serine/threonine phosphorylation within the N-terminus that is critical for β-catenin stability (Liu et al., 2002), several other phosphorylation events have been implicated in the turnover and/or subcellular localization of β-catenin. Ryo et al reported that phosphorylation of Ser246 and the subsequent conformational changes of the p-Ser-Pro bond by the prolyl isomerase Pin1 increased both total and nuclear levels of β-catenin by inhibiting its interaction with APC (Ryo et al., 2001). However, in our studies mutation of Ser246 did not have any obvious effect on either stability or activity of β-catenin in response to Wnt. Moreover, Wnt stimulation did not cause significant phosphorylation at Ser246. The discrepancy could reflect differences in the experimental systems. More recently, phosphorylation of Ser552 by Akt has been implicated in β-catenin signaling in intestinal cells (He et al., 2007). Thus, multiple signal cascades may converge on β-catenin via phosphorylation to control canonical Wnt signaling.
Functional cooperation between JNK and β-catenin has been recently reported in the intestine (Nateri et al., 2005). There, JNK-mediated phosphorylation of c-Jun induced formation of a ternary complex among c-Jun, Tcf4 and β-catenin, which in turn activated the c-Jun promoter. This result and our current finding, however, appear to be at odds with a recent report that sustained, high levels of nuclear JNK activity in early Xenopus embryos prevented nuclear accumulation of β-catenin (Liao et al., 2006). The discrepancy could indicate that the role of JNK in canonical Wnt signaling depends on the duration, degree, and/or subcellular location of its activation. In this regard, it is worth noting that in our study, the complexes among Rac1, β-catenin and JNK1/2 were only detected in the cytoplasm but not in the nucleus, and that Wnt3a specifically activated JNK2 in this context. On the other hand, Wnt7b, which did not activate canonical Wnt signaling in ST2 cells, also failed to activate JNK2 in the β-catenin complexes. One may speculate that such activation event of JNK2 could be “pathway-specific”, and may not be achieved by other mechanisms. Liao et al also reported that purified bacterially-expressed GST-β-catenin could not be phosphorylated by JNK in vitro; this result could indicate that the GST fusion protein was not properly folded, or that β-catenin alone was not an efficient substrate.
Future genetic studies are necessary to determine whether Rac1 participates in canonical Wnt signaling in other physiological settings besides the embryonic limb AER. Of note, Rac1-/- mouse embryos (Sugihara et al., 1998) share similar gastulation defects with β-catenin-/- (Huelsken et al., 2000), Wnt3-/- (Liu et al., 1999), and LRP5-/-/LRP6-/- mutants (Kelly et al., 2004). It is therefore of interest to determine whether Rac1 genetically interacts with these other components of canonical Wnt signaling during gastulation of mouse embryos.
Genetic evidence for Rac in canonical Wnt signaling in Drosophila has not been reported to date. This could be due to functional redundancy among the three Rac homologues (Rac1, 2 and Mtl) in the Drosophila genome (Hakeda-Suzuki et al., 2002). It is worth noting that triple mutant Drosophila embryos containing Rac1 null alleles were embryonic lethal although the phenotype has not been reported (Hakeda-Suzuki et al., 2002). Moreover, loss of function mutations of RacGap50C, a Rac GTPase-activating protein (GAP, negative regulator of Rac activity), ectopically activate Wg signaling in Drosophila embryos (Jones and Bejsovec, 2005). Finally, it is also possible that Rac may not participate in canonical Wnt signaling in Drosophila in the same way as described here for mammalian cells. Recently, important differences have been reported for Hedgehog signaling between fly and mammals (Svard et al., 2006; Varjosalo et al., 2006).
The role of JNK in canonical Wnt signaling in vivo remains to be further elucidated. Although the present work in ST2 cells indicates that JNK2 plays a principle role in mediating Wnt3a-induced phosphorylation of β-catenin, mouse embryos lacking both JNK1 and JNK2 do not exhibit the gastulation defect associated with loss of canonical Wnt signaling, even though they die around E11.5-12.5 (Kuan et al., 1999). It is possible that other members of the MAPK family regulate β-catenin phosphorylation in other cell types. Of note, ERK2-/- mouse embryos are embryonic lethal at E6.5 with no mesoderm formation (Yao et al., 2003); this phenotype resembles that caused by loss of canonical Wnt signaling. Future studies are warranted to determine whether ERK2 participates in canonical Wnt signaling during gastulation of mouse embryos. In Drosophila, JNK and Wingless/Armadillo signaling cascades have been shown to genetically interact in promoting ventral patterning (McEwen et al., 2000). More recently, Rac, and a Rac GAP of the chimaerin family, known as RhoGAP5A, were shown to interact genetically with ERK in regulating the distribution of Armadillo and cell number in the Drosophila eye (Bruinsma et al., 2007).
The finding that PI-3K and Rac1 participate in canonical Wnt signaling has important implications in cancer. Activating mutations of PI-3K have been frequently identified in ovarian, breast, hepatocellular and colorectal carcinomas (Lee et al., 2005; Levine et al., 2005; Samuels et al., 2004). More recently, PI-3K/Pten signaling was shown to cooperate with β-catenin in causing ovarian endometrioid adenocarcinoma in women and in mice (Wu et al., 2007). Similarly, high levels of Rac1 activation have been found in colon cancer cells containing elevated β-catenin levels, and contribute to aberrant activation of canonical Wnt signaling (Esufali and Bapat, 2004). Overall, the Wnt-PI-3K-Rac1 pathway may provide additional therapeutic targets for diminishing canonical Wnt signaling in cancer cells.
Experimental Procedures
Mouse strains and embryo analyses
Msx2-Cre (Sun et al., 2000), Rac1c/c (Gu et al., 2003) and TOPGAL (DasGupta and Fuchs, 1999) mouse strains were as reported. Lac Z staining and whole mount skeletal preparations were based on methods as previously described (Long et al., 2001). Whole-mount in situ hybridization was based on a procedure as previously described (Wilkinson and Nieto, 1993). Immunostaining for E-cadherin, and TUNEL assays (ApopTag Fluorescein In Situ Apoptosis Detection Kit, Chemicon International) were performed on paraffin sections.
The R26-Dkk1 mouse strain was generated as follows. Full-length cDNA of murine Dkk1 was first cloned into the EcoR V site of pCIG (Megason and McMahon, 2002). The resultant plasmid was then digested with Cla I and Not I to obtain the fragment containing Dkk1 cDNA followed by IRES2-GFP; the fragment was cloned into pBigT (Srinivas et al., 2001). The pBigT construct was digested with Pac I and Asc I, and the Dkk1-containing fragment was cloned into pRosa-PAS (Srinivas et al., 2001). The final construct was linearized with Swa I before electroporation into RW-4 ES cells (Murine Embryonic Stem Cell Core, Siteman Cancer Center, Washington University Medical School). ES clones were screened for homologous recombination by Southern blot analyses using a ∼500 bp Xho I-Bam HI fragment derived from the Rosa26 promoter (Kisseberth et al., 1999). Chimeric mice were generated by injection of ES cells into C57BL6 blatocysts (Tg/KO Micro-injection Core, Department of Pathology and Immunology, Washington University Medical School). Subsequent genotyping was performed by PCR using ear biopsy samples (Soriano, 1999).
Cell cultures, transfections, infections, viruses, plasmids and oligonucleotides
ST-2 cells, Wnt3a-expressing and control L cells, transfection and infection procedures, retroviruses expressing GqI, Dkk1, Dvl-2 derivatives or Wnt7b were all as previously described (Tu et al., 2007). Additional viruses were produced in the same way to express N17Rac1, N17Cdc42 (cDNA Resource Center, University of Missouri, Rolla) or V12Rac1. V12Rac1 was generated by site-directed mutagenesis (Stratagene, La Jolla, CA). Full-length cDNA for β-catenin was cloned by PCR from a mouse 15-day embryo cDNA pool Marathon-Ready (BD Biosciences Clontech, Palo Alto, CA) and mutations were introduced by site-directed mutagenesis; all variants of β-catenin were cloned in Myc-tagged pCS2+MT vector (Dr. Xi He, Harvard Medical School). All gene-specific siRNA oligonucleotides (ON-TARGETplus SMARTpool), and the control siRNA (ON-TARGETplus siCONTROL Non-targeting siRNA #1) were purchased from Dharmacon (Lafayette, CO). MTT assay kit was form ATCC (Manassas, VA).
Antibodies, proteins and chemicals
Antibodies for JNK, ERK, p38, phospho-JNK, phospho-ERK and phospho-p38 were from Cell Signaling (Beverly, CA); antibody for CREB1 was from Upstate (Charlottesville, VA). GAPDH antibody was from Chemicon (Temecula, CA); β-catenin and E-cadherin antibodies were from BD Biosciences Pharmigen (San Diego, CA). TCF4 antibody, Myc antibody and HRP-conjugated secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Alexa555- and Alexa488-conjugated secondary antibodies were from Molecular Probes (Eugene, OR). Recombinant human active JNK2α2 and mouse Wnt3a were from Invitrogen (Carlsbad,CA) and R&D Systems (Minneapolis, MN), respectively. Wortmannin, SP600125, U0126 and PD98059 were from Calbiochem (San Diego, CA).
Assays for Rac1, CDC42 and RhoA activation and AP activity
Activation of Rho family small GTPases was detected using EZ-Detect Rac1, Cdc42 and RhoA Activation Kit (Pierce Biotechnology, Rockford, IL). AP expression was assayed as previously described (Tu et al., 2007).
Cytosolic and nuclear fractionation, immunoprecipitation, Western analyses, immunofluorescence confocal microscopy
The cytosolic and nuclear fractions of cells were prepared as follows. Trypsinized cells were washed with cold Ca2+ and Mg2+ free PBS and resuspended in HLB buffer (10 mM HEPES/KOH pH7.9, 10 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT) containing Protease Inhibitor Cocktail (Roche) and Phosphatase Inhibitor Cocktail 1 and 2 (Sigma) on ice for 15 min. The cells were then dounce homogenized and centrifuged at 1,000xg at 4°C for 5 min. The supernatant was ultracentrifuged at 100,000xg at 4°C for 1 h, and the final supernatant (cytosolic fraction) was stored at -80°C until use. For preparation of the nuclear extracts, the pellet was washed with the HLB buffer twice and extracted for 30 min on ice with nuclear extraction buffer (20mM HEPES/KOH, pH7.9, 420 mM NaCl, 1.2 mM MgCl2, 0.2 mM EDTA, 25% Glycerol, Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail 1 and 2), with brief vortex every 5 min and final spin down at 15,000xg for 30 min at 4°C. The supernatant (nuclear extracts) was diluted in the same volume of nuclear dilution buffer (20 mM HEPES/KOH, pH 7.9, 80 mM NaCl, 0.5 mM EDTA, 0.5 mM DTT, Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail 1 and 2)and stored at -80°C before use. Immunoprecipitation and Western analyses were performed using standard protocols. Proteins were detected using ECL Plus Western Blotting Detection System (Amersham Biosciences, England). The intensity of protein bands was quantified using ImageJ (http://rsb.info.nih.gov/ij/). Immunocytochemistry was performed in cells cultured on chamber slides (Nalge Nunc International, Naperville, IL), using specific primary antibodies and an Alexa555-conjugated secondary antibody, followed by confocal microscopy.
Phosphorylation assays in vitro and in intact cells
For in vitro phosphorylation, cells expressing various forms of β-catenin (all Myc-tagged) following transient transfection were starved for 24 hrs, lysed and immunoprecipitated with anti-Myc-agarose beads; the beads were washed and then incubated in JNK assay buffer containing recombinant human active JNK2α2 and [γ-32P]ATP (Perkin-Elmer Life Science, Wellesley, MA) at 30 °C for 30 min, followed by boiling for 5 min. Reaction products were subjected to SDS-PAGE and transferred to a PVDF membrane, followed by autoradiography and then detection with a Myc antibody. For 32P labeling in intact cells, cells expressing various forms of β-catenin following transient transfection were starved for 24 hrs, and then cultured in phosphate- and serum-free medium (Invitrogen) containing 0.2 mCi/ml [32P]orthophosphate (GE Healthcare Bio-Science, Piscataway, NJ), with or without recombinant Wnt3a at 50 ng/ml for 1 hr. Cell lysates were immunoprecipitated with a Myc antibody (Fig. 6C) or a β-catenin antibody (Fig. 6D), subjected to SDS-PAGE and then transferred to a PVDF membrane, followed by autoradiography. Western blotting was performed with anti-Myc either on the same membrane used for autoradiography (Fig. 6C), or with parallel samples (Fig. 6D).
Supplementary Material
Acknowledgements
We are indebted to Drs. Xi He, Christof Niehrs and Walter Koch for providing reagents. We thank Mr. Congxing Lin in Dr. Liang Ma’s laboratory and Dr. Kai Yu in Dr. David Ornitz’s laboratory for technical advice on limb bud studies. The work was supported in part by NIH grants R01 DK 065789 (F.L.) and R01 DK 62757 (D.A.W.), and a NIH postdoctoral training grant 5T32AR07033 (M.J.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bafico A, Liu G, Yaniv A, Gazit A, Aaronson SA. Novel mechanism of Wnt signalling inhibition mediated by Dickkopf-1 interaction with LRP6/Arrow. Nat Cell Biol. 2001;3:683–686. doi: 10.1038/35083081. [DOI] [PubMed] [Google Scholar]
- Barrow JR, Thomas KR, Boussadia-Zahui O, Moore R, Kemler R, Capecchi MR, McMahon AP. Ectodermal Wnt3/beta-catenin signaling is required for the establishment and maintenance of the apical ectodermal ridge. Genes Dev. 2003;17:394–409. doi: 10.1101/gad.1044903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M, Paricio N, Strutt DI, Mlodzik M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell. 1998;94:109–118. doi: 10.1016/s0092-8674(00)81226-x. [DOI] [PubMed] [Google Scholar]
- Bruinsma SP, Cagan RL, Baranski TJ. Chimaerin and Rac regulate cell number, adherens junctions, and ERK MAP kinase signaling in the Drosophila eye. Proc Natl Acad Sci U S A. 2007;104:7098–7103. doi: 10.1073/pnas.0701686104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caneparo L, Huang YL, Staudt N, Tada M, Ahrendt R, Kazanskaya O, Niehrs C, Houart C. Dickkopf-1 regulates gastrulation movements by coordinated modulation of Wnt/beta catenin and Wnt/PCP activities, through interaction with the Dally-like homolog Knypek. Genes Dev. 2007;21:465–480. doi: 10.1101/gad.406007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro MN, Schwartz DR, Vonica A, Brivanlou AH, Cho KR, Varmus HE. FGF-20 and DKK1 are transcriptional targets of beta-catenin and FGF-20 is implicated in cancer and development. Embo J. 2005;24:73–84. doi: 10.1038/sj.emboj.7600460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SC, Han JK. Xenopus Cdc42 regulates convergent extension movements during gastrulation through Wnt/Ca2+ signaling pathway. Dev Biol. 2002;244:342–357. doi: 10.1006/dbio.2002.0602. [DOI] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Cong F, Varmus H. Nuclear-cytoplasmic shuttling of Axin regulates subcellular localization of beta-catenin. Proc Natl Acad Sci U S A. 2004;101:2882–2887. doi: 10.1073/pnas.0307344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DasGupta R, Fuchs E. Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development. 1999;126:4557–4568. doi: 10.1242/dev.126.20.4557. [DOI] [PubMed] [Google Scholar]
- Dong X, Mo Z, Bokoch G, Guo C, Li Z, Wu D. P-Rex1 is a primary Rac2 guanine nucleotide exchange factor in mouse neutrophils. Curr Biol. 2005;15:1874–1879. doi: 10.1016/j.cub.2005.09.014. [DOI] [PubMed] [Google Scholar]
- Eaton S, Wepf R, Simons K. Roles for Rac1 and Cdc42 in planar polarization and hair outgrowth in the wing of Drosophila. J Cell Biol. 1996;135:1277–1289. doi: 10.1083/jcb.135.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esufali S, Bapat B. Cross-talk between Rac1 GTPase and dysregulated Wnt signaling pathway leads to cellular redistribution of beta-catenin and TCF/LEF-mediated transcriptional activation. Oncogene. 2004;23:8260–8271. doi: 10.1038/sj.onc.1208007. [DOI] [PubMed] [Google Scholar]
- Fanto M, Weber U, Strutt DI, Mlodzik M. Nuclear signaling by Rac and Rho GTPases is required in the establishment of epithelial planar polarity in the Drosophila eye. Curr Biol. 2000;10:979–988. doi: 10.1016/s0960-9822(00)00645-x. [DOI] [PubMed] [Google Scholar]
- Glinka A, Wu W, Delius H, Monaghan AP, Blumenstock C, Niehrs C. Dickkopf-1 is a member of a new family of secreted proteins and functions in head induction. Nature. 1998;391:357–362. doi: 10.1038/34848. [DOI] [PubMed] [Google Scholar]
- Gu Y, Filippi MD, Cancelas JA, Siefring JE, Williams EP, Jasti AC, Harris CE, Lee AW, Prabhakar R, Atkinson SJ, et al. Hematopoietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science. 2003;302:445–449. doi: 10.1126/science.1088485. [DOI] [PubMed] [Google Scholar]
- Habas R, Dawid IB, He X. Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev. 2003;17:295–309. doi: 10.1101/gad.1022203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habas R, Kato Y, He X. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell. 2001;107:843–854. doi: 10.1016/s0092-8674(01)00614-6. [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S, Ng J, Tzu J, Dietzl G, Sun Y, Harms M, Nardine T, Luo L, Dickson BJ. Rac function and regulation during Drosophila development. Nature. 2002;416:438–442. doi: 10.1038/416438a. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- He XC, Yin T, Grindley JC, Tian Q, Sato T, Tao WA, Dirisina R, Porter-Westpfahl KS, Hembree M, Johnson T, et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat Genet. 2007;39:189–198. doi: 10.1038/ng1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson BR. Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat Cell Biol. 2000;2:653–660. doi: 10.1038/35023605. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Birchmeier W. New aspects of Wnt signaling pathways in higher vertebrates. Curr Opin Genet Dev. 2001;11:547–553. doi: 10.1016/s0959-437x(00)00231-8. [DOI] [PubMed] [Google Scholar]
- Huelsken J, Vogel R, Brinkmann V, Erdmann B, Birchmeier C, Birchmeier W. Requirement for beta-catenin in anterior-posterior axis formation in mice. J Cell Biol. 2000;148:567–578. doi: 10.1083/jcb.148.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones WM, Bejsovec A. RacGap50C negatively regulates wingless pathway activity during Drosophila embryonic development. Genetics. 2005;169:2075–2086. doi: 10.1534/genetics.104.039735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima T, Bao YC, Nomura Y, Moon Y, Tonozuka Y, Minoshima Y, Hatori T, Tsuchiya A, Kiyono M, Nosaka T, et al. Rac1 and a GTPase-activating protein, MgcRacGAP, are required for nuclear translocation of STAT transcription factors. J Cell Biol. 2006;175:937–946. doi: 10.1083/jcb.200604073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly OG, Pinson KI, Skarnes WC. The Wnt co-receptors Lrp5 and Lrp6 are essential for gastrulation in mice. Development. 2004;131:2803–2815. doi: 10.1242/dev.01137. [DOI] [PubMed] [Google Scholar]
- Kisseberth WC, Brettingen NT, Lohse JK, Sandgren EP. Ubiquitous expression of marker transgenes in mice and rats. Dev Biol. 1999;214:128–138. doi: 10.1006/dbio.1999.9417. [DOI] [PubMed] [Google Scholar]
- Krieghoff E, Behrens J, Mayr B. Nucleo-cytoplasmic distribution of beta-catenin is regulated by retention. J Cell Sci. 2006;119:1453–1463. doi: 10.1242/jcs.02864. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Lanning CC, Ruiz-Velasco R, Williams CL. Novel mechanism of the co-regulation of nuclear transport of SmgGDS and Rac1. J Biol Chem. 2003;278:12495–12506. doi: 10.1074/jbc.M211286200. [DOI] [PubMed] [Google Scholar]
- Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24:1477–1480. doi: 10.1038/sj.onc.1208304. [DOI] [PubMed] [Google Scholar]
- Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, Boyd J. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;11:2875–2878. doi: 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- Li L, Yuan H, Xie W, Mao J, Caruso AM, McMahon A, Sussman DJ, Wu D. Dishevelled proteins lead to two signaling pathways. Regulation of LEF-1 and c-Jun N-terminal kinase in mammalian cells. J Biol Chem. 1999;274:129–134. doi: 10.1074/jbc.274.1.129. [DOI] [PubMed] [Google Scholar]
- Liao G, Tao Q, Kofron M, Chen JS, Schloemer A, Davis RJ, Hsieh JC, Wylie C, Heasman J, Kuan CY. Jun NH2-terminal kinase (JNK) prevents nuclear beta-catenin accumulation and regulates axis formation in Xenopus embryos. Proc Natl Acad Sci U S A. 2006;103:16313–16318. doi: 10.1073/pnas.0602557103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002;108:837–847. doi: 10.1016/s0092-8674(02)00685-2. [DOI] [PubMed] [Google Scholar]
- Liu P, Wakamiya M, Shea MJ, Albrecht U, Behringer RR, Bradley A. Requirement for Wnt3 in vertebrate axis formation. Nat Genet. 1999;22:361–365. doi: 10.1038/11932. [DOI] [PubMed] [Google Scholar]
- Long F, Zhang XM, Karp S, Yang Y, McMahon AP. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development. 2001;128:5099–5108. doi: 10.1242/dev.128.24.5099. [DOI] [PubMed] [Google Scholar]
- Malbon CC. G proteins in development. Nat Rev Mol Cell Biol. 2005;6:689–701. doi: 10.1038/nrm1716. [DOI] [PubMed] [Google Scholar]
- Mao B, Wu W, Li Y, Hoppe D, Stannek P, Glinka A, Niehrs C. LDL-receptor-related protein 6 is a receptor for Dickkopf proteins. Nature. 2001;411:321–325. doi: 10.1038/35077108. [DOI] [PubMed] [Google Scholar]
- McEwen DG, Cox RT, Peifer M. The canonical Wg and JNK signaling cascades collaborate to promote both dorsal closure and ventral patterning. Development. 2000;127:3607–3617. doi: 10.1242/dev.127.16.3607. [DOI] [PubMed] [Google Scholar]
- Megason SG, McMahon AP. A mitogen gradient of dorsal midline Wnts organizes growth in the CNS. Development. 2002;129:2087–2098. doi: 10.1242/dev.129.9.2087. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Kawachi K, Kamakura S, Masuyama N, Yamanaka H, Matsumoto K, Kikuchi A, Nishida E. Distinct domains of mouse dishevelled are responsible for the c-Jun N-terminal kinase/stress-activated protein kinase activation and the axis formation in vertebrates. J Biol Chem. 1999;274:30957–30962. doi: 10.1074/jbc.274.43.30957. [DOI] [PubMed] [Google Scholar]
- Nateri AS, Spencer-Dene B, Behrens A. Interaction of phosphorylated c-Jun with TCF4 regulates intestinal cancer development. Nature. 2005;437:281–285. doi: 10.1038/nature03914. [DOI] [PubMed] [Google Scholar]
- Neufeld KL, Zhang F, Cullen BR, White RL. APC-mediated downregulation of beta-catenin activity involves nuclear sequestration and nuclear export. EMBO Rep. 2000;1:519–523. doi: 10.1093/embo-reports/kvd117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida A, Hiroko T, Kasai M, Furukawa Y, Nakamura Y, Suzuki Y, Sugano S, Akiyama T. DKK1, a negative regulator of Wnt signaling, is a target of the beta-catenin/TCF pathway. Oncogene. 2004;23:8520–8526. doi: 10.1038/sj.onc.1207892. [DOI] [PubMed] [Google Scholar]
- Penzo-Mendez A, Umbhauer M, Djiane A, Boucaut JC, Riou JF. Activation of Gbetagamma signaling downstream of Wnt-11/Xfz7 regulates Cdc42 activity during Xenopus gastrulation. Dev Biol. 2003;257:302–314. doi: 10.1016/s0012-1606(03)00067-8. [DOI] [PubMed] [Google Scholar]
- Ryo A, Nakamura M, Wulf G, Liou YC, Lu KP. Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat Cell Biol. 2001;3:793–801. doi: 10.1038/ncb0901-793. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Semenov MV, Tamai K, Brott BK, Kuhl M, Sokol S, He X. Head inducer Dickkopf-1 is a ligand for Wnt coreceptor LRP6. Curr Biol. 2001;11:951–961. doi: 10.1016/s0960-9822(01)00290-1. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strutt DI, Weber U, Mlodzik M. The role of RhoA in tissue polarity and Frizzled signalling. Nature. 1997;387:292–295. doi: 10.1038/387292a0. [DOI] [PubMed] [Google Scholar]
- Sugihara K, Nakatsuji N, Nakamura K, Nakao K, Hashimoto R, Otani H, Sakagami H, Kondo H, Nozawa S, Aiba A, Katsuki M. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene. 1998;17:3427–3433. doi: 10.1038/sj.onc.1202595. [DOI] [PubMed] [Google Scholar]
- Sun X, Lewandoski M, Meyers EN, Liu YH, Maxson RE, Jr., Martin GR. Conditional inactivation of Fgf4 reveals complexity of signalling during limb bud development. Nat Genet. 2000;25:83–86. doi: 10.1038/75644. [DOI] [PubMed] [Google Scholar]
- Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, Ericson J, Toftgard R, Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Townsley FM, Cliffe A, Bienz M. Pygopus and Legless target Armadillo/beta-catenin to the nucleus to enable its transcriptional co-activator function. Nat Cell Biol. 2004;6:626–633. doi: 10.1038/ncb1141. [DOI] [PubMed] [Google Scholar]
- Tu X, Joeng KS, Nakayama KI, Nakayama K, Rajagopal J, Carroll TJ, McMahon AP, Long F. Noncanonical Wnt Signaling through G Protein-Linked PKCdelta Activation Promotes Bone Formation. Dev Cell. 2007;12:113–127. doi: 10.1016/j.devcel.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varjosalo M, Li SP, Taipale J. Divergence of hedgehog signal transduction mechanism between Drosophila and mammals. Dev Cell. 2006;10:177–186. doi: 10.1016/j.devcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, Tempst P, Hawkins PT, Stephens LR. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- Welch HC, Condliffe AM, Milne LJ, Ferguson GJ, Hill K, Webb LM, Okkenhaug K, Coadwell WJ, Andrews SR, Thelen M, et al. P-Rex1 regulates neutrophil function. Curr Biol. 2005;15:1867–1873. doi: 10.1016/j.cub.2005.09.050. [DOI] [PubMed] [Google Scholar]
- Wilkinson DG, Nieto MA. Detection of messenger RNA by in situ hybridization to tissue sections and whole mounts. Methods Enzymol. 1993;225:361–373. doi: 10.1016/0076-6879(93)25025-w. [DOI] [PubMed] [Google Scholar]
- Wodarz A, Nusse R. Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- Wu R, Hendrix-Lucas N, Kuick R, Zhai Y, Schwartz DR, Akyol A, Hanash S, Misek DE, Katabuchi H, Williams BO, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11:321–333. doi: 10.1016/j.ccr.2007.02.016. [DOI] [PubMed] [Google Scholar]
- Yamanaka H, Moriguchi T, Masuyama N, Kusakabe M, Hanafusa H, Takada R, Takada S, Nishida E. JNK functions in the non-canonical Wnt pathway to regulate convergent extension movements in vertebrates. EMBO Rep. 2002;3:69–75. doi: 10.1093/embo-reports/kvf008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Li W, Wu J, Germann UA, Su MS, Kuida K, Boucher DM. Extracellular signal-regulated kinase 2 is necessary for mesoderm differentiation. Proc Natl Acad Sci U S A. 2003;100:12759–12764. doi: 10.1073/pnas.2134254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.