

Abstract
Purpose
The purpose of this project was to determine if mutations, including large insertions or deletions, in the recently identified RP31 gene topoisomerase I-binding arginine-serine rich (RS) protein (TOPORS), cause an appreciable fraction of autosomal dominant retinitis pigmentosa (adRP).
Methods
An adRP cohort of 215 families was used to determine the frequency of TOPORS mutations. We looked for mutations in TOPORS by testing 89 probands from the cohort without mutations in other known adRP genes. Mutation detection was performed by fluorescent capillary sequencing and by multiplex ligation probe amplification.
Results
Two different TOPORS mutations, p.Glu808X and p.Arg857GlyfsX9, were each identified in one proband. Patients with these mutations exhibited clinical signs typical of advanced adRP. No large deletions or insertions of TOPORS were identified in our study.
Conclusions
Point mutations and small insertions or deletions in TOPORS cause approximately 1% of adRP. Large deletions or insertions of TOPORS are not an appreciable cause of adRP. Contrary to previous reports, no distinct clinical phenotype was seen in these patients.
Introduction
Retinitis pigmentosa (RP) is a heterogeneous form of inherited blindness initially characterized by night blindness and peripheral vision loss, usually culminating in legal or complete blindness. Approximately one out of 3,000–4,000 individuals is affected with RP [1]. RP can be inherited in an autosomal dominant (adRP), autosomal recessive (arRP), or X-linked (XlRP) pattern and to date, 16 autosomal dominant, 18 autosomal recessive, and six X-linked loci have been identified, with some genes causing multiple overlapping diseases. (RetNet) [2].
Disease-associated genes have been identified for 15 of the 16 adRP loci (RetNet) [3]. The most recent disease-associated gene identified was for the RP31 form of adRP [4]. Mutations in the topoisomerase I-binding arginine-serine rich (RS) gene (TOPORS) cause RP31 [5]. Initial findings identified two different TOPORS mutations associated with disease, both of which were small insertions/deletions resulting in frame-shifts and premature termination of the protein. The mutant TOPORS protein was not detected in lymphoblast cell lines from patients with these mutations.
Examination of additional adRP probands for mutations in TOPORS will provide information on mutation frequency and may potentially provide additional biologic information regarding the disease mechanism. Since haploinsufficiency is believed to be the likely disease mechanism associated with TOPORS mutations, it is possible that large deletions, or copy number variants (CNV) in TOPORS, may also be a cause of adRP. This project examines a well defined adRP cohort using both standard sequencing methodology and multiplex ligation probe amplification (MLPA) to look for CNVs [6,7].
Methods
Autosomal dominant retinitis pigmentosa cohort and controls
The cohort of 215 adRP probands used in this study has been described in detail previously [6-8]. This cohort is a set of 215 families which, based on pedigree analyses, have a high likelihood of having adRP. Each proband has been previously tested for mutations in the complete coding region of CA4, CRX, FSCN2, IMPDH1, NRL, PRPF31, RDS, RHO, ROM1, and RP9. Samples were also screened for mutations in mutational “hot spots” of RP1, PRPF3, PRPF8, and NR2E3. Likely disease-causing mutations have been identified in 126 of the 215 families. Probands from the remaining 89 families were tested in this study. A set of 90 unrelated normal control samples obtained from the Centre dEtude du Polymorphisme Humain were also tested for the presence of the two mutations identified in the cohort samples [9].
This study was performed in accordance with the Declaration of Helsinki, and informed consent was obtained from all participants. This research was approved by the Committee for the Protection of Human Subjects at the University of Texas Health Science Center at Houston and by the respective human subjects’ review boards at each participating institution.
Mutation detection
Sequencing analyses
PCR product sequencing was employed to screen patient DNA for mutations in the entire coding region and flanking intron/exon junction of TOPORS. PCR amplification and sequencing were performed as previously described using the primers in Table 1 [6]. Briefly, exons 1 and 2 were each amplified using 30–50 ng of genomic DNA and AmpliTaq Gold (Applied Biosystems, Foster City, CA) in a 12.5 μl reaction for 35 cycles. Exon 3 was amplified in two pieces using 100 ng of genomic DNA and either AmpliTaq Gold or HotStarTaq DNA polymerase (Qiagen, Valencia, CA) with Q-solution, in a 25 μl reaction for 35–40 cycles.
Table 1. Primers used in polymerase chain reaction amplification and sequencing.
| Exon | Amplification primers (5’-3’) | Annealing temperature | Sequencing primers (5’-3’) |
| 1 | ACGTAAGAAGCGGAAGATCG | 63 °C | Same as amplification |
| GCCTGGGAGGTTACTGTAAGG | |||
| 2 | GTGGGTCTC GCT CTC TGC | 63 °C | Same as amplification |
| CCCATTGTTCCGAATCTCAC | |||
| 3A | TCAAGGTCTTTATTTGCATTTTTG | 52 °C | TCAAGGTCTTTATTTGCATTTTTG |
| GCTTCTTCTGGACCAACTGC | AGAACAACAACTCCACCG | ||
| GCCTTCACAGATTAGTCCC | |||
| GAGAAACGATCTACATCATTGTC | |||
| AGTTGGCCTCCTTACTGCAA | |||
| GACCACTCCTGTACACAGCGAAAAC | |||
| TTCTGGGGTCCTCTCAGCTA | |||
| GGCTTCTTCTGGACCAACTGC | |||
| 3B | TAGCTGAGAGGACCCCAGAA | 58 °C | AGTTGGTCCAGAAGAAGCCA |
| GGAGGAAGAGAGTTTTCACCAA | TACAAAACACGGCATTTGGA | ||
| AAGACCCGGAGCCTAAGTGT | |||
| GATGAAGATTTTTGGTAATGACTG |
The above PCR and sequencing primers were used to identify TOPORS mutations in our cohort of adRP patients. For exons 1 and 2, the same primers were used for amplification and sequencing reactions. For exons 3A and 3B, several nested sequencing primers were used to span each amplified PCR product.
PCR products were treated with ExoSapIt (USB, Cleveland, OH) and sequenced unidirectionally with BigDye v1.1 (Applied Biosystems) and the primers described in Table 1. Sequence reactions were purified using BigDye® Xterminator Kit (Applied Biosystems) and the manufacturer’s protocol. Purified reactions were run on an ABI 3100-Avant Genetic Analyzer (Applied Biosystems) and analyzed using SeqScape Software (Applied Biosystems).
Multiplex ligation probe amplification analyses
MLPA analyses were performed as previously described using eight probe pairs designed to span each of the TOPORS amplification primers (Table 2), seven control probe pairs, and the EK1 kit (MRC-Holland, Amsterdam, The Netherlands) [7]. Briefly, probes were selected based on the recommendations of MRC-Holland and Raw-Probe Software (MRC-Holland). All half probes were synthesized by Sigma Genosys (The Woodlands, TX) and desalted at the time of synthesis.
Table 2. Probes used in multiplex ligation probe amplification.
| Probe set | Probe location* | 5′ half probe (5′-3′) | Phosphorylated 3′ half probe (5′-3′) |
| Amp1 Front | 32542621–32542674 | GGGTTCCCTAAGGGTTGGA TGGCGGGTACACCCAGC AGCCCTTAC | pGTAAGAAGCGGAAGATCGTATCCT CCAGTCTAGATTGGATCTTGCTGG CAC |
| Amp 1 Back | 32542362–32542421 | GGGTTCCCTAAGGGTTGGA GGCAGCAGTCCGCGGGA GCTGGCGGGAG | pCTGCGGGCCTTACAGTAACCTCCC AGGCGGTGTCTAGATTGGATCTTGCTGGCAC |
| Amp 2 Front | 32540968–32541035 | GGGTTCCCTAAGGGTTGGA CTGGGGGGTCTCGCTCTC TGCCCTGCTTCCGAG | pCTGCCATTGGTGATGAGCCCTTTG CGTCACATCTAGATTGGATCTTGC TGGCAC |
| Amp 2 Back | 32540731–32540800 | GGGTTCCCTAAGGGTTGGA CAGCCAGGCCTGCGCCG GCATCCTCCGAG | pGTGAGTGAGATTCGGAACAAT GGG ACGCGGGGGTCGGAAGGTCTAGATTGGATCTTGCTGGCAC |
| Amp 3A Front | 32534380–32534475 | GGGTTCCCTAAGGGTTGGA CAGTGCCCCTTTATAAA ATAAAACAAAAGTAATGG GTCACTTAAGTATTTTCAC | pCAAAATAAGTTTCAAGGTCTTTAT TTGCATTTTTGTTGAGACTCTAGATTGGATCTTGCTGGCAC |
| Amp 3A Back | 32532520–32532601 | GGGTTCCCTAAGGGTTGGA GAGACAAAAAGAGATCA AGAACTAGAGATAGCAG TTGGTC | pCAGAAGAAGCCAAACTCTGTCTCT AAGTAGTGAAAGCACAAGTCTAGATTGGATCTTGCTGGCAC |
| Amp 3B Front | 32533017–32533090 | GGGTTCCCTAAGGGTTGGA GTGTCATTGTTGGGTTTGTT AAACCACTAGCTGAGAG | pGACCCCAGAACTTGTTGAACTGTC CTCTGATTCTGAGTCTAGATTGGATCTTGCTGGCAC |
| Amp 3B Back | 32531257–32531342 | GGGTTCCCTAAGGGTTGGA GGAAAAAGGAAGAATGT CGTCTACTGCAGTCTATT TAAAGAT | pGACATTTGGTGAAAACTCTCTTCC TCCTTACAATATTTTAAATGTCTAGATTGGATCTTGCTGGCAC |
Half probes were designed to anneal to the equivalent genomic sequence as the sequencing amplification primers described in Table 1. Each probe sequence contains a universal half probe sequence which is underlined. The asterisk indicates the position on chromosome 9 based on the University of California Santa Cruz human genome assembly of March 2006 (hg18).
Probe cocktails were hybridized overnight with 25–50 ng of genomic DNA, ligated, and then PCR amplified according to the DNA detection-quantification protocol recommended by MRC-Holland. PCR product was diluted in deionized formamide (Applied Biosystems) containing GeneScan-500 LIZ size standards (Applied Biosystems) and run on a 3100-Avant Genetic Analyzer. Dosage quotients (DQs) were calculated for each TOPORS probe as described by Stern et al. using GeneMapper (Applied Biosystems) and Excel (Microsoft, Redmond, WA) software [10]. A DQ of 1.0 indicated the presence of two alleles while a 0.5 or 1.5 suggested that either a deletion or duplication of the target sequence, respectively.
Results
Sequencing analyses
We tested genomic DNA from 89 adRP probands for mutations in TOPORS using fluorescent capillary sequencing. This procedure detected likely disease-causing mutations in two of the probands tested. Each of these mutations was heterozygous, consistent with autosomal dominant inheritance. A 1 bp deletion at nucleotide 2,569 was detected in the first proband from family UTAD102 (c.2569delA; p.Arg857GlyfsX9). This deletion caused a frame-shift at amino acid residue 857, and was predicted to result in the addition of eight incorrect amino acids followed by premature termination. Unfortunately, no additional family members were available for testing. Analysis of 180 chromosomes from normal controls failed to find this DNA change.
The second TOPORS mutation was a c.G2422T nonsense change, which resulted in a p.Glu808X in family RFS169. Analysis of this mutation in eight additional family members demonstrated that this mutation tracks with disease (Figure 1). This mutation was also not found in the 180 normal chromosomes tested. Both of the mutations identified in this study, like those identified previously by Chakarova and colleagues [5], resulted in premature termination of the TOPORS protein.
Figure 1.

Pedigrees of families with TOPORS mutations. A: This family has the p.Arg857GlyfsX9 (c.2569delA) mutation. B: RFS169. This family has the p.Glu808X (c.2422C>T) mutation. Circles indicate females; squares indicate males. Black filled symbols are affected individuals, open symbols are unaffected individuals, and the “Q” indicated an individual in New York who reports being asymptomatic. “E”s indicate individuals who had eye examination at either the Retina Foundation of the Southwest or the Jules Stein Eye Institute. “M”s indicate individuals for whom ophthalmic medical records were reviewed. Plus signs show individuals whose DNA tested positive for the family's mutation; minus signs are individuals whose DNA tested negative for the family's mutations.
During our analyses we also identified two missense changes not found in database of single nucleotide polymorphisms (dbSNP). The first change, a p.Pro20Ser (c.C58T), was found in four apparently unrelated Caucasian probands. Additional family members from one family were tested for the presence of the variant. The p.Pro20Ser variant was not found in four affected individuals or obligate carriers in this family and hence was not considered disease-causing. The second variant, p.Thr782Ala (c.A2344G), was found in two Caucasian probands. Testing of additional family members showed that this variant also did not track with disease and therefore, is benign.
Multiplex ligation probe amplification
Work recently published by Chakarova et al. [5] suggested that haploinsufficiency is the disease mechanism of TOPORS mutations. This is based on protein analysis of the two previously identified TOPORS mutations that, like the ones found in this study, result in a premature protein termination. The protein analyses of Chakarova et al. [5] failed to detect the mutant protein in lymphoblast cell lines from either mutation.
Given that haploinsufficiency is a likely disease mechanism for TOPORS mutations, it is possible that a gross deletion or CNV of TOPORS would also lead to retinal disease. To determine if CNVs are a common cause of RP, we performed MLPA analyses of TOPORS using a series of eight custom probe sets designed in our laboratory. These probes were designed to overlap with the original amplification primers used in sequence analysis such that any failure to amplify would also likely be detected. MLPA analyses of TOPORS in the 89 individuals from our adRP cohort did not detect CNVs.
Clinical description
UTAD102
The prospectus was a 52-year-old Hispanic male with only hand motion vision in both eyes. He had two brothers and three children diagnosed with RP (Figure 1A). Extensive bone-spicule-like pigment deposits and severely attenuated blood vessels were seen bilaterally, consistent with end stage RP. Optic disc pallor and macular retinal pigment epithelium atrophy were also present. Goldman visual fields were not possible due to the patient’s poor vision.
RFS169
The prospectus was a 31-year-old Caucasian male whose major complaint was difficulty going down steps and curbs in dim light or at night. He was aware that his peripheral vision was poor and reported frequent inability to locate objects. His sister, mother, maternal aunt, maternal grandfather, maternal great-grandfather, and maternal great uncle had been diagnosed with RP (Figure 1B). He reported that his mother had severe night blindness at age 20 and was now 60 years old with extremely poor vision.
Visual acuity was 20/20–2 OD and 20/20–2 OS. Slit lamp findings were normal bilaterally. Ophthalmoscopy revealed clear vitreous ocular uterque (OU) and normal discs. Both maculas were normal without edema. The midperiphery contained numerous bone-spicule-like pigment deposits. The retinal arterioles were slightly narrowed by comparison to the veins. Static perimetry was obtained with a Humphrey Field Analyzer (Humphrey Instruments, San Leandro, CA), using programs 30–2 and 60–2. Sensitivity was minimally disturbed in the fovea and central 15 °. Sensitivity was zero at most locations beyond 7.5 ° eccentricity. The 60–2 field showed a region of preserved function in the lower temporal field.
Visual thresholds following 45 min of dark adaptation were elevated by 1.5 log unit. Full-field electroretinograms (ERGs) showed that the International Society for Clinical Electrophysiology of Vision (ISCEV)-standard rod response was not detectable. The maximum rod photoresponse to a 4.2 log scot td-sec flash was 10 μV, compared to a lower limit of normal of 155 μV. Cone b-wave amplitude to 31 Hz flicker was 6.4 μV, compared to a lower limit of normal of 35 μV. Cone b-wave implicit time was delayed by 9.3 msec. The maximum cone photoresponse obtained in the presence of a 3.2 log td background was 7.7 μV, compared to a lower limit of normal of 33.7 μV.
The sister of the prospectus was 41 years old at the time of examination. She too was aware of night vision and side vision impairment. In addition, she complained of poor central vision. Ophthalmoscopy revealed clear vitreous OU and normal discs. Both maculae were normal without edema. The midperiphery contained numerous bone-spicule-like pigment deposits. The retinal arterioles were slightly narrowed by comparison to the veins (Figure 2).
Figure 2.
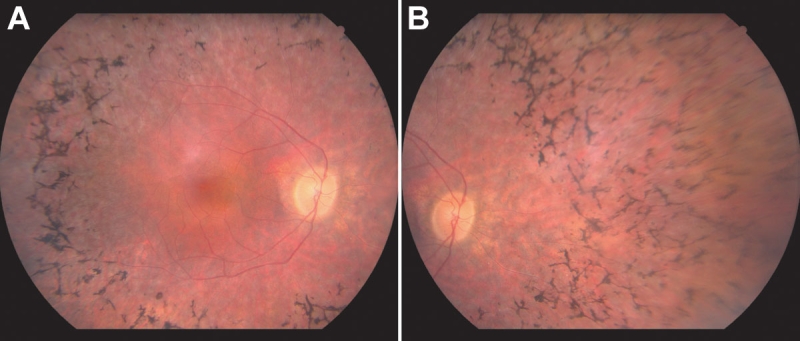
Fundus photographs from RFS169. Right midpheripheral fundus (A) and right peripheral fundus (B) photographs from the 41-year-old sister of the proband from family RFS169. The midperiphery of both eyes contained numerous bone-spicule-like pigment deposits and the retinal arterioles were slightly narrowed by comparison to the veins. There was no evidence of a perivascular cuff of retinal pigment epithelium atrophy around the superior and inferior arcades in this family.
Visual acuity was 20/20–2 OD and 20/20–2 OS. Humphrey perimetric values could only be obtained from the central four locations on the 30–2 field; all locations were decreased in sensitivity by at least 20 dB with the exception of the fovea, where sensitivity was within the normal range. Full-field ERGs showed that the ISCEV-standard rod response was not detectable, as was the maximum rod photoresponse. Cone b-wave amplitude to 31 Hz flicker was 0.5 μV, compared to a lower limit of normal of 35 μV. Cone b-wave implicit time was within the normal range.
Medical record examination also provided some detail regarding the clinical details of the nuclear family found at the right of the pedigree (Figure 1B). The oldest affected male cousin of the prospectus was last examined at age 42. At that time his visual acuity OU was 20/20. He was reported to have severe field constriction but no fields were available. Fundus drawings indicated crescent-like areas of atrophy around the arcades. The younger brother of this individual was last examined when he was 30 years old. His acuity was 20/30 in each eye. Humphrey fields measured less than 10 degrees in each eye, and standard ERGs (no computer averaging) were not detectable. Fundus appearance was typical for RP; specifically there was no note of atrophy around the arcade vessels.
The youngest affected brother in this nuclear family was last examined at age 32. His visual acuity was 20/150 in each eye. He had a long history of keratoconus with corneal grafts in each eye. Due to these corneal problems, there was no fundus or visual field information available.
Medical records were not immediately available from the mother of these three brothers. Results from a self-reported questionnaire, completed at age 66, indicated that she did not have any trouble with her vision other than glasses needed for acuity correction. Specifically, she reported no trouble seeing at night or with her peripheral vision. This suggests that she is an asymptomatic carrier of the disease-causing mutation found in her three affected sons.
Discussion
Based on our analyses, mutations in TOPORS cause approximately 1% of adRP. Further, these mutations are most likely to be nonsense changes or small insertion/deletions that lead to premature termination of the protein [5]. No CNVs were identified in this study, making it unlikely that TOPORS CNVs cause an appreciable fraction of adRP. Since our adRP cohort is composed primarily of probands of Western European origin, it is possible that the TOPORS mutation frequency, like other adRP gene mutation frequencies, could be different in other populations [3]. Additional studies will be needed to address this question.
Chakarova et al. [5] reported finding a unique clinical phenotype in the large family that originally mapped the RP31 locus. In four children they observed a perivascular cuff of retinal pigment epithelium atrophy in the superior and inferior arcades that progressed into pigmentary retinopathy with choroidal sclerosis. The unique perivascular cuff was not seen in any of our examined patients, but its absence could easily be due to the later age at which our patients were examined. Several members of the original RP31 family were also reported to be asymptomatic despite carrying the TOPORS mutation [4]. One member from the RFS169 family discussed in this study is known to carry the mutation and has reported being asymptomatic (Figure 1B).
With the addition of TOPORS to the list of adRP-asssociated genes, mutations can now be identified in 60% of individuals with adRP (Figure 3) [6-8]. Mutations in the remaining 40% of affected individuals remain to be identified. It is clear that there are still additional adRP genes to be identified.
Figure 3.

Frequency of autosomal dominant retinitis pigmentosa mutations found in the autosomal dominant retinitis pigmentosa cohort by gene. Gene abbreviations: rhodopsin (RHO); peripherin 2 (PRPH2); pre-mRNA processing factor 31 homolog (PRPF31); retinitis pigmentosa 1 (RP1); pre-mRNA processing factor 8 homolog (PRPF8); inosine monophosphate dehydrogenase 1 (IMPDH1); retinitis pigmentosa GTPase regulator (RPGR); nuclear receptor subfamily 2, group E, member 3 (NR2E3); pre-mRNA processing factor 3 homolog (PRPF3); topoisomerase I-binding arginine-serine rich gene (TOPORS); cone-rod otx-like photoreceptor homeobox transcription factor (CRX); retinal outer segment membrane protein 1 (ROM1). Testing identified mutations in 60% of our autosomal dominant retinitis pigmentosa cohort of 215 families. Mutations have yet to be identified in the remaining 40%.
Acknowledgments
We thank Jill Oversier for her invaluable help contacting the adRP families and Catherine Spellicy for technical assistance. This work was supported by grants from the Foundation Fighting Blindness, The William Stamps Farish Fund, The Gustavus and Louise Pfeiffer Research Foundation, the Herman Eye Fund, and National Eye Institute grants EY007142 and EY005235.
References
- 1.Haim M. Epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl. 2002;(233):1–34. doi: 10.1046/j.1395-3907.2002.00001.x. [DOI] [PubMed] [Google Scholar]
- 2.Heckenlively J, Daiger S. 2002 Hereditary retinal and choroidal degenerations, pp 3555–3593 in Emery and Rimoin’s Principles and Practices of Medical Genetics, Vol. 3. Chapter 137. edited by. Rimoin DL, Connor JM, Pyeritz RE, and Korf BR. Churchill Livingstone, Los Angeles. [Google Scholar]
- 3.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol. 2007;125:151–8. doi: 10.1001/archopht.125.2.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papaioannou M, Chakarova CF, Prescott DC, Waseem N, Theis T, Lopez I, Gill B, Koenekoop RK, Bhattacharya SS. A new locus (RP31) for autosomal dominant retinitis pigmentosa maps to chromosome 9p. Hum Genet. 2005;118:501–3. doi: 10.1007/s00439-005-0063-3. [DOI] [PubMed] [Google Scholar]
- 5.Chakarova CF, Papaioannou MG, Khanna H, Lopez I, Waseem N, Shah A, Theis T, Friedman J, Maubaret C, Bujakowska K, Veraitch B, Abd El-Aziz MM. Prescott de Q, Parapuram SK, Bickmore WA, Munro PM, Gal A, Hamel CP, Marigo V, Ponting CP, Wissinger B, Zrenner E, Matter K, Swaroop A, Koenekoop RK, Bhattacharya SS. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am J Hum Genet. 2007;81:1098–103. doi: 10.1086/521953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Lewis RA, Garcia CA, Ruiz RS, Blanton SH, Northrup H, Gire AI, Seaman R, Duzkale H, Spellicy CJ, Zhu J, Shankar SP, Daiger SP. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: a screen of known genes in 200 families. Invest Ophthalmol Vis Sci. 2006;47:3052–64. doi: 10.1167/iovs.05-1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sullivan LS, Bowne SJ, Seaman CR, Blanton SH, Lewis RA, Heckenlively JR, Birch DG, Hughbanks-Wheaton D, Daiger SP. Genomic rearrangements of the PRPF31 gene account for 2.5% of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:4579–88. doi: 10.1167/iovs.06-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gire AI, Sullivan LS, Bowne SJ, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP. The Gly56Arg mutation in NR2E3 accounts for 1–2% of autosomal dominant retinitis pigmentosa. Mol Vis. 2007;13:1970–5. [PubMed] [Google Scholar]
- 9.Dausset J, Cann H, Cohen D, Lathrop M, Lalouel JM, White R. Centre d'etude du polymorphisme humain (CEPH): collaborative genetic mapping of the human genome. Genomics. 1990;6:575–7. doi: 10.1016/0888-7543(90)90491-c. [DOI] [PubMed] [Google Scholar]
- 10.Stern RF, Roberts RG, Mann K, Yau SC, Berg J, Ogilvie CM. Multiplex ligation-dependent probe amplification using a completely synthetic probe set. Biotechniques. 2004;37:399–405. doi: 10.2144/04373ST04. [DOI] [PubMed] [Google Scholar]