

Abstract
H-Dmt-Tic-NH-CH2-Bid (UFP-502) was the first δ opioid agonist prepared from the Dmt-Tic pharmacophore. It showed interesting pharmacological properties, such as stimulation of mRNA BDNF expression, and antidepression. To evaluate the importance of 1H-benzimidazol-2-yl (Bid) in the induction of δ agonism, it was substituted by similar heterocycles: The substitution of NH(1) by O or S, transforms the reference δ agonist into δ antagonists. Phenyl ring of benzimidazole is not important for δ agonism; in fact 1H-imidazole-2-yl retains δ agonist activity.
1. Introduction
Extensive structure-activity studies on the prototype δ -opioid receptor antagonist, H-Dmt-Tic-OH,a, 1 revealed that even minor chemical modifications changed its pharmacological profile,2 including enhanced δ -antagonism,3 the appearance of mixed μ -agonism/ δ-agonism,4 as well as formation of mixed μ-agonism/ δ -antagonism,4μ -agonism,5 μ-antagonism,5 and δ agonism.4, 6 δ -Opioid receptor agonists are known to produce many pharmacological effects in rodents, including analgesia,7 antidepressant,8 neuroprotection/neurogenesis,9 and anti-Parkinson activities.10 Our first reported potent and selective δ -opioid agonist derived from the δ -antagonist Dmt-Tic pharmacophore; it is characterized by the presence of Bid (1H-benzimidazole-2-yl) at its C-terminus (H-Dmt-Tic-NH-CH2-Bid).4 It is endowed with antidepressant-like effects in rodents, with a propensity to induce convulsions to a lower degree than non peptide -agonists.11, 12 With the aim of evaluating the importance of Bid in the induction of δ -agonism, it was substituted by related heterocycles, and an homocycle, as follows: 1, benzoxazol-2-yl (Boa);13 2, benzothiazol-2-yl (Bta);13, 14 3, 1H-indol-2-yl (Indl);15 4, 2,3-dihydro-1H-inden-2-yl (Indn);16 5, 4-phenyl-1H-imidazol-2-yl (ImidPh);17 and 6, 1H-imidazol-2-yl (Imid).18 Structures of new compounds and their bioactivities are reported in Fig 1. and Table 1, respectively.
Fig. 1.

Structures of new compounds 1–6.
Table 1.
Receptor Binding Affinities and Functional Bioactivities of Compounds 1–6.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Receptor affinity a (nM) | Selectivity | Functional bioactivity (nM) | ||||||
| Comp. | Structure | R | Kiδ | Kiμ | Kiμ/Kiδ | MVD (IC50)b | MVD (p A2)c | GPI (IC50)b |
| Ref. | H-Dmt-Tic-NH-CH2-Bid |  |
0.035d | 0.50d | 14d | 0.94±0.21 | 35.5±7.3 | |
| 1 | H-Dmt-Tic-NH-CH2-Boa |

|
0.283±0.048 (3) | 1.42±0.25 (6) | 5.0 | 9.42 | 169.9±32.2 | |
| 2 | H-Dmt-Tic-NH-CH2-Bta |

|
0.145±0.018 (3) | 1.06±0.11 (4) | 7.3 | 9.37 | 129.1±21.5 | |
| 3 | H-Dmt-Tic-NH-CH2-Indl |

|
0.066±0.013 (3) | 0.7±0.18 (4) | 10.6 | 9.45 | 208.9±48.1 | |
| 4 | H-Dmt-Tic-NH-CH2-Indn |
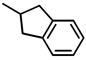
|
0.199±0.032 (3) | 0.16±0.019 (4) | 1.2# | 129.0±23.9 | 28.41±1.74 | |
| 5 | H-Dmt-Tic-NH-CH2-ImidPh |
|
0.443±0.13 (3) | 6.74±0.98 (5) | 15 | 9.40 | 153.5±10.4 | |
| 6 | H-Dmt-Tic-NH-CH2-Imid |

|
0.114±0.017 (4) | 1.18±0.12 (6) | 10 | 3.63±0.52 | 111.3±37.8 | |
μ selectivity Kiδ/ Kiμ.
The Ki values (nM) were determined according to Cheng and Prusoff.20 The mean ± SE with n repetitions in parenthesis is based on independent duplicate binding assays with five to eight peptide doses using several different synaptosomal preparations.
Agonist activity was expressed as IC50 obtained from dose-response curves. These values represents the mean ± SE for at least four tissue samples. DPDPE and PL-017 were the internal standards for MVD (δ-opioid receptor bioactivity) and GPI (μ-opioid receptor bioactivity) tissue preparation, respectively.
The pA2 values of opioid antagonists against the agonists (deltorphin II and endomorphin-2) were determined by the method of Kosterlitz and Watt.21
Data taken from Balboni et al.4
2. Chemistry
Pseudopeptides (1–6) were prepared in solution by peptide synthetic methods. Simply, Boc-Dmt-Tic-OH19 was coupled with H2N-CH2-hetero-/homo-cycle via WSC/HOBt. Final N-terminal Boc deprotection with TFA and purification by preparative HPLC, gave compounds (1–6). The intermediates (benzo[d]oxazol-2-yl)methanamine (H2N-CH2-Boa) and (benzo[d]thiazol-2-yl)methanamine (H2N-CH2-Bta) were prepared according to the procedure of Nestor et al.13 Mixed carbonic anhydride coupling of Z-Gly-OH with o-aminophenol gave the crude intermediate amide, which was converted without purification to the desired benzoxazole (Z-NH-CH2-Boa) by cyclization and dehydration in refluxing propionic acid (~140°C). N-terminal Z deprotection was accomplished by catalytic hydrogenation (H2; Pd/C). Z-NH-CH2-Bta was prepared in a similar manner using the disulfide of o-aminothiophenol which reacts more effectively than the monomeric o-aminothiophenol). The dimeric intermediate amide was reduced with Zn/AcOH to the sulfhydril compound, which underwent cyclization to Z-NH-CH2-Bta upon treatment with TEA in dioxane. N-terminal Z-deprotection was accomplished by HBr/AcOH treatment. H2N-CH2-Indl and H2N-CH2-Indn were prepared according to procedures described by Wright et al. 15 and Shinozaki, et al., 16 respectively. H2N-CH2-IndPh was prepared according to Poitout et al. starting from Boc-Gly-OH instead of Boc-Trp-OH.17 Boc-Gly-OH was treated with cesium carbonate followed by condensation with phenacyl bromide. Cyclization of the resulting ketoester using ammonium acetate in refluxing xylene yielded the desired Boc-NH-CH2-ImidPh which was finally Boc-deprotected upon TFA treatment. Finally, H2N-CH2-Imid was prepared as reported by Bastiaansen et al.18
3. Results and Discussion
3.1. Receptor Affinity Analysis
Receptor binding data for μ- and δ-receptors and selectivity (Kiμ/Kiδ or Kiδ/ Kiμ )are reported in Table 1. All new compounds (1–6) had subnanomolar affinity for δ-opioid receptors (Kiδ=0.066–0.443 nM); nonetheless, their averaged affinity was 6 fold less than the reference compound (H-Dmt-Tic-NH-CH2-Bid). As expected, the lack of a free carboxylic function induces an increase in the μ-receptor affinity (Kiμ = 0.16–6.74 nM).5 None of these new compounds exhibited high receptor selectivity; (Kiμ Kiδ= 5–15) in comparison with the reference; only compound (4) doesn’t follow this trend showing an almost complete lack of selectivity (Kiδ Kiμ= 1.2).
3.2. Functional Bioactivity
Compounds (1–6) were tested in the electrically stimulated MVD and GPI pharmacological assays for intrinsic functional bioactivity (Table 1). As often seen, especially in the Dmt-Tic pharmacophore field, a close correlation between binding and functional bioactivity data is often lacking. We and other investigators have previously discussed this discrepancy; unfortunately, until now we have neither definitive nor comprehensive answers for these observations.5 The substitution of Bid with benzoxazole (Boa), benzothiazole (Bta), indole (Indl) and 4-phenyl-imidazole heterocycles transforms the potent and moderately selective reference -agonist (MVD; IC50 = 0.94 nM) into potent -antagonists (MVD, pA2 = 9.37–9.45). Compound 1–6) were tested in the electrically stimulated MVD and GPI pharmacological assays for intrinsic functional bioactivity (Table 1). As often seen, especially in the Dmt-Tic pharmacophore field, a close correlation between binding and functional bioactivity data is often lacking. We and other investigators have previously discussed this discrepancy; unfortunately, until now we have neither definitive nor comprehensive answers for these observations.5 The substitution of Bid with benzoxazole (Boa), benzothiazole (Bta), indole (Indl) and 4-phenyl-imidazole heterocycles transforms the potent and moderately selective reference -agonist (MVD; IC50 = 0.94 nM) into potent -antagonists (MVD, pA2 = 9.37–9.45). Compound 4, containing the 2,3-dihydro-1H-indene nucleus (Indn) exhibited a δ -agonist activity that was 137-fold lower than the reference, but maintained a similar μ -agonist activity. Finally, compound 6, characterized by the presence of the only imidazole (Imid) ring instead of the benzimidazole (Bid), substantially maintained the δ - and μ -agonist activities (3.86 and 3.14 fold lower than the reference, respectively). In general, all compounds (except 4) decreased in μ -agonist activity (3.1–5.9 fold) which could be useful to improve their selectivity as δ -agonists or antagonists.
4. Conclusion
As confirmation that even minor chemical modifications change the pharmacological profile of the opioid Dmt-Tic pharmacophore; this study demonstrates that the transformation of a potent δ -agonist into potent δ -antagonists can be affected by simply changing N1-H in the benzimidazole ring by O, S, or by N3 deletion. Surprisingly, compound 4 shows μ agonist activity comparable to the reference, accomplished by a weak δ agonist activity; its behaviour is reversed in comparison with the reference. Finally, compounds 5 and 6 containing the imidazole heterocycle with (5) or without a phenyl ring (6), are characterized by a completely different behaviour; in fact the presence of the phenyl ring induces a potent antagonist activity (5) while its absence (6) yield potent δ agonist activity. The conclusions drawn from this series of compounds are as follows: i) Bid is important, but not essential for δ agonist activity; since the substitution of N1-H by O or S results in δ antagonism. ii) Deletion of N3 in the benzimidazole ring provides δ -antagonism. iii) Finally and more important, the removal of the phenyl ring from the benzimidazole function (Bid) to form an imidazole, maintains the δ -agonism of the reference compound H-Dmt-Tic-NH-CH2-Bid (UFP-502). The new δ -agonist H-Dmt-Tic-NH-CH2-Imid could be a useful tool in future pharmacological studies; for example, preliminary data obtained in animal models for Parkinson’s disease with δ -opioid agonists containing the Dmt-Tic pharmacophore, provide evidence of an interesting activity profile that differs from that of SNC-80.10
5. Experimental
5.1. Chemistry
5.1.1. General Methods
Crude compounds were purified by preparative reversed-phase HPLC [Waters Delta Prep 4000 system with Waters Prep LC 40 mm Assembly column C18 (30 x 4 cm, 15 μm particle size)] and eluted at a flow rate of 20 mL/min with mobile phase solvent A (10% acetonitrile + 0.1% TFA in H2O, v/v), and a linear gradient from 10 to 60% solvent B (60%, acetonitrile + 0.1% TFA in H2O, v/v) in 30 min. Analytical HPLC analyses were performed with a Beckman System Gold (Beckman ultrasphere ODS column, 250 x 4.6 mm, 5 μm particle size). Analytical determinations and capacity factor (K’) of the products used HPLC in solvents A and B programmed at flow rate of 1 mL/min with linear gradient from 0 to 100% B in 25 min. Analogues had less than 1% impurities when monitored at 220 and 254 nm.
TLC was performed on precoated plates of silica gel F254 (Merck, Darmstadt, Germany): (A) 1-butanol/AcOH/H2O (3:1:1, v/v/v); (B) CH2Cl2/toluene/methanol (17:1:2). Ninhydrin (1% ethanol, Merck), fluorescamine (Hoffman-La Roche) and chlorine spray reagents. Melting points were determined on a Kofler apparatus and are uncorrected. Optical rotations were assessed at 10 mg/mL in methanol with a Perkin-Elmer 241 polarimeter in a 10 cm water-jacketed cell. Molecular weights of the compounds were determined by a MALDI-TOF analysis (Hewlett Packard G2025A LD-TOF system mass spectrometer) and α-cyano-4-hydroxycinnamic acid as a matrix. 1H NMR (δ ) spectra were measured, when not specified, in DMSO-d6 solution using a Bruker AC-200 spectrometer, and peak positions are given in parts per million downfield from tetramethylsilane as internal standard.
5.2. Peptide Synthesis
5.2.1. Benzyl (benzo[d]oxazol-2-yl)methylcarbamate (Z-NH-CH2-Boa)
A solution of Z-Gly-OH (4.18 g, 20 mmol) and TEA (2.8 mL, 20 mmol) in dry THF (70 mL) was treated at −20°C with isobutyl chloroformate (2.6 mL, 20 mmol). After 30 min at −20°C, a solution of o-hydroxyaniline (2.4 g; 22 mmol) in THF (50 mL) was added. The reaction mixture was stirred while slowly warming to room temperature (1 h). The solution was partitioned between 5% NaHCO3 and AcOEt. The organic layer was washed with 5% NaHSO4, H2O, 5% NaHCO3, H2O, and brine. The AcOEt layer was dried (Na2SO4) and filtered. The filtrate was concentrated to yield the intermediate amide as a yellow oil; that was directly cyclized without purification by refluxing in propionic acid for 4 h. The solution was concentrated in vacuo, and the residual oil was subjected to silica gel chromatography with a linear gradient from CH2Cl2 to CH2Cl2/Et2O (3:1): yield 2.37 g (42%); Rf(B) 0.73; HPLC K′ 6.31; oil; m/z 283 (M+H)+; 1H-NMR (DMSO-d6) δ 4.18–4.23 (d, 2H), 5.34 (s, 2H), 7.19–7.26 (m, 9H).
5.2.2. H2N-CH2-Boa
A mixture of Z-NH-CH2-Boa (1.13 g, 4 mmol) and 400 mg of 10% Pd/C in acetic acid (20 mL) was treated with H2 for 1 h at atmospheric pressure. The catalyst was filtered through Celite, and the filtrate was concentrated in vacuo to yield a pale yellow oil which was partitioned between 5% NaHCO3 and AcOEt. The organic layer was washed with 5% NaHCO3, H2O, and brine. The AcOEt layer was dried (Na2SO4), filtered and evaporated to dryness. The residual oil was subjected to silica gel chromatography with a linear gradient from CH2Cl2 to CH2Cl2/Et2O (3:1): yield 0.53 g (89%); Rf(B) 0.34; HPLC K′ 3.41; mp 120–122 °C; m/z 149 (M+H)+; 1H-NMR (DMSO-d6) δ 3.87–3.93 (d, 2H), 7.24–7.28 (m, 4H).
5.2.3. Boc-Dmt-Tic-NH-CH2-Boa
To a solution of Boc-Dmt-Tic-OH19 (0.14 g, 0.3 mmol) and H2N-CH2-Boa (0.04 g, 0.3 mmol) in DMF (10 mL) at 0 °C, HOBt (0.05 g, 0.33 mmol), and WSC (0.06 g, 0.33 mmol) were added. The reaction mixture was stirred for 3 h at 0 °C and 24 h at room temperature. After DMF was evaporated, the residue was dissolved in EtOAc and washed with citric acid (10% in H2O), NaHCO3 (5% in H2O), and brine. The organic phase was dried (Na2SO4) and evaporated to dryness. The residue was precipitated from Et2O/Pe (1:9, v/v): yield 0.15 g (82%); Rf(B) 0.75; HPLC K′ 6.02; mp 140–142 °C; [α]20D -20.8; m/z 600 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 2.35 (s, 6H), 2.92–3.17 (m, 4H), 4.41–4.92 (m, 6H), 6.29 (s, 2H), 6.96–7.26 (m, 8H).
5.2.4. TFA.H-Dmt-Tic-NH-CH2-Boa (1)
Boc-Dmt-Tic-NH-CH2-Boa (0.12 g, 0.2 mmol) was treated with TFA (1 mL) for 0.5 h at room temperature. Et2O/Pe (1:1, v/v) were added to the solution until the product precipitated: yield 0.12 g (97%); Rf(A) 0.36; HPLC K′ 4.36; mp 150–152 °C; [α]20D -21.5; m/z 500 (M+H)+; 1H-NMR (DMSO-d6) δ 2.35 (s, 6H), 2.92–3.17 (m, 4H), 3.95–4.92 (m, 6H), 6.29 (s, 2H), 6.96–7.26 (m, 8H). Anal. Calcd. for C31H31F3N4O6: C, 60.78; H, 5.10; N, 9.15. Found: C, 60.62; H, 5.03; N, 8.98.
5.2.5. N-(benzyloxycarbonyl)-glycine o-mercaptoanilide, disulfide derivative
A solution of Z-Gly-OH (1.57 g, 7.5 mmol) and TEA (1.05 mL, 7.5 mmol) in dry THF (25 mL) was treated at −20°C with isobutyl chloroformate (0.98 mL, 7.5 mmol). After 30 min at −20°C, a solution of o-aminophenyl disulfide (0.93 g; 3.75 mmol) in THF (25 mL) was added. The reaction mixture was stirred while slowly warming to room temperature (4 h). The solvent was evaporated at reduced pressure, and the residue was slurried in EtOH. The solid was filtered, washed with EtOH and dried in vacuo: yield 1.77 g (75%); Rf(B) 0.82; HPLC K′ 6.54; mp 160–162 °C; m/z 632 (M+H)+; 1H-NMR (DMSO-d6) δ 3.87–3.92 (d, 2H), 5.34 (s, 2H), 7.19–7.48 (m, 9H).
5.2.6. Benzyl (benzo[d]thiazol-2-yl)methylcarbamate (Z-NH-CH2-Bta)
The above disulfide derivative (1.5 g, 2.38 mmol) in glacial acetic acid (200 mL) was warmed to 50 °C. Zn powder (4 g, 60 mmol) was added slowly while most of the disulfide dissolved. The Zn was filtered, the solution was concentrated to dryness. The residue was dissolved in dioxane (200 mL), and the pH was adjusted to 10 with TEA. The solution was stirred under N2 at room temperature overnight. After solvent evaporation, the residue was purified by column chromatography as reported above. For Z-NH-CH2-Boa: yield 1 g (71%); Rf(B) 0.71; HPLC K′ 6.12; mp 93–95 °C; m/z 299 (M+H)+; 1H-NMR (DMSO-d6) δ 4.18–4.23 (d, 2H), 5.34 (s, 2H), 7.19–8.23 (m, 9H).
5.2.7. H2N-CH2-Bta
Z-NH-CH2-Bta (0.8 g, 2.68 mmol) was treated with 4N HBr/AcOH (30 mL) for 1.5 h at room temperature. The mixture was concentrated in vacuo and purified by column chromatography as reported for H2N-CH2-Boa. The purified intermediate was deprotonated with 5% NaHCO3 as reported for H2N-CH2-Boa: yield 0.39 g (88%); Rf(B) 0.32; HPLC K′ 3.19; mp 125–127 °C; m/z 165 (M+H)+; 1H-NMR (DMSO-d6) δ 3.87–3.93 (d, 2H), 7.55–8.23 (m, 4H).
5.2.8. Boc-Dmt-Tic-NH-CH2-Bta
This compound was obtained by condensation of Boc-Dmt-Tic-OH with H2N-CH2-Bta via WSC/HOBt as reported for Boc-Dmt-Tic-NH-CH2-Boa: yield 0.14 g (83%); Rf(B) 0.71; HPLC K′ 5.89; mp 143–145 °C; [α]20D -19.3; m/z 616 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 2.35 (s, 6H), 2.92–3.17 (m, 4H), 4.41–4.92 (m, 6H), 6.29 (s, 2H), 6.96–8.23 (m, 8H).
5.2.9. TFA.H-Dmt-Tic-NH-CH2-Bta (2)
Boc-Dmt-Tic-NH-CH2-Bta was treated with TFA as reported for TFA.H-Dmt-Tic-NH-CH2-Boa: yield 0.1 g (93%); Rf(A) 0.34; HPLC K′ 4.22; mp 155–157 °C; [α]20D -18.7; m/z 516 (M+H)+; 1H-NMR (DMSO-d6) δ 2.35 (s, 6H), 2.92–3.17 (m, 4H), 3.95–4.92 (m, 6H), 6.29 (s, 2H), 6.96–8.23 (m, 8H). Anal. Calcd. for C31H31F3N4O5S: C, 59.23; H, 4.97; N, 8.91. Found: C, 59.52; H, 5.13; N, 8.71.
5.2.10. Boc-Dmt-Tic-NH-CH2-Indl
This compound was obtained by condensation of Boc-Dmt-Tic-OH with H2N-CH2-Indl15 via WSC/HOBt as reported for Boc-Dmt-Tic-NH-CH2-Boa: yield 0.11 g (84%); Rf(B) 0.67; HPLC K′ 5.78; mp 147–149 °C; [α]20D -20.8; m/z 598 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 2.35 (s, 6H), 2.92–3.17 (m, 4H), 4.41–4.92 (m, 6H), 6.13–6.29 (m, 3H), 6.96–7.08 (m, 8H).
5.2.11. TFA.H-Dmt-Tic-NH-CH2-Indl (3)
Boc-Dmt-Tic-NH-CH2-Indl was treated with TFA as reported for TFA.H-Dmt-Tic-NH-CH2-Boa: yield 0.09 g (91%); Rf(A) 0.30; HPLC K′ 4.16; mp 149–151 °C; [α]20D -18.1; m/z 498 (M+H)+; 1H-NMR (DMSO-d6) δ 2.35 (s, 6H), 2.92–3.17 (m, 4H), 3.95–4.92 (m, 6H), 6.13–6.29 (m, 3H), 6.96–7.08 (m, 8H). Anal. Calcd. for C32H33F3N4O5: C, 62.94; H, 5.45; N, 9.18. Found: C, 63.15; H, 5.59; N, 9.29.
5.2.13. Boc-Dmt-Tic-NH-CH2-Indn
This compound was obtained by condensation of Boc-Dmt-Tic-OH with H2N-CH2-Indn16 via WSC/HOBt as reported for Boc-Dmt-Tic-NH-CH2-Boa: yield 0.14 g (87%); Rf(B) 0.74; HPLC K′ 6.21; mp 139–141 °C; [α]20D -21.4; m/z 599 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 2.35 (s, 6H), 2.66–3.17 (m, 11H), 4.41–4.92 (m, 4H), 6.29 (s, 2H), 6.96–7.20 (m, 8H).
5.2.14. TFA.H-Dmt-Tic-NH-CH2-Indn (4)
Boc-Dmt-Tic-NH-CH2-Indn was treated with TFA as reported for TFA.H-Dmt-Tic-NH-CH2-Boa: yield 0.10 g (90%); Rf(A) 0.35; HPLC K′ 4.44; mp 140–142 °C; [α]20D -19.8; m/z 499 (M+H)+; 1H-NMR (DMSO-d6) δ 2.35 (s, 6H), 2.66–3.95 (m, 12H), 4.41–4.92 (m, 3H), 6.29 (s, 2H), 6.96–7.20 (m, 8H). Anal. Calcd. for C33H36F3N3O5: C, 64.80; H, 5.93; N, 6.87. Found: C, 64.67; H, 5.76; N, 6.73.
5.2.15. tert-butyl (4-phenyl-1H-imidazol-2-yl)methylcarbamate (Boc-NH-CH2-ImidPh)
A solution of Boc-Gly-OH (2.87 g, 16.4 mmol) and cesium carbonate (2.7 g, 8.3 mmol) in EtOH (50 mL) was shaken for 30 min at room temperature, and then evaporated under reduced pressure. To the resulting salt in DMF (60 mL) was added 2-bromoacetophenone (3.26 g, 16.4 mmol). The mixture was stirred for 1 h at room temperature under argon and then concentrated under reduced pressure. AcOEt (40 mL) was added, the mixture filtered, and the CsBr washed with AcOEt. The filtrate was then concentrated under reduced pressure. A solution of the resulting oil and ammonium acetate (2.5 g, 32 mmol) in xylene (200 mL) was refluxed for 45 min. Excess NH4OAc and H2O were removed using a Dean-Stark trap. The mixture was then cooled to room temperature, diluted with AcOEt (100 mL) and washed with H2O, 5% NaHCO3 and brine. The organic phase was dried (Na2SO4), and concentrated to dryness. The residue was precipitated from Et2O/Pe (1:1): yield 2.6 g (58%); Rf(B) 0.57; HPLC K′ 5.68; mp 181–183 °C; m/z 274 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 4.18–4.23 (d, 2H), 6.9–7.48 (m, 6H).
5.2.16. H2N-CH2-ImidPh
Boc-NH-CH2-ImidPh (2 g, 7.33 mmol) was treated with TFA (4 mL) for 0.5 h at room temperature. Et2O/Pe (1:1, v/v) were added to the solution until the product precipitated. The solid was filtered and dissolved in AcOEt for deprotonation with 5% NaHCO3 as reported for H2N-CH2-Boa: yield 1.14 g (90%); Rf(B) 0.25; HPLC K′ 2.87; mp 121–123 °C; m/z 174 (M+H)+; 1H-NMR (DMSO-d6) δ 3.87–3.93 (d, 2H), 6.9–7.48 (m, 4H).
5.2.17. Boc-Dmt-Tic-NH-CH2-ImidPh
This compound was obtained by condensation of Boc-Dmt-Tic-OH with H2N-CH2-ImidPh via WSC/HOBt (except washing with aqueous citric acid solution) as reported for Boc-Dmt-Tic-NH-CH2-Boa: yield 0.11 g (80%); Rf(B) 0.58; HPLC K′ 5.65; mp 148–150 °C; [α]20D -22.5; m/z 625 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 2.35 (s, 6H), 2.92–3.17 (m, 4H), 4.41–4.92 (m, 6H), 6.29 (s, 2H), 6.9–7.48 (m, 10H).
5.2.18. 2TFA.H-Dmt-Tic-NH-CH2-ImidPh (5)
Boc-Dmt-Tic-NH-CH2-ImidPh was treated with TFA as reported for TFA.H-Dmt-Tic-NH-CH2-Boa: yield 0.08 g (93%); Rf(A) 0.29; HPLC K′ 4.12; mp 152–154 °C; [α]20D -23.7; m/z 525 (M+H)+; 1H-NMR (DMSO-d6) δ 2.35 (s, 6H), 2.92–3.17 (m, 4H), 3.95–4.92 (m, 6H), 6.29 (s, 2H), 6.9–7.48 (m, 10H). Anal. Calcd. for C35H35F6N5O7: C, 55.93; H, 4.69; N, 9.32. Found: C, 55.77; H, 4.62; N, 9.18.
5.2.19. Boc-Dmt-Tic-NH-CH2-Imid
This compound was obtained by condensation of Boc-Dmt-Tic-OH with H2N-CH2-Imid18 via WSC/HOBt (except washing with aqueous citric acid solution) as reported for Boc-Dmt-Tic-NH-CH2-Boa: yield 0.09 g (83%); Rf(B) 0.43; HPLC K′ 5.01; mp 153–155 °C; [α]20D -23.6; m/z 549 (M+H)+; 1H-NMR (DMSO-d6) δ 1.40 (s, 9H), 2.35 (s, 6H), 2.92–3.17 (m, 4H), 4.41–4.92 (m, 6H), 6.29 (s, 2H), 6.87–7.02 (m, 6H).
5.2.20. 2TFA.H-Dmt-Tic-NH-CH2-Imid (6)
Boc-Dmt-Tic-NH-CH2-Imid was treated with TFA as reported for TFA.H-Dmt-Tic-NH-CH2-Boa: yield 0.07 g (94%); Rf(A) 0.25; HPLC K′ 3.97; mp 156–158 °C; [α]20D -24.1; m/z 449 (M+H)+; 1H-NMR (DMSO-d6) δ 2.35 (s, 6H), 2.92–3.17 (m, 4H), 3.95–4.92 (m, 6H), 6.29 (s, 2H), 6.87–7.02 (m, 6H). Anal. Calcd. for C29H31F6N5O7: C, 51.56; H, 4.63; N, 10.37. Found: C, 51.74; H, 4.79; N, 10.18.
5.3. Pharmacology
5.3.1. Radioreceptor binding assays
Opioid receptor affinity were determined under equilibrium conditions [2.5 h at room temperature (23 °C)] in a competition assay using brain P2 synaptosomal membranes prepared from Sprague-Dawley rats.22, 23 Synaptosomes were preincubated to remove endogenous opioid peptides and stored at −80 °C in buffered 20% glycerol.22, 24 Each analogue was analyzed in duplicate assays using five to eight dosages and three to five independent repetitions with different synaptosomal preparations (n values are listed in Table 1 in parenthesis and results are mean ± SE). Unlabeled peptide (2 μM) was used to determine non-specific binding in the presence of 1.9 nM [3H]deltorphin II (45.0 Ci/mmol, Perkin Elmer, Boston, MA; KD = 1.4 nM) for δ -opioid receptors and 3.5 nM [3H]DAMGO (50.0 Ci/mmol), Amersham Bioscience, Buckinghamshire, U. K.; KD = 1.5 nM) for μ-opioid receptors. Glass fibre filters (Whatman GFC) were soaked in 0.1% polyethylenimine in order to enhance the signal-to-noise ratio of the bound radiolabeled-synaptosome complex, and the filters were washed thrice in ice-cold buffered BSA.22 The affinity constants (Ki) were calculated according to Cheng and Prusoff.20
5.3.2. Biological activity in isolated tissue preparation
The myenteric plexus longitudinal muscle preparations (2–3 cm segments) from the small intestine of male Hartley strain guinea pigs (GPI) measured μ-opioid receptor agonism, and a single mouse vas deferens (MVD) was used to determine δ -opioid receptor agonism as described previously.25 The isolated tissues were suspended in organ baths containing balanced salt solutions in a physiological buffer, pH 7.5. Agonists were tested for the inhibition of electrically evoked contraction and expressed as IC50 (nM) obtained from the dose-response curves. The IC50 values represent the mean ± SE of five or six separate assays. δ -antagonist potencies in the MVD assay were determined against the δ -agonist DPDPE; μ-antagonism in the GPI assay used the μ-agonist PL-017 and both are expressed as pA2 determined using the Schild Plot.26
Acknowledgments
This research was supported in part by the University of Cagliari, University of Ferrara and the Intramural Research Program of NIH and NIEHS. The authors appreciate the professional expertise and assistance of the library staff at NIEHS.
Abbreviations
In addition to the IUPAC-IUB Commission on Biochemical Nomenclature (J. Biol. Chem. 1985, 260, 14-42), this paper uses the following additional symbols and abbreviations
- AcOEt
ethyl acetate
- AcOH
acetic acid
- Bid
1H-benzimidazol-2-yl
- Boa
benzoxazol-2-yl
- Boc
tert-butyloxycarbonyl
- Bta
benzothiazol-2-yl
- DAMGO
- DEL C
deltorphin II (H-Tyr-D-Ala-Phe-Asp-Val-Val-Gly-NH2)
- DMF
N,N-dimethylformamide
- DMSO-d6
hexadeuteriodimethyl sulfoxide
- Dmt
2′,6′-dimethyl-L-tyrosine
- DPDPE
- GPI
guinea-pig ileum
- HOBt
1-hydroxybenzotriazole
- HPLC
high performance liquid chromatography
- Imid
1H-imidazol-2-yl
- ImidPh
4-phenyl-1H-imidazol-2-yl
- Indl
1H-indol-2-yl
- Indn
2,3-dihydro-1H-inden-2-yl
- MALDI-TOF
matrix assisted laser desorption ionization time-of-flight
- MVD
mouse vas deferens
- NMM
4-methylmorpholine
- pA2
negative log of the molar concentration required to double the agonist concentration to achieve the original response
- PL-017
- TEA
triethylamine
- TFA
trifluoroacetic acid
- Tic
1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
- TLC
thin-layer chromatography
- WSC
1-ethyl-3-[3′ -dimethyl)aminopropyl]-carbodiimide hydrochloride
- Z
benzyloxycarbonyl
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Salvadori S, Attila M, Balboni G, Bianchi C, Bryant SD, Crescenzi O, Guerrini R, Picone D, Tancredi T, Temussi PA, Lazarus LH. Mol Med. 1995;1:678–689. [PMC free article] [PubMed] [Google Scholar]
- 2.Bryant SD, Jinsmaa Y, Salvadori S, Okada Y, Lazarus LH. Biopolymers. 2003;71:86–102. doi: 10.1002/bip.10399. [DOI] [PubMed] [Google Scholar]
- 3.Salvadori S, Balboni G, Guerrini R, Tomatis R, Bianchi C, Bryant SD, Cooper PS, Lazarus LH. J Med Chem. 1997;40:3100–3108. doi: 10.1021/jm9607663. [DOI] [PubMed] [Google Scholar]
- 4.Balboni G, Guerrini R, Salvadori S, Bianchi C, Rizzi D, Bryant SD, Lazarus LH. J Med Chem. 2002;45:713–720. doi: 10.1021/jm010449i. [DOI] [PubMed] [Google Scholar]
- 5.Balboni G, Onnis V, Congiu C, Zotti M, Sasaki Y, Ambo A, Bryant SD, Jinsmaa Y, Lazarus LH, Trapella C, Salvadori S. J Med Chem. 2006;49:5610–5617. doi: 10.1021/jm060741w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balboni G, Salvadori S, Guerrini R, Negri L, Giannini E, Jinsmaa Y, Bryant SD, Lazarus LH. J Med Chem. 2002;45:5556–5563. doi: 10.1021/jm020336e. [DOI] [PubMed] [Google Scholar]
- 7.Dai X, Cui SG, Li SR, Chen Q, Wang R. Behav Brain Res. 2007;182:21–27. doi: 10.1016/j.bbr.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 8.Jutkiewicz EM. Mol Interv. 2006;6:162–169. doi: 10.1124/mi.6.3.7. [DOI] [PubMed] [Google Scholar]
- 9.Narita M, Kuzumaki N, Miyatake M, Sato F, Wachi H, Seyama Y, Suzuki TR. J Neurochem. 2006;97:1494–1505. doi: 10.1111/j.1471-4159.2006.03849.x. [DOI] [PubMed] [Google Scholar]
- 10.Hill MP, Hille CJ, Brotchie JM. Drug News Perspect. 2000;13:261–268. [PubMed] [Google Scholar]
- 11.Torregrossa MM, Jutkiewicz EM, Mosberg HI, Balboni G, Watson SJ, Woods JH. Brain Res. 2006;1069:172–181. doi: 10.1016/j.brainres.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vergura R, Valenti E, Hebbes CP, Gavioli EC, Spagnolo B, McDonald J, Lambert DG, Balboni G, Salvadori S, Regoli D, Calò G. Peptides. 2006;27:3322–3330. doi: 10.1016/j.peptides.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 13.Nestor JJ, Jr, Horner BL, Ho TL, Jones GH, McRae G, Vickery BH. J Med Chem. 1984;27:320–325. doi: 10.1021/jm00369a016. [DOI] [PubMed] [Google Scholar]
- 14.Baudet P, Otten C. Helv Chim Acta. 1970;53:1683–1693. doi: 10.1002/hlca.19700530428. [DOI] [PubMed] [Google Scholar]
- 15.Wright WB, Jr, Brabander HJ. J Med Chem. 1968;11:1164–1167. doi: 10.1021/jm00312a014. [DOI] [PubMed] [Google Scholar]
- 16.Shinozaki K, Sato H, Iwakuma T, Sato R, Kurimoto T, Yoshida K. Bioorg Med Chem Lett. 1999;9:401–406. doi: 10.1016/s0960-894x(99)00014-1. [DOI] [PubMed] [Google Scholar]
- 17.Poitout L, Roubert P, Contour-Galcéra M-O, Moinet C, Lannoy J, Pommier J, Plas P, Bigg D, Thurieau C. J Med Chem. 2001;44:2990–3000. doi: 10.1021/jm0108449. [DOI] [PubMed] [Google Scholar]
- 18.Bastiaansen LAM, Godefroi EF. J Org Chem. 1978;43:1603–1604. [Google Scholar]
- 19.Salvadori S, Guerrini R, Balboni G, Bianchi C, Bryant SD, Cooper PS, Lazaarus LH. J Med Chem. 1999;42:5010–5019. doi: 10.1021/jm990165m. [DOI] [PubMed] [Google Scholar]
- 20.Cheng YC, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 21.Kosterlitz HW, Watt AJ. Br J Pharmacol. 1968;33:266–276. doi: 10.1111/j.1476-5381.1968.tb00988.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazarus LH, Salvadori S, Santagada V, Tomatis R, Wilson WE. J Med Chem. 1991;34:1350–1355. doi: 10.1021/jm00108a017. [DOI] [PubMed] [Google Scholar]
- 23.Lazarus LH, Salvadori S, Attila M, Grieco P, Bundy DM, Wilson WE, Tomatis R. Peptides. 1993;14:21–28. doi: 10.1016/0196-9781(93)90006-3. [DOI] [PubMed] [Google Scholar]
- 24.Lazarus LH, Wilson WE, de Castglione R, Guglietta A. J Biol Chem. 1989;264:3047–3050. [PubMed] [Google Scholar]
- 25.Sasaki Y, Sasaki A, Niizuma H, Goto H, Ambo A. Bioorg Med Chem. 2003;11:675–678. doi: 10.1016/s0968-0896(02)00601-6. [DOI] [PubMed] [Google Scholar]
- 26.Arunlakshana Q, Schild HO. Br J Pharmcol. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]