

Abstract
The contribution of hypoxia to cisplatin-induced renal tubular injury is controversial. Because the hypoxia-inducible factor (HIF) pathway is a master regulator of adaptation to hypoxia, we measured the effects of cisplatin on HIF accumulation in vitro and in vivo, and tested whether hypoxic preconditioning is protective against cisplatin-induced injury. We found that cisplatin did not stabilize HIF-1α protein in vitro or in vivo under normoxic conditions. However, hypoxic preconditioning of cisplatin-treated proximal tubular cells in culture reduced apoptosis in an HIF-1α-dependent fashion and increased cell proliferation as measured by BrdU incorporation. In vivo, rats preconditioned with carbon monoxide before cisplatin administration had significantly better renal function than rats kept in normoxic conditions throughout. Moreover, the histomorphological extent of renal damage and tubular apoptosis was reduced by the preconditional treatment. Therefore, development of pharmacologic agents to induce renal HIF might provide a new approach to ameliorate cisplatin-induced nephrotoxicity.
Ischemia and toxicity are considered the main pathophysiological factors that lead to the development of acute kidney injury. A substance that is well known to induce toxic kidney injury is cis-diammino-dichloroplatinum (cisplatin).1 Cisplatin is widely used as a chemotherapeutic agent in the treatment of solid tumors, but its application is frequently limited because of nephrotoxic side effects, which occur even after a single dose of cisplatin in up to 28% to 36% of cancer patients.2 Renal cisplatin toxicity is associated with the induction of apoptotic cell death, inflammation, and necrosis predominantly of the S3 segment of the proximal tubule.3–5 In addition to the toxic damage of tubular cells, reduced renal blood flow6 with subsequent hypoxia of the outer medulla may contribute to cisplatin-induced renal injury.7 Because the S3 segment has a low capacity for anaerobic glycolysis despite high metabolic demands, a decline in oxygen tension in the outer medulla, where oxygen tensions are already physiologically low, could rapidly lead to energy deprivation and thereby contribute to cellular injury.8
A central cellular mechanism of adaptation to low oxygen tensions operates through activation of the hypoxia-inducible transcription factor (HIF). The oxygen-regulated α-subunit of HIF dimerizes with the aryl hydrocarbon receptor nuclear translocator (ARNT) and transcriptionally induces a broad spectrum of target genes involved in glycolysis, pH regulation, vascular tone, or erythropoiesis.9,10 The HIF-α subunit is constitutively expressed but rapidly degraded by the ubiquitin-proteasome system in the presence of oxygen. Up-regulation of HIF in different models of acute renal injury, such as renal segmental infarction,11 ischemia-reperfusion,12,13 and radiocontrast-induced injury,14 suggests that the renal HIF system plays an important role in the protection against injury. In support of this concept, we have recently been able to show that induction of HIF before ischemia-reperfusion ameliorates renal injury and function.15 One important question in this context is whether the protective effect of the induction of HIF target genes is limited to hypoxic injuries or may also extend to different nonhypoxic mechanisms of cellular damage.
Cisplatin-induced renal injury appears to be an attractive and clinically important model to test this question, but conflicting data exist so far about the role of HIF and hypoxia in cisplatin-induced tubular toxicity. One report demonstrated HIF-independent protective effects of hypoxia when cells were simultaneously exposed to cisplatin.16 In contrast, Tanaka et al.17 showed that cisplatin itself increased HIF-1α activation in the rat kidney and injection of cobalt chloride, potentially through HIF-α, reduced the number of apoptotic cells. However, the effect of hypoxic preconditioning in cisplatin-induced renal injury has not yet been studied. We thus investigated in vitro and in vivo whether HIF is induced by cisplatin, if hypoxic preconditioning has protective effects on cellular injury induced by cisplatin and whether such an effect is mediated by HIF.
Results
HIF-1α Protein Is Not Induced by Cisplatin in Proximal Tubular Cells
The effects of cisplatin on the HIF-system were tested in the immortalized human renal proximal tubular cell line HKC-8.18 No cellular accumulation of HIF-1α protein was observed under normoxic conditions and after 8 h incubation with increasing cisplatin concentrations (10 to 100 μM) (Figure 1, A). To test HIF-1α transactivation, HKC-8 cells were transfected with an HIF-1α-sensitive luciferase reporter construct.19 Increasing doses of cisplatin led to a dose-dependent decrease of reporter gene activity compared with untreated or vehicle-treated cells, which confirmed that cisplatin does not activate HIF (Figure 1, B). HKC-8 cells, exposed to the chemical HIF inducer 2′2-dipyridyl (DP) for 8 h, showed a marked HIF-1α protein accumulation. Preincubation with increasing cisplatin concentrations reduced DP-induced HIF signals dose-dependently (Figure 1, C). Removal of cisplatin before DP stimulation led to decreased HIF protein levels that were comparable to those under continued cisplatin exposure (Figure 1, D). Thus, HIF-1α protein and HIF transactivation are not induced but rather suppressed by cisplatin in renal tubular cells in vitro.
Figure 1.
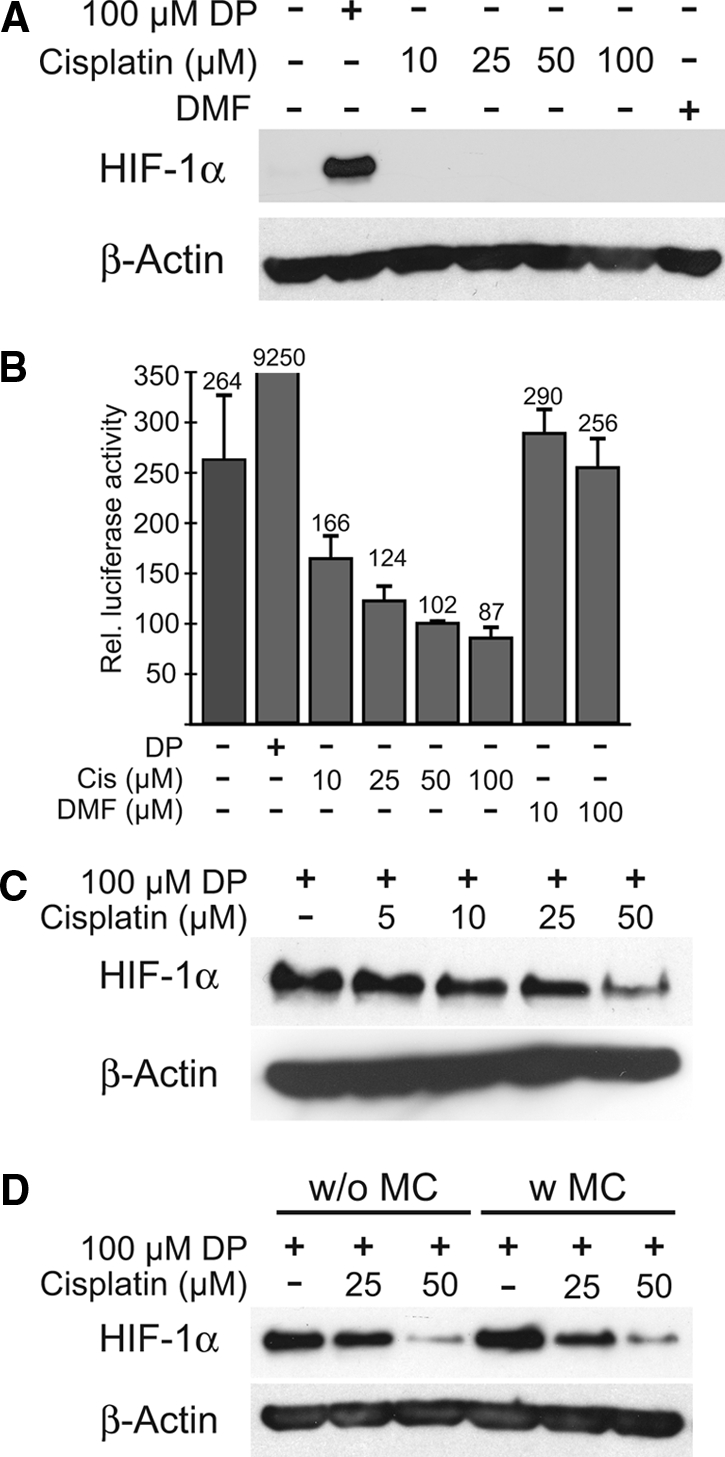
Cisplatin does not induce HIF-1α protein and does not transactivate an HIF-1α responsive reporter construct. (A) No induction of HIF-1α protein in the renal proximal tubular cell line HKC-8 was detectable with different concentrations of cisplatin or vehicle (DMF). The chemical HIF-inducer 2,2′dipyridyl (DP) was used as a positive control. (B) Accordingly, an HIF-1α responsive luciferase reporter construct was not activated but suppressed by different concentrations of cisplatin in transient transfection assays in HKC-8 cells. DP was used as a positive control for HIF-1α activation (normalized to β-galactosidase, mean ± SD, n = 3). (C) Pretreatment with cisplatin before HIF-1α stabilization with DP reduced HIF protein levels dose-dependently in HKC-8 cells. This reduction was irrespective of ongoing cisplatin exposure (w/o MC, without medium change; D, left 3 lanes) or of cisplatin withdrawal (w MC, with medium change; D, right 3 lanes). Representative immunoblots of 3 independent experiments are shown. β-actin was used as loading control.
Cisplatin Treatment of HKC-8 Cells Reduces HIF-1α Target Gene Expression
To test for effects of cisplatin on the transcriptional activity of HIF in HKC-8 cells, we performed RNase protection assays for HIF-1α and target genes. mRNA levels of the HIF-1 target glucose transporter-1 (Glut-1) were induced by the chemical HIF-inducer DP. Increasing concentrations of cisplatin decreased Glut-1 mRNA expression as well as HIF-1α mRNA expression (Figure 2, A). Pretreatment with cisplatin for 8 h almost completely suppressed the Glut-1 mRNA induction by DP (Figure 2, B). Because cisplatin was removed before DP stimulation, this indicates that the inhibitory effect persisted beyond the exposure. The internal loading control (U6-small nuclear RNA) was also suppressed at high doses of cisplatin, which presumably reflected a reduced total transcriptional activity and/or a loss of viability of cells under these conditions. Taken together, these data indicate that in cultured renal cells HIF target gene activation is markedly suppressed by cisplatin.
Figure 2.
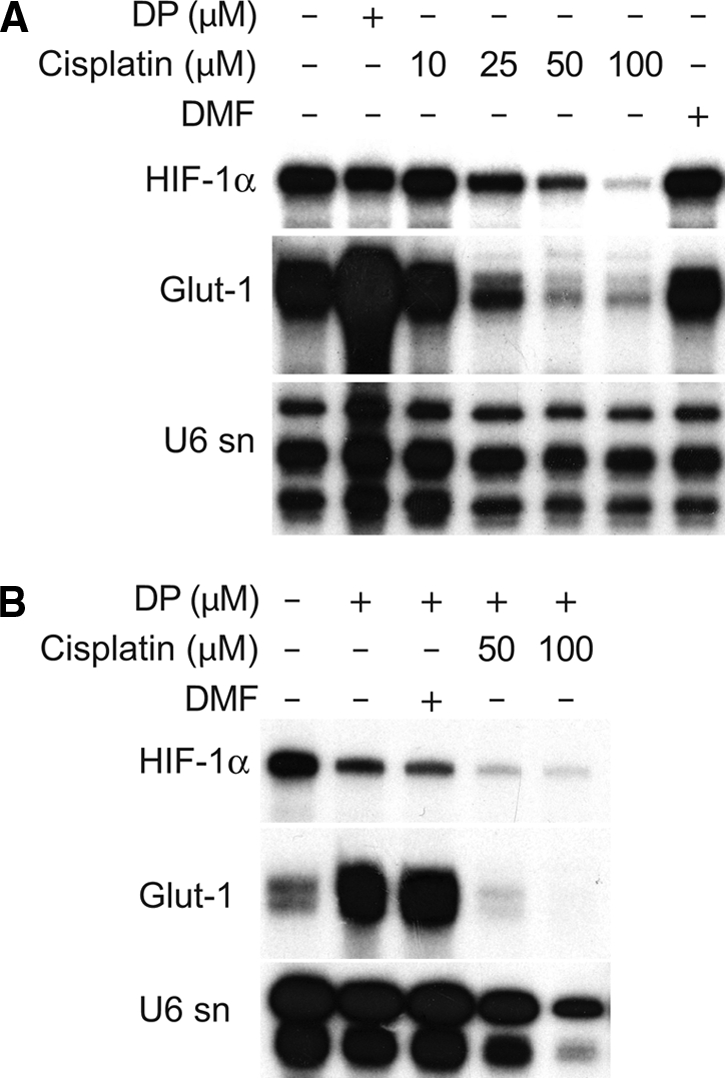
HIF-1α and HIF-1α target gene mRNA levels are reduced by cisplatin. (A) RNase protection assays show that HIF-1α mRNA was reduced dose-dependently in HKC-8 cells after cisplatin treatment compared with controls or vehicle (DMF)-treated cells. Expression of the HIF target gene Glut-1 was also reduced by cisplatin. DP treatment demonstrated up-regulation of Glut-1 mRNA. (B) Up-regulation of Glut-1 is reduced dose-dependently after HKC-8 cells were pretreated with cisplatin, demonstrating the impaired responsiveness of the HIF system in the presence of cisplatin. Representative data of 3 independent experiments. U6sn mRNA was used as loading control.
Hypoxic Preconditioning Reduces Cisplatin-Induced Apoptosis in Proximal Tubular Cells
Preconditional HIF induction was recently demonstrated to be protective against ischemic renal injury.15 We therefore investigated whether this principle could also be applied to cisplatin toxicity. Caspase-3-dependent apoptosis induced by cisplatin was quantified by measuring cleavage of the fluorogenic substrate of caspase-3, DEVD-AMC. In HKC-8 cells, 50 μM cisplatin for 8 h increased the caspase-3 activity 6.96 ± 1.1-fold compared with untreated cells. Hypoxic preconditioning at 1% oxygen for 12 h before cisplatin treatment significantly reduced this increase to 4.81 ± 0.7-fold (n = 3, P < 0.05), indicating an antiapoptotic effect (Figure 3, A). To rule out cell line-specific effects, we also used isolated primary mouse proximal tubular cells (mPTs); 50 μM cisplatin increased caspase-3 activity 6.36 ± 0.1-fold in mPTs, which was significantly reduced to 3.89 ± 0.4-fold by hypoxic preconditioning (n = 3, P < 0.05), thus confirming results obtained in HKC-8 cells (Figure 3, A). To investigate the role of HIF-1α in the protective effects of hypoxic preconditioning, HIF-1α knockout murine embryonic fibroblasts (MEFHIF1α−/−) were compared with corresponding wild-type MEFs. Hypoxic preconditioning significantly reduced cisplatin-induced caspase-3 activity in wild-type MEFs (from 3.1 ± 0.2-fold to 2.1 ± 0.4-fold; n = 3, P < 0.05; Figure 3, B), whereas it had no effect in MEFHIF1α−/− cells (6.2 ± 0.6-fold versus 6.8 ± 1.1-fold; n = 3, P = not significant). Moreover, the increase in caspase-3 activity after cisplatin exposure was higher in HIF-deficient than in wild-type MEFs, suggesting that basal HIF activation is already protective against cisplatin. Simultaneous exposure to cisplatin and hypoxia also attenuated apoptosis, but independent of the HIF-1α genotype of the cells (Figure 3, B). We conclude that a functional HIF pathway is required for the anti-apoptotic effects of hypoxic preconditioning in vitro.
Figure 3.

Hypoxic preconditioning reduces cisplatin-induced apoptosis in tubular cells and embryonic fibroblasts. (A) HKC-8 cells (left) and mouse primary proximal tubular cells (mPT, right) were incubated with 1% O2 before stimulation with 50 μM cisplatin. In both cell types, hypoxic preconditioning reduced relative fluorometric caspase-3 activity significantly, indicating an anti-apoptotic effect. (B) No reduction in cisplatin-induced caspase-3 activity after hypoxic preconditioning was observed in HIF-1α-deficient murine embryonic fibroblasts (MEFHIF-1α−/−) compared with wild-type MEFs, which indicates that the observed cytoprotective effect was dependent on the presence of functional HIF-1α. When cisplatin was applied simultaneously to hypoxia, apoptosis rates were comparably attenuated, but effects were seen in both genotypes, indicating that HIF induction plays a role in preconditional but not in simultaneous application of hypoxia. Data are mean of 3 independent experiments ± SD. *P < 0.05.
Hypoxic Preconditioning Prevents Cisplatin-Induced Reduction of DNA Synthesis but Does Not Change DNA Content
To evaluate effects of hypoxic preconditioning before cisplatin administration on cellular proliferation rates, BrdU incorporation was investigated in HKC-8 cells. BrdU incorporation was not reduced by hypoxia (Figure 4, C), suggesting that hypoxia did not suppress proliferation. Distribution of the cells in G1, S, and G2 phase of the cell cycle was also not significantly altered by hypoxia alone (Figure 4, F). Hypoxic preconditioning significantly increased the number of BrdU positive cells after cisplatin treatment (33.8 ± 9.5% versus 12.2 ± 5%, n = 3, P < 0.05; Figure 4, B, D, and F), indicating increased proliferation and viability. Proportions of cells in G2/M phase were significantly reduced in cisplatin-treated samples independent of hypoxic preconditioning (Figure 4, F). Taken together, hypoxic preconditioning does not induce cell cycle arrest in HKC-8 cells and the protective effect of hypoxia becomes apparent in the increased ability of cells to proliferate after cisplatin exposure.
Figure 4.

Increased BrdU incorporation in HKC-8 cells pretreated with hypoxia. (A, C) HKC-8 cells grown in hypoxia did not exhibit reduced BrdU incorporation compared with normoxic controls. Hypoxic preconditioning before incubation with 50 μM cisplatin significantly increased proliferation (D) compared with unconditioned cells (B), indicating increased cellular viability and proliferation after hypoxic preconditioning. (A through E) Representative pictures of BrdU incorporation. (F) Hypoxia did not change the percentage of cells in different cell cycle phases compared with normoxia. Cells in G2 phase were reduced in cisplatin-treated cells independent of hypoxic preincubation. Statistical analysis of BrdU-positive cells and DNA content of 3 independent experiments. Data are mean ± SD. *P < 0.05.
Renal Cisplatin Injury Is Not Associated With Marked Tissue Hypoxia and Does Not Induce HIF-1α in Rats
To corroborate the in vitro results that cisplatin does not induce HIF-1α in renal tubular cells, cisplatin (8 mg/kg body weight) was injected intraperitoneally into rats and HIF-protein was studied by immunohistochemistry after 1, 24, 72, and 120 h. In line with previous studies,13 little or no HIF-1α staining was observed in normoxic control animals. Treatment with 0.1% carbon monoxide (CO) led to a robust induction of HIF-1α (Figure 5, B). Corresponding to in vitro experiments, HIF-1α was not detectable in cisplatin injured rat kidneys at 120 h (Figure 5, C) and at all other time points examined (not shown).
Figure 5.

HIF-1α protein is not detectable by immunohistochemistry in kidneys of cisplatin-treated rats. In the kidney cortex of untreated rats, HIF-1α (A) and pimonidazole (E) was not detectable. Ten-hour exposition to 0.1% CO led to a marked increase in HIF-1α-positive tubular cells, in particular in the outer medulla (B). In cisplatin-treated animals, no induction of HIF-1α protein occurred at 120 h (C) and at all earlier time points investigated (1 h, 24 h, and 72 h, not shown). In contrast, some regional accumulation of the hypoxic marker pimonidazole was visible after 120 h in perinecrotic areas (F). Preconditional CO treatment before cisplatin administration did not increase HIF-1α protein or pimonidazole staining after 120 h (D and G).
To test if cisplatin induces tissue hypoxia, pimonidazole was injected 30 min before death. Pimonidazole accumulates in hypoxic tissues20 and was detectable in the renal medulla of normoxic kidneys, which is known to have physiologically low oxygen tensions13 (not shown). Pimonidazole did not accumulate in the untreated kidney cortex (Figure 5, E) and not 1 h and 72 h after cisplatin. First after 120 h pimonidazole staining was slightly increased in perinecrotic regions of the tubuli (Figure 5, F), indicating some degree of regional tissue hypoxia at a late time point and possibly secondary to the toxic injury. Neither HIF-1α nor pimonidazole was detectable in animals preconditioned with CO for 10 h before cisplatin administration at the endpoint after 120 h (Figure 5, D and G).
Preconditioning With Carbon Monoxide Ameliorates Renal Morphology and Reduces Apoptosis in Cisplatin-Treated Rats
To investigate whether the protection by hypoxic preconditioning in vitro could be transferred to the in vivo situation, we exposed rats to 0.1% CO for 10 h before cisplatin injection. CO treatment reduces tissue oxygen availability by blocking the oxygen carrying capacity of hemoglobin and has previously been shown to robustly induce HIFα in renal tubular cells in vivo (Figure 5, B).13 After 10 h, CO exposure was stopped and cisplatin was administered. 120 h after injection of cisplatin, hematoxylin and eosin and periodic acid-Schiff-stained cross sections of the left kidney were analyzed. Cisplatin administration induced acute tubular injury with a histomorphological injury score of 1.25 ± 0.81. Animals preconditioned with 0.1% CO exhibited a clear preservation of epithelial structure and morphologic injury scores were reduced to 0.38 ± 0.59 (P < 0.05 versus control animals, n = 10 per group) (Figure 6, A through C).
Figure 6.

CO preconditioning improves renal morphology and reduces apoptosis after cisplatin-induced ARF. The extent of acute tubular necrosis (ATN) was decreased in CO-preconditioned rats with cisplatin-induced ARF compared with control animals in hematoxylin and eosin-stained renal cross sections (A, B). The blinded ATN scoring (C) revealed a significantly lower score for CO-treated animals (*P < 0.05 versus control). Tubular apoptosis was also reduced in renal sections of CO-preconditioned rats (E) compared with unconditioned animals (D). Blinded analysis of sections demonstrated that the number of TUNEL-positive cells was significantly reduced (F, *P < 0.05 versus control).
Apoptosis is a critical pathophysiological event in cisplatin-induced renal failure. We therefore evaluated terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining of the kidneys 120 h after cisplatin administration. In control animals with cisplatin injury, a mean of 218 ± 14 TUNEL-positive cells were detectable in 10 randomly selected fields. TUNEL-positive cells were significantly reduced to 101 ± 45 in CO-preconditioned animals (P < 0.05; Figure 6, D through F). Caspase-3 staining also showed a clear trend toward reduced apoptosis; however, the difference was not statistically significant (not shown). Taken together, preconditioning with CO resulted in significantly less tissue damage and apoptosis in vivo in cisplatin-induced renal injury.
Preconditioning With Carbon Monoxide Preserves Renal Function in Cisplatin-Induced Acute Renal Failure (ARF) in Rats
To evaluate if the preserved tissue morphology after preconditioning with CO correlated with preserved kidney function, we analyzed serum creatinine and urea after 0, 24, 72, and 120 h. Serum creatinine levels rose from 0.2 ± 0.02 mg/dl to 0.77 ± 0.49 mg/dl (n = 10) in cisplatin-treated animals at 72 h and subsequently decreased again. This increase was markedly blunted by CO pretreatment (0.34 ± 0.21 mg/dl at 72 h, P < 0.05) (Figure 7, A). Serum urea levels supported the creatinine, data which were also reduced in the CO-treated group compared with control animals (39.9 ± 37.6 mg/dl at 72 h versus 78 ± 14.7 mg/dl, P < 0.05) (Figure 7, B). Thus, together with the morphologic data, these findings demonstrate a clear protection against cisplatin-induced kidney injury by CO preconditioning in vivo.
Figure 7.
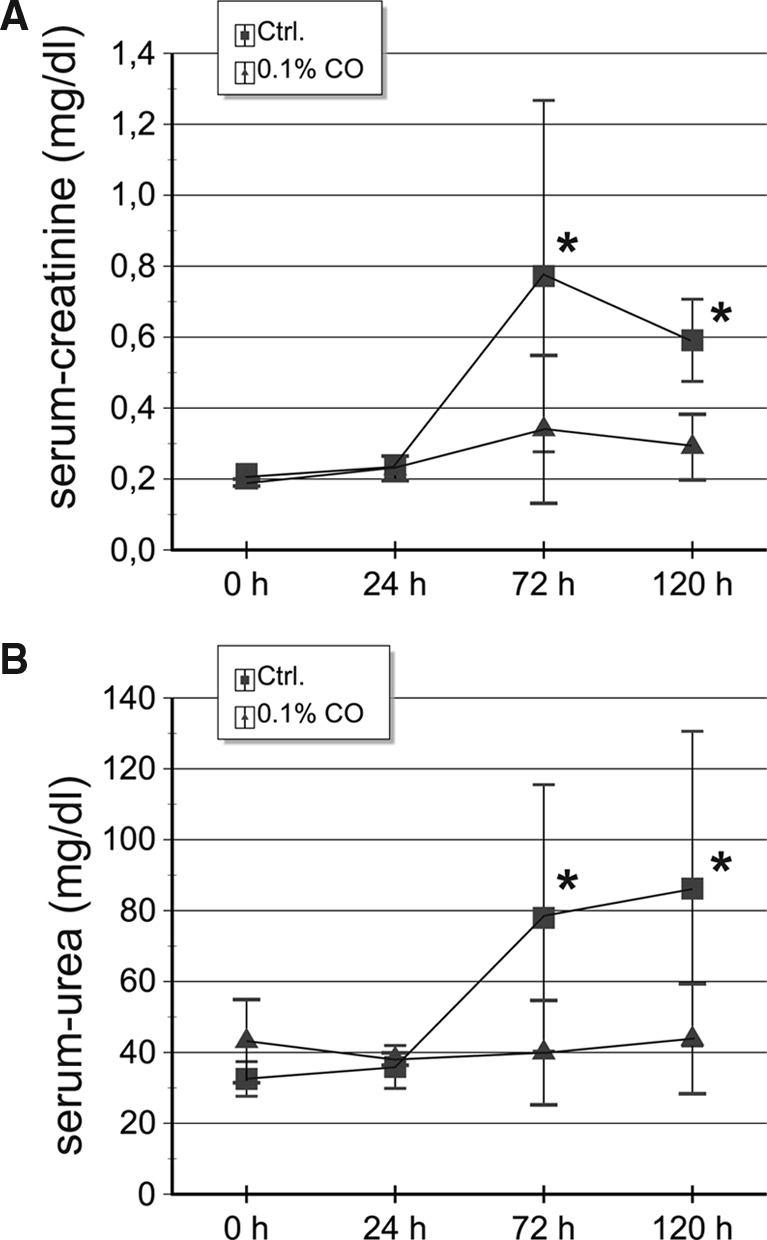
CO preconditioning preserves serum creatinine and urea levels after cisplatin-induced ARF. Exposure to 0.1% CO before cisplatin-induced ARF results in a protection of renal function with significantly lower levels of serum creatinine (A) and serum urea (B) compared with control animals at 72 h and 120 h after cisplatin injection. At 24 h, creatinine and urea parameters were not different between groups. Data are mean ± SD (n = 10). *P < 0.05 versus control.
Discussion
The main finding of this study is that preconditional HIF induction can protect renal tubular cells against cisplatin, which does not by itself induce but rather impairs the HIF system. This conclusion is based on the combined and consistent results of in vitro and in vivo experiments, showing a protective effect of preconditional HIF up-regulation, independent of whether this was induced by hypoxia in vitro or by functional anemia in vivo.
In previous studies, conflicting results were reported with regard to the effect of cisplatin on HIF accumulation in cells and tissues. For instance, in rat hepatoma cells, cisplatin has been described to induce HIF protein levels regardless of prevailing oxygen tensions.21 In contrast, Tanaka et al. did not observe any HIF activation in an immortalized rat proximal tubule cell line.17 In the present study, results of HIF immunoblots, reporter gene assays, and gene expression analysis indicate that cisplatin suppresses the HIF system in renal cells. In line with these findings, another study demonstrated that the HIF target erythropoietin was down-regulated by cisplatin in Hep3B cells, which was attributed to decreased DNA binding activity of HIF.22 Thus, this study provides further insights into the interactions of cisplatin and the HIF system and implicates that suppression by cisplatin can occur at various levels of regulation. It has been reported that cisplatin induces generation of reactive oxygen species,23 and reactive oxygen species have also been implicated in stabilization of HIF-1α.24 However, we did not observe HIF stabilization by cisplatin. Therefore, other effects, such as repression of transcription and translation by cisplatin, may override a potential reactive oxygen species-mediated HIF stabilization, which results in an overall reduction in HIF mRNA and protein levels.
In contrast to Tanaka et al.,17 we were unable to detect any HIF-1α activation or significant pimonidazole binding in the kidneys of cisplatin-treated animals. Differences in the experimental protocols, such as the doses of cisplatin used, the animal strains, pimonidazole application protocols (30 min before death in our versus 3 d in their study), and the techniques used for the detection of HIF activation (HIF immunostaining in our wild-type animals versus HRE reporter activation in transgenic rats) may contribute to these discrepancies.
With respect to the possible mechanisms by which HIF target genes apparently provide protection, various pathophysiological mechanisms of cisplatin have to be considered, which may contribute to its tubular toxicity.25 In addition to a direct DNA damage,26 these include generation of free oxygen radicals,23 caspase activation,4 or activation of TNFα-dependent apoptosis pathways.27 Recently, inhibition of the cyclin-dependent kinase-2 (cdk-2) has been identified to be protective in cisplatin-induced cellular injury.28,29 In MEFs, hypoxia significantly reduced proliferation, decreased cdk-2 activity, and increased p21 expression in an HIF-1α-dependent manner.30 Therefore, it could be argued that the protective effects of preconditional HIF activation in cisplatin-induced renal injury described here may also be mediated by the cessation of cell growth. However, BrdU incorporation assays in HKC-8 cells indicated that hypoxic preconditioning did not alter cell proliferation and cell cycle distribution compared with normoxia. Moreover, it significantly increased the percentage of proliferating cells compared with cisplatin treatment without preconditioning. Whether the higher proliferation rates after hypoxic preconditioning only result from better cell survival and less cell damage or represent a causative factor for the protective role of hypoxia cannot clearly be answered.
Additional reports demonstrated beneficial effects of known HIF-downstream gene products such as vascular endothelial growth factor31 and heme-oxygenase 132 in cisplatin-induced injury. Daily administration of very high doses of the HIF target erythropoietin to cisplatin-treated rats improved creatinine clearance33 or increased tubular regeneration.6 Although it is possible that each of these HIF target genes play an important role in mediating the protective effect of preconditional HIF induction, we hypothesize that activating both isoforms of the transcription factor HIF itself is superior to the administration of single-target gene products. Activation of HIF-1α and HIF-2α contributes to the induction of a broad array of different adaptive genes, although the role of the latter in the hypoxic response is less completely understood.
HIF is considered as a central mechanism of adaptation to hypoxia, but reduced oxygen availability affects cellular function in multiple ways. In the present study, we used mouse embryonic fibroblasts lacking functional HIF-1α as a model to investigate its contribution to the protective effects of hypoxic preconditioning. Because only cells that express HIF-1α protein were protected against cisplatin injury, we conclude that HIF is critical for the observed effects. Wang et al.16 observed an HIF-independent suppression of apoptosis when cisplatin-exposed immortalized rat kidney proximal tubular cells were simultaneously kept under hypoxic conditions (2% oxygen). The protective effect of this experimental design was attributed to suppressed p53 levels resulting from inhibition of the respiratory chain complex III. In keeping with their results, we also observed reduced caspase-3 levels independent of HIF-1α expression when cells were exposed to cisplatin under simultaneous hypoxia (Figure 3, B). Importantly, we observed an HIF-dependent effect of hypoxia when applied preconditionally. This demonstrates that both HIF-dependent and HIF-independent effects of hypoxia can mitigate toxic injury. Preliminary experiments, studying the effect of hypoxic preconditioning in a model of gentamycin toxicity, are suggesting that this protective effect might not be not specific for cisplatin.
One of the limitations of the present study is the lack of specific compounds for preconditional HIF stabilization. New prolyl-hydroxylase inhibitors are presently under investigation, which inhibit HIF degradation under normoxic conditions and thereby induce HIF target genes.34 We have unfortunately not been able so far to prove the protective effect of a prolyl hydroxylase inhibitor in the cisplatin model in vivo because of the restricted availability of such compounds. Because functional anemia induced by CO has previously been demonstrated to stabilize HIF, we used CO for preconditional experiments. In ischemia reperfusion experiments, using the same protocol for CO exposure as in this study, CO preconditioning and a new prolyl hydroxylase inhibitor led to comparable levels of HIF induction and preservation of renal function.15 A recent study suggested that HIF-1α, induced by CO in macrophages, is a critical factor for the protective response of CO.35 On the other hand, CO has also been implicated to be involved in cytoprotection at low concentrations by mechanisms other than HIF activation, such as MAPK36 and PPAR-γ activation.37 In addition, CO releasing molecules have also been successfully tested for cytoprotection in cisplatin-mediated cell injury.38 Therefore, we can not definitely rule out that effects other than HIF stabilization contribute to the in the protective effect of CO observed in our experiments.
Our findings may have significant clinical implications. While hypoxic exposure per se appears unfeasible and cobalt chloride, another inducer of the HIF system used in experimental models,17 is too toxic for application in humans, the prolyl hydroxylase inhibitors offer significant potential for organ protection and could be used to mitigate kidney injury during subsequent cisplatin administration. A potential caveat of such an approach is that up-regulation of the HIF system would not be confined to the kidney and might also interfere with the desired chemotherapeutic effects of cisplatin on tumor cells. A future challenge may therefore be to develop compounds that preferentially induce HIF in the kidney. Taken together, our data provide evidence that HIF is an attractive target for protection against not only hypoxic but also nonhypoxic kidney injuries.
Concise Methods
Cells and Reagents
The human proximal tubular cell line HKC-8 was generously provided by L. Racusen (Baltimore, MD). MEFs were a gift of G. Semenza (Baltimore, MD). Cell culture reagents were from Invitrogen (Karlsruhe, Germany) and Biochrom (Berlin, Germany). All other chemicals were from Sigma (Taufkirchen, Germany). The reporter plasmid 6xHRE/tk/luc was a kind gift from P. Ratcliffe (Oxford, United Kingdom).
Cell Culture
HKC-8 cells were cultured in DMEM/Ham's F-12 supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin, 5 μg/ml insulin, 5 μg/ml transferrin, and 5 ng/ml selenium. MEFs were grown in Dulbecco's modified Eagle's medium with 5% fetal calf serum. Cisplatin was dissolved in dimethylformamide (DMF).
Isolation of Murine Proximal Tubular Cells
Primary murine proximal tubular cells were isolated from the kidney cortex of 4-wk-old mice essentially as described39 and cultured in serum-free DMEM/Ham's F-12 supplemented with 5 μg/ml insulin, 5 μg/ml transferrin, and 5 ng/ml selenium.
HIF Protein Extraction and Immunoblotting
For HIF protein expression, HKC-8 cells were treated with various concentrations of cisplatin (5 to 100 μM) or vehicle (DMF) for 8 h in the absence or presence of the iron chelator 2,2′dipyridyl (100 μM) (ICN, Costa Mesa, CA). Preparation of cell lysates and immunoblotting were performed as described previously40 with antibodies against HIF-1α (Alexis Biochemicals, Loerrach, Germany).
Luciferase Reporter Gene Assays
HKC-8 cells were transfected with the HIF-1α-responsive 6xHRE/tk/luc reporter plasmid, which contains six HRE binding motifs of the mouse phosphoglycerate kinase, which control the expression of firefly luciferase19 and with a pCMV-β-galactosidase expression vector using Fugene (Roche, Mannheim, Germany). After stimulation with cisplatin, vehicle or 100 μM DP for 18 h, luciferase activities were determined using the luciferase assay system (Promega, Mannheim, Germany) and were normalized to the respective β-galactosidase expression.
RNA Preparation and RNase Protection Assay
Total RNA was extracted with RNAzol-B (Biozol, Eching, Germany) and analyzed by RNase protection assay as described previously41; 40 μg of total RNA (1 μg for U6-snRNA) was hybridized to [32P]-labeled antisense RNA probes of HIF-1α, glucose transporter-1, and U6-small nuclear RNA.
Measurement of Caspase-3 Activity
After hypoxic preconditioning for 12 h with 1% O2 in a Jouan IG 750 incubator (Thermo Electron, Dreieich, Germany), cells were incubated with 50 μM cisplatin for 12 h in serum containing medium. They were lysed in a buffer containing 100 mM HEPES, 10% sucrose, 0.1% Chaps, and 1 mM EDTA; 30 μg of protein was incubated with 13 μM DEVD-AMC (Biomol, Hamburg, Germany), the fluorogenic substrate of caspase-3. Cleaved AMC substrate was detected by a fluorometer (Tecan, Crailsheim, Germany) using 360 nm excitation and 465 nm emission wavelength.
BrdU Incorporation and Cell Cycle Analysis
Cell cycle analysis was performed with a modified protocol for flow cytometry.42 DNA content was detected by propidium iodide staining, and DNA synthesis by BrdU incorporation. Cells were either incubated at normoxia or 1% O2 for 12 h before cisplatin (50 μM) was added to the medium for 6 h. A BrdU pulse (50 μM) was performed for 30 min, then cells were harvested and fixed overnight in 65% methanol. After washing with PAB buffer (2% bovine serum albumin, 0.01% sodium azide in phosphate-buffered saline) RNAse A was added (81 U/ml) for 12 min at 37°C and 40 min at 20°C. Then cells were incubated for 4 min in pepsin-HCL solution (0.0093N HCl and 0.33 mg/ml pepsin) at 37°C. The enzyme reaction was stopped with ice cold PAB buffer, and cells were washed and resuspended in 2N HCl for 10 min at room temperature. Cells were incubated with anti-BrdU antibody (DAKO, Glostrop, Denmark), followed by incubation with an FITC-conjugated rabbit anti-mouse IgG antibody (DAKO). After removal of nonbound antibody, propidium iodide (50 μg/ml in phosphate-buffered saline) was added and analyzed by FACS (EPICX XL, Beckman-Coulter, Krefeld, Germany) using EXPO32 ADC software (Beckman-Coulter). Cell cycle analysis was performed with Multicycle software (Phoenix Flow Systems, San Diego, CA). Data presented are the means of 3 independent experiments.
Animals
Animal experiments were approved by the institutional review board for the care of animal subjects and were performed in accordance with National Institutes of Health guidelines. Male Sprague-Dawley rats (Charles River, Sulzfeld, Germany) were used at weights of 200 to 230 g. For the isolation of proximal tubular cells, C57BL/6 mice were purchased from Jackson Laboratories, Sulzfeld, Germany.
Experimental Groups and Protocol of Cisplatin-Induced Acute Renal Injury
To induce acute renal injury in rats, 8 mg/kg bw cisplatin was injected intraperitoneally. Serum creatinine and urea were determined at baseline (0 h) and at 24 h, 72 h, and at the end of the experiments (120 h). Animals (n = 10) were either preconditioned with 0.1% CO for 10 h to activate HIF and HIF target genes as described previously15 or were breathing room air before cisplatin injection. For immunohistochemistry, animals were killed at 1, 24, 72, and 120 h after cisplatin administration.
Immunohistochemistry and Pimonidazole Detection
Immunohistochemistry was essentially performed as described15 with a monoclonal antibody against HIF-1α (Novus Biologicals, Littleton, CO). For signal amplification, a catalyzed signal amplification system (CSA-Kit, Dako, Hamburg, Germany) was used. To detect tissue hypoxia,20 pimonidazole was injected intravenously (60 mg/kg body weight) 30 min before death. Signal detection was performed using an anti-pimonidazole antibody (Natural Pharmacia International, Belmont, MA).13
Histomorphological Scoring of Acute Tubular Injury
Renal cross sections were stained with hematoxylin and eosin and periodic acid-Schiff. Samples were analyzed for tubular cell necrosis, tubular dilation, and intratubular cell detachment (×200 magnification) and were all evaluated in a blinded manner by a nephropathologist. Abnormalities were graded by a semiquantitative score from 0 to 4+: 0, no abnormalities; 1+, changes affecting less than 25% of the sample; 2+, changes affecting 25% to 50%; 3+, changes affecting 50% to 75%; 4+, changes affecting more than 75%.
TUNEL Assay
Apoptotic cells were detected by the TUNEL assay as described previously.43 Cells were regarded as TUNEL-positive if their nuclei stained black and displayed typical apoptotic morphology with chromatin condensation. The number of apoptotic cells was counted in 10 randomly selected visual fields of blinded samples, using ×400 magnification.
Statistics
Unless indicated otherwise, data represent the means of 3 independent experiments ± SD. A P value <0.05 was considered significant. Statistical analyses were performed using t test, covariate analysis, Mann-Whitney-U test, and Kruskal-Wallis test calculated with SPSS Software for Windows (version 13.0).
Disclosures
None.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft, Sonderforschungsbereich 423, Kidney Injury: Pathogenesis and Regenerative Mechanisms, and the Interdisciplinary Center for Clinical Research of the University of Erlangen-Nuremberg.
The authors thank Andrea Kosel, Andrea Luedke, Brigitte Rogge, Hans Fees, and Johannes Schoedel for excellent technical assistance.
Published online ahead of print. Publication date available at www.jasn.org.
REFERENCES
- 1.Arany I, Safirstein RL: Cisplatin nephrotoxicity. Semin Nephrol 23: 460–464, 2003 [DOI] [PubMed] [Google Scholar]
- 2.Lebwohl D, Canetta R: Clinical development of platinum complexes in cancer therapy: an historical perspective and an update. Eur J Cancer 34: 1522–1534, 1998 [DOI] [PubMed] [Google Scholar]
- 3.Cummings BS, Schnellmann RG: Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways. J Pharmacol Exp Ther 302: 8–17, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Kaushal GP, Kaushal V, Hong X, Shah SV: Role and regulation of activation of caspases in cisplatin-induced injury to renal tubular epithelial cells. Kidney Int 60: 1726–1736, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Lau AH: Apoptosis induced by cisplatin nephrotoxic injury. Kidney Int 56: 1295–1298, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Bagnis C, Beaufils H, Jacquiaud C, Adabra Y, Jouanneau C, Le Nahour G, Jaudon MC, Bourbouze R, Jacobs C, Deray G: Erythropoietin enhances recovery after cisplatin-induced acute renal failure in the rat. Nephrol Dial Transplant 16: 932–938, 2001 [DOI] [PubMed] [Google Scholar]
- 7.Winston JA, Safirstein R: Reduced renal blood flow in early cisplatin-induced acute renal failure in the rat. Am J Physiol 249: F490–F496, 1985 [DOI] [PubMed] [Google Scholar]
- 8.Eckardt KU, Bernhardt WM, Weidemann A, Warnecke C, Rosenberger C, Wiesener MS, Willam C: Role of hypoxia in the pathogenesis of renal disease. Kidney Int Suppl 99: 46–51, 2005 [DOI] [PubMed] [Google Scholar]
- 9.Pugh CW, Ratcliffe PJ: Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 9: 677–684, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Schofield CJ, Ratcliffe PJ: Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol 5: 343–354, 2004 [DOI] [PubMed] [Google Scholar]
- 11.Rosenberger C, Griethe W, Gruber G, Wiesener M, Frei U, Bachmann S, Eckardt KU: Cellular responses to hypoxia after renal segmental infarction. Kidney Int 64: 874–886, 2003 [DOI] [PubMed] [Google Scholar]
- 12.Matsumoto M, Makino Y, Tanaka T, Tanaka H, Ishizaka N, Noiri E, Fujita T, Nangaku M: Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J Am Soc Nephrol 14: 1825–1832, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Rosenberger C, Mandriota S, Jurgensen JS, Wiesener MS, Horstrup JH, Frei U, Ratcliffe PJ, Maxwell PH, Bachmann S, Eckardt KU: Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol 13: 1721–1732, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Rosenberger C, Heyman SN, Rosen S, Shina A, Goldfarb M, Griethe W, Frei U, Reinke P, Bachmann S, Eckardt KU: Up-regulation of HIF in experimental acute renal failure: evidence for a protective transcriptional response to hypoxia. Kidney Int 67: 531–542, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Bernhardt WM, Campean V, Kany S, Jurgensen JS, Weidemann A, Warnecke C, Arend M, Klaus S, Gunzler V, Amann K, Willam C, Wiesener MS, Eckardt KU: Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol 17: 1970–1978, 2006 [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Biju MP, Wang MH, Haase VH, Dong Z: Cytoprotective effects of hypoxia against cisplatin-induced tubular cell apoptosis: involvement of mitochondrial inhibition and p53 suppression. J Am Soc Nephrol 17: 1875–1885, 2006 [DOI] [PubMed] [Google Scholar]
- 17.Tanaka T, Kojima I, Ohse T, Inagi R, Miyata T, Ingelfinger JR, Fujita T, Nangaku M: Hypoxia-inducible factor modulates tubular cell survival in cisplatin nephrotoxicity. Am J Physiol Renal Physiol 289: F1123–F1133, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Racusen LC, Monteil C, Sgrignoli A, Lucskay M, Marouillat S, Rhim JG, Morin JP: Cell lines with extended in vitro growth potential from human renal proximal tubule: characterization, response to inducers, and comparison with established cell lines. J Lab Clin Med 129: 318–329, 1997 [DOI] [PubMed] [Google Scholar]
- 19.Warnecke C, Zaborowska Z, Kurreck J, Erdmann VA, Frei U, Wiesener M, Eckardt KU: Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is an HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J 18: 1462–1464, 2004 [DOI] [PubMed] [Google Scholar]
- 20.Kennedy AS, Raleigh JA, Perez GM, Calkins DP, Thrall DE, Novotny DB, Varia MA: Proliferation and hypoxia in human squamous cell carcinoma of the cervix: first report of combined immunohistochemical assays. Int J Radiat Oncol Biol Phys 37: 897–905, 1997 [DOI] [PubMed] [Google Scholar]
- 21.Yang ZF, Poon RT, To J, Ho DW, Fan ST: The potential role of hypoxia inducible factor 1alpha in tumor progression after hypoxia and chemotherapy in hepatocellular carcinoma. Cancer Res 64: 5496–5503, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Horiguchi H, Kayama F, Oguma E, Willmore WG, Hradecky P, Bunn HF: Cadmium and platinum suppression of erythropoietin production in cell culture: clinical implications. Blood 96: 3743–3747, 2000 [PubMed] [Google Scholar]
- 23.Matsushima H, Yonemura K, Ohishi K, Hishida A: The role of oxygen free radicals in cisplatin-induced acute renal failure in rats. J Lab Clin Med 131: 518–526, 1998 [DOI] [PubMed] [Google Scholar]
- 24.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT: Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 275: 25130–25138, 2000 [DOI] [PubMed] [Google Scholar]
- 25.Siddik ZH: Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene 22: 7265–7279, 2003 [DOI] [PubMed] [Google Scholar]
- 26.Leibbrandt ME, Wolfgang GH, Metz AL, Ozobia AA, Haskins JR: Critical subcellular targets of cisplatin and related platinum analogs in rat renal proximal tubule cells. Kidney Int 48: 761–770, 1995 [DOI] [PubMed] [Google Scholar]
- 27.Ramesh G, Reeves WB: TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 110: 835–842, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price PM, Yu F, Kaldis P, Aleem E, Nowak G, Safirstein RL, Megyesi J: Dependence of cisplatin-induced cell death in vitro and in vivo on cyclin-dependent kinase 2. J Am Soc Nephrol 17: 2434–2442, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu F, Megyesi J, Safirstein RL, Price PM: The involvement of the Cdk2–E2f1 pathway in cisplatin cytotoxicity in vitro and in vivo. Am J Physiol Renal Physiol 293: F52–F59, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS: Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol 23: 359–369, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanellis J, Fraser S, Katerelos M, Power DA: Vascular endothelial growth factor is a survival factor for renal tubular epithelial cells. Am J Physiol Renal Physiol 278: F905–F915, 2000 [DOI] [PubMed] [Google Scholar]
- 32.Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A: Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis. Am J Physiol Renal Physiol 278: F726–F736, 2000 [DOI] [PubMed] [Google Scholar]
- 33.Vaziri ND, Zhou XJ, Liao SY: Erythropoietin enhances recovery from cisplatin-induced acute renal failure. Am J Physiol 266: F360–F366, 1994 [DOI] [PubMed] [Google Scholar]
- 34.Macdougall IC, Eckardt KU: Novel strategies for stimulating erythropoiesis and potential new treatments for anaemia. Lancet 368: 947–953, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Chin BY, Jiang G, Wegiel B, Wang HJ, Macdonald T, Zhang XC, Gallo D, Cszimadia E, Bach FH, Lee PJ, Otterbein LE: Hypoxia-inducible factor 1alpha stabilization by carbon monoxide results in cytoprotective preconditioning. Proc Natl Acad Sci U S A 104: 5109–5114, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mishra S, Fujita T, Lama VN, Nam D, Liao H, Okada M, Minamoto K, Yoshikawa Y, Harada H, Pinsky DJ: Carbon monoxide rescues ischemic lungs by interrupting MAPK-driven expression of early growth response 1 gene and its downstream target genes. Proc Natl Acad Sci U S A 103: 5191–5196, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bilban M, Bach FH, Otterbein SL, Ifedigbo E, de Costa d'Avila, J, Esterbauer H, Chin BY, Usheva A, Robson SC, Wagner O, Otterbein LE: Carbon monoxide orchestrates a protective response through PPARgamma. Immunity 24: 601–610, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Tayem Y, Johnson TR, Mann BE, Green CJ, Motterlini R: Protection against cisplatin-induced nephrotoxicity by a carbon monoxide-releasing molecule. Am J Physiol Renal Physiol 290: F789–F94, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Taub M, Sato G: Growth of functional primary cultures of kidney epithelial cells in defined medium. J Cell Physiol 105: 369–378, 1980 [DOI] [PubMed] [Google Scholar]
- 40.Wiesener MS, Turley H, Allen WE, Willam C, Eckardt KU, Talks KL, Wood SM, Gatter KC, Harris AL, Pugh CW, Ratcliffe PJ, Maxwell PH: Induction of endothelial PAS domain protein-1 by hypoxia: characterization and comparison with hypoxia-inducible factor-1alpha. Blood 92: 2260–2268, 1998 [PubMed] [Google Scholar]
- 41.Maxwell PH, Pugh CW, Ratcliffe PJ: Inducible operation of the erythropoietin 3′ enhancer in multiple cell lines: evidence for a widespread oxygen-sensing mechanism. Proc Natl Acad Sci U S A 90: 2423–2427, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diermeier S, Schmidt-Bruecken E, Kubbies M, Kunz-Schughart LA, Brockhoff G: Exposure to continuous bromodeoxyuridine (BrdU) differentially affects cell cycle progression of human breast and bladder cancer cell lines. Cell Prolif 37: 195–206, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hughes J, Johnson RJ, Mooney A, Hugo C, Gordon K, Savill J: Neutrophil fate in experimental glomerular capillary injury in the rat: emigration exceeds in situ clearance by apoptosis. Am J Pathol 150: 223–234, 1997 [PMC free article] [PubMed] [Google Scholar]