

Abstract
Toxicoproteomics uses the discovery potential of proteomics in toxicology research by applying global protein measurement technologies to biofluids and tissues after host exposure to injurious agents. Toxicoproteomic studies thus far have focused on protein profiling of major organs and biofluids such as liver and blood in preclinical species exposed to model toxicants. The slow pace of discovery for new biomarkers, toxicity signatures and mechanistic insights is partially due to the limited proteome coverage derived from analysis of native organs, tissues and body fluids by traditional proteomic platforms. Improved toxicoproteomic analysis would result by combining higher data density LC-MS/MS platforms with stable isotope labelled peptides and parallel use of complementary platforms. Study designs that remove abundant proteins from biofluids, enrich subcellular structures and include cell specific isolation from heterogeneous tissues would greatly increase differential expression capabilities. By leveraging resources from immunology, cell biology and nutrition research communities, toxicoproteomics could make particular contributions in three inter-related areas to advance mechanistic insights and biomarker development: the plasma proteome and circulating microparticles, the adductome and idiosyncratic toxicity.
Keywords: toxicoproteomics, toxicology, toxicogenomics, biomarkers, mass spectrometry, proteomics, idiosyncratic toxicity, adductome
TOXICOPROTEOMICS
Toxicoproteomics uses the discovery potential of proteomics in toxicology research by applying global protein measurement technologies to biofluids and tissues after host exposure to injurious agents [1]. Hosts include preclinical species, environmental species and organisms, human subjects and patients with disease. Injurious agents are often novel, small mass chemical entities but can also include recombinant proteins or other biological agents designed to maintain health or treat disease.
Although toxicoproteomics was initiated under the umbrella of toxicogenomics and proteomics, it has emerged as its own discipline. Toxicoproteomics is defined by goals of furthering mechanistic understanding of how specific exposures alter protein expression, protein behaviour and host response to cause injury and disease, and also by an evolving research agenda that is equipped with the tools of proteomics, bioinformatics and other enabling technologies. In addition, biomarker and toxicity signature discovery is very high in the minds of those who use proteomics in toxicology to assess drug exposure, efficacy and toxicity in the pharmaceutical arena or environmental hazard evaluation for public health [2, 3]. Finally, an overarching principle among discovery technologies is that eventual placement of protein changes within biochemical pathways and processes will improve our understanding of larger biochemical systems and signalling networks. Systems biology has come to represent this wider integration of functional genomics disciplines such as transcriptomics, proteomics, interactomics and metabolomics among organisms [4, 5]. Concepts and definitions in toxicoproteomics are summarized in Table 1.
Table 1.
Concepts in toxicoproteomics
| Concepts | |
|---|---|
| Toxicoproteomics | The application of global protein measurement technologies to toxicology research. |
| Aims | Discovery of |
| • Mechanistic insights by identifying key modified proteins in acute chemical and drug-induced injury and as contributors to long-term development of disease. | |
| • Biomarker and toxicity signature development to better describe and measure toxicity. | |
| • Systems biology approach to toxicity by placing altered proteins in affected pathways, biochemical systems and signalling networks. | |
| Biomarker | Singular measure of a protein, enzyme activity, or small molecule associated with health, disease or toxicity in a biological sample. |
| Toxicity Signature | Distinct set of expressed proteins, or genes, that distinguish between health or toxicity in a sample; requires validation and can be generalized to causal or relevant biological processes. |
| Tier I Analysis | Tier I analysis in toxicoproteomics is protein profiling to identify and quantify proteins in a specific spatial location. |
| Tier II Analysis | Tier II analysis globally screens protein structural and behavioural properties such as functions, interactions, 3-dimensional structure or post-translational modifications. |
| Plasma Proteome | Soluble protein fraction of blood that suspends red blood cells, leucocytes and platelets. Serum proteome represents those soluble proteins remaining after clot formation. |
| Microparticles | Intact vesicles (ectosomes) derived from cell membranes of 0.2−2μM in size that are found in blood. |
| Protein Adductome | Set of proteins with residues or chemical groups that are capable of `adduct' formation (covalent binding) by small foreign molecules such as drugs or chemicals. |
| Idiosyncratic Toxicity | Unexpected and relatively rare host responses to therapeutic treatment or chemical exposure of unknown mechanism. |
CHALLENGES OF THE FIELD
Several reviews of toxicoproteomics, or about proteomics applied to toxicological settings, have been written in the past several years. These authors describe toxicoproteomics as a new field involving a relationship with toxicologic pathology and toxicogenomics [1, 6]; explore its importance in developing serum protein pattern diagnostics [7], delineate new biomarkers and toxicity signatures [2], highlight the field's achievements and limitations in biomarker development [8] and review discovery of early markers in drug toxicity [9]. Reported data from toxicoproteomic studies in these literature reviews were categorized by (i) pharmaceutical or chemical toxicant under study; (ii) the model organism or clinical subjects involved; (iii) target organs analysed such as liver, kidney, brain and heart; (iv) biofluids analysed such as serum, plasma, urine or cerebral spinal fluid; (v) proteomic method of analysis that was conducted such as 2D gel, differential gel electrophoresis (DIGE) isotope coded affinity tags (ICAT), antibody array; (vi) the use of in vivo or in vitro model systems; and (vii) the number of differentially expressed and identified proteins. While specific proteins were identified as differentially expressed in each of the cited investigations, these reviews noted that validation and follow-up studies to confirm either individual proteins or sets of proteins, as ‘biomarkers’ were extremely limited. There was a general consensus among reviews of toxicoproteomics studies [1, 2, 6–9] about common aims of the field. First, the discovery potential of proteomics technologies can be exploited to find new biomarkers or toxicity signatures during preclinical safety assessment or hazard evaluation and in diagnosing and treating human disease. Second, toxicoproteomics can be used to achieve a better understanding of molecular mechanisms underlying chemically-induced toxicity in preclinical and experimental settings. Third, toxicoproteomics can integrate with data from other Omics technologies, bioinformatics, imaging and computation tools and toxicogenomics databases for a systems biology approach to predictive mechanistic toxicology. These collective aims represent a practical stratification of the discovery to knowledge process that often begins with biomarker(s) development that leads to an improved understanding of toxicity mechanisms. As multiple consequences of mechanistic research mature and expand into a larger context of systems biology, initial molecular toxic insults are translated into a predictable series of downstream events that form a visible phenotype of toxicity.
A limited number of citations have been categorized as ‘toxicoproteomics’ in citation databases or have been included in the title of professional societies, university departments, organizations and commercial entities. So, it appears the field is still early in its development. However, the value of a scientific discipline is not only measured by citations to the field but also by its ability to organize groups and resources for suitable, substantive, and specific research questions. The intent of this review is to examine relevant proteomic platforms and considerations in toxicoproteomics studies and then to suggest three areas of research that are consistent with goals of the field and involve the plasma proteome, the adductome and idiosyncratic toxicity. These three research areas are inter-related, take advantage of the unique capabilities of proteomic analysis, are important areas for toxicology research and would further our understanding of injurious agents and how they affect biological systems during toxicity and disease.
DISCIPLINES AND PLATFORMS FOR TOXICOPROTEOMICS RESEARCH
The complexities of protein properties and structures have led to different groupings of proteomic analysis that bring focus to global protein analysis studies. These groupings represent various disciplines of proteomics as shown in Figure 1 and provide a means to categorize much of toxicoproteomics research. Proteomics in global protein analysis mode involves separation and identification platforms that are often used in protein mapping studies to ennumerate all proteins that can be identified in a sample derived from a specific location within the host. However, the inherently comparative nature of toxicoproteomics studies makes only the subset of proteins that change upon chemical exposure of greatest interest rather than a more exhaustive analysis to know the totality of proteins. Further, it is requisite to be able to accurately measure the amounts of differentially expressed proteins. Measurement of change can either be relative compared with a reference or control sample (fold change) or as absolute protein concentrations, which is much more challenging to measure. Protein profiling that determines relative changes in protein expression produced by chemicals agents, pharmaceuticals or processes that cause injury, represents the first level of toxicoproteomics and most frequent type of analysis. Therefore, the First Tier in toxicoproteomics is to determine individual protein identities (mass fingerprint or amino acid sequence), to measure relative (or absolute) quantities of proteins and their spatial location within cell(s), tissues and biofluids of interest.
Figure 1.
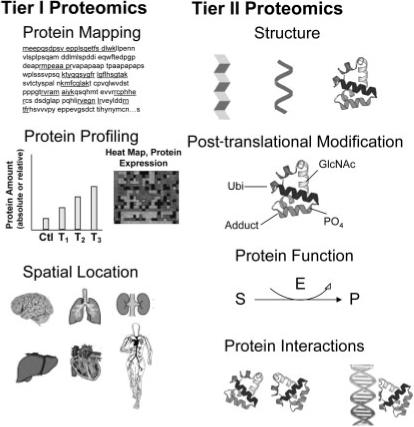
Disciplines of toxicoproteomics to study effects of drug, chemical, disease or environmental stressor exposure. Proteomic analysis attempts to describe various protein attributes in a global manner. Tier I Proteomic Analysis involves protein mapping or profiling. Protein mapping for identification reflects the property of primary amino acid sequence; quantitations of all proteins from a defined space are inherent in Protein Profiling; and isolation or enrichment of proteins from a particular spatial location within cells or tissues help characterize the organism's phenotype. Tier II Proteomic Analysis involves global determination of individual protein attributes (behaviour and structure) regarding their three-dimensional structures, PTMs, functional capabilities and interactions and complexation with other biomolecules. Further explanations follow. In Protein Mapping, the underlined portions of an individual protein represent tryptic peptides for amino acid sequencing for identification by MALDI or MS/MS. In Protein Profiling, changing levels of individual proteins (bar graph) I or groups of proteins (cluster analysis) are measured over treatment (T1, T2, T3) or time. A proteome of interest occupies a specific spatial location for analysis and may comprise a subcellular organelle, tissue or organ. Protein structure may represent the β-pleated sheet or α-helix to form tertiary or quaternary protein folding. Specific post-translational moieties such as ubiquitin (Ubi), phosphorylation (PO4), glycosylation (ClcNAc), chemical adduct or many others are covalently bound to specific amino acid residues on the protein that impart important functional and biophysical properties. Protein Function may be: enzymatic such as enzymatic (E) conversion of substrate (S) to product (P); structural providing form and shape; translocational across cells or tissues; signalling and transduction; or many other utilities to be carried out within cells and tissues. Protein interactions may occur between other proteins, between DNA and proteins, or between other biomolecules.
A second level of proteomic analysis, or Tier II, globally screens for protein functions, protein interactions, three-dimensional structure and specific post-translational modifications (PTMs). The origin of these groupings directly reflect those properties of proteins that relate to their function (i.e. enzymatic, structural and photosensitive), their abilities to interact or bind with other proteins or macromolecules (i.e. DNA, lipids), their complex biophysical structure, and finally the post-translational changes at amino acid specific residues (i.e. phosphorylation, glycosylation and nitrosylation). These First and Second Tiers of proteomic analysis reflect intrinsic attributes of proteins that factor into toxicoproteomic analysis [10] as shown in Figure 1.
Proteomic platforms vary greatly in their respective abilities to deliver data on any one particular property or upon all protein attributes simultaneously during one analysis. The proteomes of most cells, tissues and organs are so vast that, unlike whole genome queries, proteomes cannot be completely analysed by existing proteomic platforms. By default, toxicoproteomic studies most often analyse only a portion of the proteome contained in biological samples. A frequent strategy to broaden protein coverage is to take steps prior to analysis to reduce sample complexity (analyse a portion of the proteome or ‘subproteome’) by procedures such as subcellular fractionation, affinity or adsorptive chromatography or electrophoretic separation. It is also important to recognize that toxicoproteomic analysis may be conducted as an independent activity or alternately as a component of a large, formalized gene expression project for which the study design, type of experimental subjects and the availability or amount of biological specimens may greatly impact sample preparation procedures and proteomic platform selection [6].
Proteomic platforms in Figure 2 involving gel, affinity, adsorptive and liquid chromatography [(LC) including high performance LC and others] combined with mass spectrometry (MS) have been extensively reviewed in the literature [11, 12]. These involve 1D as SDS-PAGE separation (1D-MS) or two dimensional gel electrophoresis (2D-MS), DIGE 2D-MS and variants of single and multidimensional LC offline and online with MS like multidimensional protein identification technology (MuDPIT) and which can be coupled with the use of stable isotopes in LC-MS/MS based platforms including ICAT, isobaric tags for relative and absolute quantitation (iTRAQ) and stable isotope labelling by amino acids in cell culture (SILAC). Use of sensitive instruments like LC-FT-ICR can generate ‘accurate mass tags’ [13] and combinations of microaffinity columns [capture by anti-peptide antibodies (CAPA)] joined with stable isotope standards (SIS) comprise new separation platforms like SISCAPA (stable isotope standards and capture by anti-peptide antibodies) to capture analytes prior to LC-MS/MS [14]. The most common form of retentate chromatography MS is surface enhanced laser desorption ionization (SELDI). Antibody arrays are also becoming increasing popular platforms. Typically, most toxicoproteomics studies involve protein profiling using a single platform, the most popular of which is 2D gels combined with MS identification. Considering the complexities of a proteome, the use of complementary multiple proteomic platforms that can bring more data to bear upon toxicologic problems will likely be more prevalent in the future.
Figure 2.
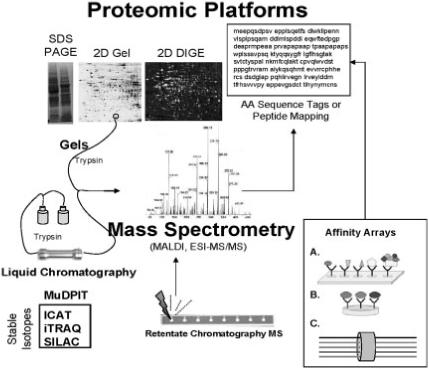
Proteomic platforms for toxicoproteomics studies. Proteomic platforms represent strategies for global separation and identification of proteins. Separations are generally accomplished by gel electrophoresis in toxicoproteomic studies although more recent studies incorporate liquid chromatography-based (LC) platforms such as linear column gradients or multidimensional chromatography (MuDPIT). Use of stable isotopes greatly facilitates protein quantitation (ICAT = isotope coded affinity tags; iTRAQ = isobaric tags for relative and absolute quantitation; SILAC = stable isotope labelling by amino acids in cell culture). Mass spectrometry predominates as a means of protein identification. Identification occurs by peptide mapping or amino acid (AA) sequencing. Retentate chromatography mass spectrometry has been used for rapid profiling of biofluid samples using chemically reactive surfaces for separation and MALDI for generating protein mass spectra (i.e. SELDI technology). Alternatives for MS-based proteomics involve affinity arrays such as (A) antibody arrays, (B) antibody multiplexing and (C) fluorescently tagged antibody bound bead suspensions (i.e. Luminex technology).
IMPROVEMENTS IN TOXICOPROTEOMICS STUDIES
The application of proteomics has generated a heightened sense of optimism in many established scientific disciplines of basic biology, medicine, toxicology and pharmaceutical development. The discovery potential and breadth of coverage of an organism's genome or at least that part of the genome capable of responding to drugs, chemicals and environmental stressors, is a major reason for such optimism. The imprecise meaning of the term ‘biomarker’ and the extensive validation requirements are well recognized [15, 16]. The many subcategories of biomarkers that include biologic, surrogate, prognostic, diagnostic and bridging biomarkers [15, 17], demonstrate the nuanced meanings of the term. Gene or protein expression changes that form an ‘expression pattern’, ‘cluster’, or ‘toxicity signature’ or ‘group of biomarkers’ are terms in frequent but loosely defined use. Despite the imprecision of these terms, discrete sets of transcripts from DNA microarray studies are being discovered in ‘context specific settings’ such as drug-induced hepatic phospholipidosis [18] or steatosis [19, 20]. The expectations are that toxicoproteomics will play a similar role in biomarker and toxicity signature development.
Breakthroughs in biomarker discovery and improvements over traditional measures in preclinical assessment from proteomic analysis have lagged behind gene expression profiling technologies. One fundamental reason is DNA technologies’ ability to exploit the knowledge of whole genome sequence and apply it in mass parallel formats such as cDNA or oligonucleotide microarrays. Generally, MS based platforms in proteomics involve protein or peptide separations that are combined with de novo amino acid sequencing which is a comparatively slower and more labour-intensive process. Most importantly, the accuracies of mass determination of fragmentation ions into amino acid sequences is not absolute so that identifications are usually qualified by statistical confidence [21, 22]. However, improved platforms for proteomics are in dynamic development and applications to toxicology settings are still being explored to match preclinical and clinical samples with appropriately sensitive platforms for differential protein expression. For example, a major step in addressing sample throughput issues and bioinformatics bottlenecks in MS are developments of stable isotope, proteotypic peptides that represent unique gene products [23]. One such study described use of multiple reaction monitoring (MRM) for 137 MRM's from human plasma that spanned 4.5 orders of magnitude [24]. Many of the published toxicoproteomics reports mentioned in toxicoproteomic reviews have not reached the point of validation. To date, many toxicoproteomics studies have served as proof-of-principle experiments that examine a well-characterized toxicant(s) and associate proteomics data output with known toxicological endpoints (i.e. serum and urine chemistries, histopathology). Many of these initial studies in toxicoproteomics do not have demonstrable dose-response relationships or time-courses. Differential protein expressions are often not accompanied by confirmation analysis via ELISA, western blot, immunohistochemistry or functional assay (i.e. enzymatic activity). Two other areas also show slow progress in toxicoproteomics research. One area is the follow up, ‘hypothesis-driven research’ that further exploits discovery findings and establishes causal-linkage of toxicant exposure and effect. The other area is in ‘validation studies’ of proposed biomarkers using independent and blinded study samples. However, the full cycle of discovery, focused confirmation analysis and hypothesis-testing for causality can be achievable as demonstrated by a number of published examples [25–28]. Validation studies to determine the general applicability of each biomarker represent a more long-term commitment to the field.
More robust study designs in toxicoproteomics experiments (and ‘Omics’ experiments in general) could involve inclusion of multiple doses, several time points, positive and negative control compounds, non-toxic chemical isomers, single and multiple dosing, confirmation of results and validation in blinded samples. Admittedly, limitations in resources and the realities of incremental research objectives preclude such ambitious elements in one study and likely account for an uneven application of such principles in many published toxicoproteomics studies. Such shortcomings, in part, reflect the state-of-the-art for toxicoproteomics since long data analysis times for interpreting mass spectra, large data volumes per experiment, statistical analyses and bioinformatics challenges in deriving biological meaning from complex datasets have made many toxicoproteomics studies cumbersome and time-consuming. The need to organize, integrate and communicate data within organizations and across many laboratories [29] underscores the continuing need for international proteomic data standards [30], accessible databases [31] and proteoinformatic tools [32] to most efficiently extract biological meaning from toxicoproteomics experiments [33, 34].
SPECIFIC RESEARCH OBJECTIVES IN TOXICOPROTEOMICS
An important research objective of toxicoproteomics is the development of new biomarkers of toxicity and chemically-induced disease. Although proteomic analysis of affected target tissues and organs are the most proximal injury sites for determining mechanism and accompanying relevant biomarkers, biofluids such as blood remain the most accessible and practical tissue for repeated, non-surgical sampling and analysis. Blood is the common biofluid, which contacts all organs of the body. Among the biofluids produced by each organ, the soluble portion of the blood isolated as either the serum or the plasma proteomes can uniquely reveal signs of specific organ toxicity or pathology from the peptides and proteins passively leaked or actively secreted during dysfunction. Three areas deserve discussion that are relevant for toxicoproteomic analysis of the blood proteome: (i) interference of abundant proteins during proteomic analysis of plasma or serum; (ii) presence of soluble microparticles; and (iii) combining proteomic and transcriptomic analysis of blood.
PLASMA PROTEOME, MICROPARTICLES AND BLOOD-OMICS
Proteomic analysis of native serum or plasma derived from whole blood presents a tremendous challenge in separation and detection of informative proteins. Abundant proteins such as albumin, immunoglobulins, transferrin and others create the bulk of biologically important but minimally informative proteins that change with organ or tissue specific injury while proteins of interest may span up to 10 orders of magnitude lower [35]. The types of proteins that inhabit the plasma proteome include tissue-secreted and carrier proteins, immunoglobulins, coagulatory proteins, homones, cytokines and other short-distance ligands, tissue leakage products, aberrant secreted proteins, foreign or infectious proteins [35], circulating, cell-free nucleic acids [36] and circulating microparticles [37].
The removal of abundant proteins greatly increases the number of detectable and identifiable proteins and should be a major consideration in the design of toxicoproteomic profiling studies. For example, immunoaffinity subtraction of nine abundant soluble proteins (∼85% of total protein) from human blood allowed 2D gel separation of 3700 separable features of which 1800 were identified and 325 were distinct [38]. Only a few hundred proteins were separable by 2D gel analysis without abundant protein removal. A review of four early proteome mapping studies of human plasma using various separation and enrichment methods found a non-redundant list of 1175 unique proteins [39] suggesting that the plasma protein subset of interest for potential biomarkers was in low abundance, comprising <10% of total plasma proteins. Many immunosubtraction matrices and devices to remove abundant plasma proteins are now commercially available. Evaluation of two different immunoaffinity fractionation columns for the top-6 or the top-12 proteins in plasma were investigated by a two-dimensional peptide separation strategy, utilizing chromatographic separation techniques, combined with tandem mass spectrometry (MS/MS). This method was employed for proteomic analysis of column retentate and flowthrough fractions [40]. A total of 2401 unique plasma proteins were identified with the additional finding of 40 unique proteins that were bound to plasma albumin. More recently, a method for immunosubtraction combined with multidimensional liquid chromatographic separation and MS was used to comprehensively map the plasma proteome from five healthy human subjects and could reproducibly assign 2928 plasma proteins [41]. Comparison of these assignments with those found in other maps suggested good agreement for the first several hundred proteins compared with other datasets.
Although mapping studies are impressive in their growing coverage of the human plasma and serum proteomes, toxicoproteomics could add biological meaning to that fraction of plasma or serum proteome that is found altered after exposure to injurious agents or chemical-induced models of disease. The timing during chemical exposure for finding altered plasma proteins either prior to onset of injury or during recovery would be of additional interest. Further, it is of great importance that additional methods be applied to validate the presence of plasma and serum biomarkers identified by MS. A limited but growing number of toxicoproteomic studies have been conducted in the rat serum and plasma proteomes noting differential protein expression in response to an estrogen receptor-β agonist [42], acetaminophen [43], bisphosphonates [44] and coenzyme-Q [45].
Another interesting phenomenon for biomarker development that might be exploited by toxicoproteomics could involve a focus upon blood microparticles during injury and recovery after chemical exposure. Microparticles are intact vesicles derived from cell membranes of 0.2−2 uM in size that are found in blood [37]. Membrane activation processes and apoptosis can create microparticles from cell types as diverse as platelets, endothelial cells, monocytes and granulocytes. Many normal cell types, including tumour cells [46], release vesicles by ectocytosis either spontaneously or in response to various stimuli [47, 48]. Review of various methods for microparticle isolation shows that flow-cytometry has been widely adopted while ultracentrifugation in <2 h provides sufficient amounts for proteomic analysis [37]. Detection of these emitted membrane vesicles have wide application in immune and cancer diseases and have physiologic roles in coagulation, vascular function, angiogenesis, wound healing, inflammation and development [49, 50]. The proteome of human platelet microparticles have recently been mapped [51] and the likelihood seems high that microparticle changes in response to chemical exposure and injury represent a prime opportunity for facile enrichment and discovery of new biomarkers during toxicoproteomic studies.
Formulation of a strategy to integrate changes in protein, toxicology and pathology data is a major objective for toxicoproteomics research [52]. One such strategy is to combine parallel proteomic analyses on affected target tissue and blood, such as serum or plasma, in preclinical species that might be guided by preceding data generated from transcriptomic studies upon the same tissues and whole blood [52]. Transcriptomic analysis of whole blood shows promise in reflecting toxicity and pathology of affected tissues affected by chemical exposure to activators of innate immunity such as lipopolysaccharide [53, 54] or environmental agents such as benzene [55]. Some studies estimate that expression of a large proportion of ∼80% of the genes encoded in the human genome are expressed in the peripheral blood transcriptome [56]. The advantage of an integrated toxicoproteomic and toxicogenomic approach to peripheral blood, called Blood-Omics, would be to bring more information to bear upon drug-related problems or disease states in order to identify the affected, biochemical and regulatory pathways that might lead to biomarker discovery in surrogate species and humans. Indeed, there is considerable interest in developing in silico methods to reconstruct regulatory networks, signalling cascades and metabolic pathways based on combining proteomics and transcriptomics data [57].
THE ADDUCTOME
Protein profiling studies measure the changing levels of expression over the course of time and exposure and represent the First Tier in toxicoproteomic analysis. At this stage of technological development, even the most exhaustive protein profiling analysis cannot yet produce the same level of gene expression coverage as transcriptomics because of technical limitations and the greater complexity of most proteomes. A Second Tier of analysis that could be used in toxicoproteomics is based upon those special properties of proteins that transcriptomics cannot directly measure and only imperfectly predict which are PTMs, three dimensional structure, protein interactions and biologic and kinetic functions. It is this second tier of analysis that toxicoproteomics could seek to exploit. For example, proteomic analysis of protein phosphorylation, oxidation and nitrosylation are relatively well-developed research areas that are of universal importance in signal transduction events [58]. Though many PTMs of proteins result from specific enzymatic activities or react with endogenous ligands or metabolic byproducts, the adducts (covalent bonds) formed with small foreign molecules such as drugs or chemicals represent a special type of PTM that often has dire consequences for protein function such as enzymatic inactivation, differences in protein structure as well as altered interactions of adducted proteins with other proteins or important biomolecules such as nucleic acids. Biotransformations of foreign or endogenous substances sometimes produce reactive intermediates or electrophiles that may form protein adducts. A comprehensive determination of bioactivation pathways of organic functional groups on xenobiotics and pharmaceutical reagents has been extensively catalogued in an effort to guide drug design [59]. Those proteins and specific sites covalently bound by reactive chemicals or their intermediates constitute a special subset of proteins of interest to toxicoproteomics called the protein ‘adductome’.
Bioactivation of xenobiotics into reactive intermediates is a matter of concern for pharmaceutical development [60] and in environmental safety [61, 62]. Current strategies to monitor bioactivation during drug development often involve the use of radiolabelled reagents to track protein adduct formation in liver microsomal preparations with and without the cytochrome P450 cofactor, NADPH; the addition of nucleophiles like glutathione (GSH) or cyanide to trap metabolic intermediates generated during in vitro metabolism; and determining protein adduct formation in liver and other tissues after in vivo exposure in rodents [60]. As a matter of practical concern, measurement of total protein adducts is an invaluable tool in pharmaceutical development to direct chemical synthesis for active and safe pharmaceuticals. However, associating the exact nature of protein adducts with toxicity as a definable goal of toxicoproteomics would not be possible without the innovations in MS over the last 10 years. Identification of adducted proteins and exact structural information about adducted chemical groups and amino acid residues has only been achievable with the introduction of high-resolution MS instruments. Groundbreaking work on the protein adductome has been performed with model biotin-tagged model electrophiles 1-biotinamido-4-(4′-[maleimidoethylcyclohexane]-carboxamido)butane (BMCC) and N-iodoacetyl-N-biotinylhexylenediamine (IAB) in human liver microsomes [63]. These authors identified 376 microsomal cysteine thiol targets in 263 proteins by LC-MS/MS analysis. Protein adducts at 90 specific cysteine sites in 70 proteins (∼25% total adducts) were targeted by both electrophiles but an equally interesting finding was selective adduction of microsomal proteins by BMCC and IAB. These results hold the promise that differing target selectivities can eventually be correlated with differences in toxicity. The authors predict that analysis of selected microsomal protein adduction reactions as well as other subcellular sites such as cytoplasm and nuclei [64] will provide a more specific indication of potential toxicity than bulk covalent binding of radiolabelled compounds [63].
Bromobenzene is a model, non-genotoxic hepatotoxin that has been studied for decades but the identities of adducted proteins are steadily being compiled to determine how particular adducted proteins might produce parenchymal damage compared with other chemically-bound proteins thought to be either protective or irrelevant to toxicity. In one study 14C-bromobenzene was administered to metabolically-induced rats and liver cytosolic proteins were separated by 2D gel electrophoresis to show 110 proteins that were radiolabelled of the 836 separated polypeptides from which 50% were identified by MALDI-MS [65]. Some of the highly adducted proteins included albumin, thioredoxin, selenium binding protein-2, fatty acid-binding protein, GSH S-transferase, regucalcin, aldehyde dehydrogenase, triosephosphate isomerase and others. A Target Protein Database has been constructed which contains 121 identified protein adducts targeted by 16 different drugs and chemicals [66].
Acetaminophen is a frequently used and safe analgesic but higher doses in susceptible individuals can result in liver toxicity, which has been highly correlated to binding of the N-acetyl-p-benzoquinoneimine (NAPQI) reactive intermediate to critical biomolecules. However, the precise connection between acetaminophen toxicity and protein adduction has not been conclusively resolved. Immunochemical detection of the highest concentration of 3-(cystein-S-yl)-acetaminophen protein adducts has been shown in plasma membranes and mitochondria [67]. Adducts to albumin [68] and glyceraldehyde-3-phosphate dehydrogenase were demonstrated where the latter has been shown to be enzymatically inactivated [69]. Targeted LC-MS-MS analysis of microsomal glutathione-S-transferase cysteine-50, a frequent adduct site of model electrophiles, has been used to detect a time-dependent adduction by the reactive acetaminophen metabolite, NAPQI, during microsomal incubations [63]. In addition, the lability of some acetaminophen adducts may explain their unusual accessibility to sensitive portions of the cell such as mitochondria. A labile ipso adduct between GSH and the quinone imine intermediate, NAPQI, has been described as capable of reverting back to NAPQI allowing it to migrate from its site of formation in the endoplasmic reticulum to other cellular compartments where it can either oxidize protein thiols or covalently modify them [70]. Furthermore, circulating albumin adducts of acetaminophen found by LC-MS/MS have been suggested as a potential biomonitoring tool for its detection in vivo and for signalling formation of reactive intermediates of drugs and drug candidates in the preclinical and clinical development phase [68]. In similar adduct studies, 18 14C-protein adducts of pulmonary toxicant, naphthalene, have been identified from 2D gel separated microsomes including protein disulfide isomerase (PDI), ER-60 protease, α-actin, mouse urinary proteins and cytochrome b5 reductase [71] and over 20 proteins from primate respiratory tissue after in vivo exposure [72].
Hepatic bioactivation of monocrotaline produces a phenotype similar to pulmonary hyptertension after injury to endothelial cells lining arterioles in the lung. Several 14C-protein adducts from exposed human pulmonary artery endothelia were separated by 2D gels and identified by MALDI, suggesting that adducts and toxicity are likely initiated at the plasma membrane and proceed into the cytosol and membranes of the endoplasmic reticulum and mitochondria [73]. In brain, cumulative neurotoxicity by acrylamide exposure linked to nerve terminal damage in the central nervous system and peripheral nervous system has been linked to formation of sulfhydryl adducts on cysteine residues of many proteins. In a sophisticated proteomic analysis of synaptosomes labelled with ICAT stable isotopic reagents, investigators identified acrylamide adducts on v-ATPase, dompamine transporter, N-ethylmaleimide sensitive factor and others that were progressively adducted with increasing exposure and therefore, are likely to be neurologically relevant targets [74].
In addition to protein adduct identification, innate, small molecule electrophiles such as aldehydes generated from phospholipids during oxidative stress can reveal dysfunctional biochemical processes brought about by disease or chemical exposures [75]. Recently published work proposes that myocardial dysfunction produced by acrolein and related aldehydes may be symptomatic of toxicological states associated with ambient environment, occupational exposures or drug toxicity [76]. Proteomic approaches were used to study cardiovascular risk in a mouse model of atherosclerosis that suggests a protective role for plasma apolipoprotein-AI (apo-AI) in minimizing the body burden of nitrative oxidants [77]. Using affinity enrichment and site-specific adduct mapping, researchers identified unique specific protein targets for nitration in the plasma of apo-AI null mice that were not present in plasma of wild type mice. Nitrated proteins in aortic tissue quantified by LC with online electrospray ionization tandem MS (LC/ESI/MS/MS) were 6-fold higher in apo-AI null mice compared with wild type mice. Proteomic results were corroborated by immunohistochemical and high-resolution immunoelectron microscopic evaluation of the lesions, which revealed abundant staining for nitrated proteins in the aortic root lesions of apo-AI null mice compared with wild type.
Therefore, an increasing number of proteins and their specific functional groups are being identified as targets for covalent binding by various chemicals, therapeutics, environmental agents and innate metabolic processes (i.e. nitration, aldehydes). Building an understanding of the protein adductome in toxicoproteomic studies could lead to a new set of biomarkers and biomonitoring targets. Undoubtedly some proteins serve as a protective function as abundant facile targets for adduct formation that might spare more critical, slow turnover proteins. It is the association of those critical protein targets necessary for cell function or inappropriate activation for death that will then be the next step for improved understanding of cytotoxicity and mechanisms of organ-specific injury.
IDIOSYNCRATIC TOXICITY
The potential contribution of toxicoproteomics to biomarker development in the plasma/serum proteomes and the adductome would be incomplete without discussion of idiosyncratic drug and chemical reactions. Idiosyncratic reactions are unexpected and relatively rare host responses to therapeutic treatment or chemical exposure of unknown mechanism. They account for only about 6−10% of all adverse reactions to drugs but they are a great cause for concern in therapy and drug development because of their high morbidity and mortality [78, 79]. Phenotypes for each response vary with the organ and tissue. For example, the drug hypersensitivity syndrome associated with aromatic anti-epileptic drugs like phenytoin, carbamazepine, phenobartbital and lamotrigine, is characterized by fever, skin rash which may be as severe as epidermal necrolysis and varied internal organ involvement [80]. Drug-induced liver injury at 5−90 days post-drug exposure broadly involves hepatocellular injury with serum ALT release, cholestatic damage evidenced by rising serum alkaline phosphatase or a mixed reaction, but may also present as part of the drug hypersensitivity syndrome (also known as ‘immune-mediated’ liver injury) or may occur as mitochondrial injury accompanied by increased serum ALT, lactic acidosis and microvesicular steatosis [81]. Generally, idiosyncratic reactions are unpredictable, may or may not be dose-related and typically occur after a latent period of exposure. The complex multifactorial nature and underlying complexities of immune activation probably account for the paucity of relevant preclinical animal models for studying drug-induced idiosyncratic responses.
Based on current animal models and clinical investigations, several hypotheses have emerged to explain severe drug reactions involving immunosensitization after therapeutic treatment: the Hapten Hypothesis which involves protein adducts that act as haptens which are recognized as foreign entities by the host to elicit an immune response; the Danger Hypothesis, that similarly proposes recognition of protein adducts as haptens but also requires the need for accompanying injury to activate the immune system; and the Pharmacological Interaction Hypothesis in which the drug or chemical interacts with HLA receptors directly without the need for metabolic activation and adduct formation [79]. One review briefly describes mechanisms of immunosensitization of the adaptive arm of immunity by a two-stage activation process; first by creation of a hapten-carrier (chemical adducted protein); and second by creation of an adjuvant or ‘danger’ signal [82]. Concurrent release of DNA or histones from damaged cells can act as an adjuvant signal for protein-adducts formed during chemical-induced damage and inflammation. Immune-mediated hypersensitivity is broadly divided into two classes involving recognition of antigenic determinants by B cell receptors or through T cell receptors whereby T cells recognize antigens that have been phagocytized, modified and presented by antigen presenting cells (APCs) [83]. The B and T cell segments of adaptive immunity cooperate since B cells can act as APCs or recognize processed antigens, and T cells can act as helper cells towards B cells.
Phenotypic outcomes of drug-induced immunosensitization often vary greatly because activation of adaptive immune cells results in selection of a unique mix of effector mechanisms of the innate immune system such as granulocytes like neutrophils, mast cells, eosinophils, or natural killer cells; complement factors; certain cytokines like INFs and TNF; and specialized cytotoxic T cells [82]. Thus, biotransformation of pharmaceutics or environmental contaminants responsible for the formation of protein adducts and a subsequent recognition and complex response of the immune system are intimately tied to many types of idiosyncratic reactions.
The liver is a frequent target of idiosyncratic drug injury in part because it is a primary organ for biotransformation of xenobiotics and intermediary metabolism [81]. Biotransformation often leads to reactive intermediates or other active metabolites that may eventually form adducts within liver tissue or metabolites that form adducts at distal sites within the host. Such protein adducts, as was discussed earlier, may be formed during the normal metabolism and clearance of drugs and yet be completely innocuous, neither interfering with liver function nor surpassing this organ's protective and repair responses. Alternately, adducts may act as haptens provoking an immune response in susceptible individuals. The high metabolic capacity makes liver a candidate organ for study of idiosyncratic reactions.
Like any complex tissue, multiple cells types and architecture are important for hepatic function. While the liver primarily consists of parenchymal cells, several non-parenchymal cells are also important for liver function and may participate in some toxic and pathologic conditions to significantly alter hepatic response and the proteomic protein profile. Such non-parenchymal cells include stellate cells, pit cells, Kupffer cells (fixed tissue macrophage of the liver), biliary cells and endothelial cells. Isolation of these cells in their resting and activated states may reveal protein profiles that contribute to liver organ toxicity that would be very difficult to observe in a mixed populated dominated by hepatocytes during toxicoproteomic analysis of whole organ homogenates. The differing cell types comprising liver and their respective cell biologies may also be specific targets for toxicity and should be carefully considered in toxicoproteomics analysis.
Recent studies [84, 85] underscore the fascinating prospect that genetic polymorphisms are a potentially critical factor that contribute to liver idiosyncratic responses in addition to biotransformation, adduct formation, antigen presentation and immune cell activation. The case of troglitazone serves as an example. Troglitazone is a thazolidinedione insulin sensitizer and antidiabetic therapeutic that was withdrawn after discovery of a high incidence of idiosyncratic drug-induced liver injury resulting in abrupt liver failure after several months of therapy. One study tested the hypothesis that a genetic defect in mitochondrial antioxidant defenses might render experimental animals more susceptible to chronic exposure of troglitazone by comparing SOD2 heterozygote mice (+/− superoxide dismutase2) to wild type mice [85]. Investigators found that prolonged treatment of SOD2+/− heterozygote mice with troglitazone induced a mitochondrial oxidant stress that correlated with liver injury in mice. Similar strides are being made in the clinic to link genetic defects with therapeutic toxicity. For example, a patient with multiple drug sensitivities was genotyped at multiple gene foci to reveal homozygosity for N-acetyltransferase 2, NAT2★tB, for slow acetylator status, and homozygous null for both GSH S-transferase isoforms, GSTM1 and GSTT1 [84]. Mitochrondrial DNA sequencing of the same patient showed a mutation in the ND4 subunit of respiratory complex I (C1) which has been shown to increase the production of reactive oxygen species and chronic oxidative stress. The authors proposed the superimposition of oxidative stress by a mitochondrial mutation in C1 complex combined with a deficiency for clearing reactive metabolites from the double null GSH S-transferase mutations as a genetic susceptibility that favours accumulation of drug-protein adducts and may therefore underlie some idiosyncratic drug reactions. Gene polymorphisms that might contribute to higher levels of adduct formation that produce toxicity directly or exceed a threshold for antigen formation and immune activation point to exciting new areas of research for toxicoproteomic study.
CHANGING FACE OF TOXICOLOGY
The discipline of toxicology is changing. Global gene and protein expression technologies and large-scale metabolite assessment are part of the new developments in toxicology. Conventional toxicity testing of pharmaceuticals, industrial chemicals and hazardous compounds is generally evaluated by a battery of standardized tests conducted in a range of surrogate test organisms [86]. Gene expression analyses using DNA arrays are currently entering into a phase of standardization that is amenable to toxicology testing [87] and large studies have been conducted to determine cross-lab consistency of results [88]. However, toxicoproteomic analysis is not yet sufficiently comprehensive or massively parallel in proteome coverage to function in a standardized platform.
In preclinical toxicity testing, the exposure phenotypes of altered organ or body function, affected biochemistry, development of disease or malignancy supported by histopathology are the basis for the No Observable Effect Level (NOEL) or Lowest Observable Effects Level (LOEL) for regulation and assessment of human risk. The introduction of genomic, proteomic and metabolomic technologies (‘panomics’) are generally viewed as a significant next step for better defining the molecular basis of adverse effects and injury [31]. However, the direct and successful applications of global expression technologies toward better mechanistic understanding of chemical-induced injuries have proven to be challenging. Improved approaches now incorporate dose-response transcriptomic data that strive for a consistent match of particular cellular processes with estimated chemical exposure at target tissue sites [89]. The expectation is that new biomarkers or toxicity signatures, such as those recently reported for chemically-induced renal failure [90] and microbial [91] models of infection, will emanate from global query technologies to better define injury and disease.
These new proteomic and genomic approaches to toxicity assessment underlie a continued drive for better molecular understanding of chemical and pharmaceutical-induced injury. On a more expansive level, toxicoproteomics and toxicogenomics represent efforts for an improved understanding of long-term environmental exposures (chemical, microbial, nutritional and biophysical factors) as contributors to disease. Despite the progress made in defining genomes and genes of humans and preclinical species, there are no generalized answers that point to the overriding importance of either genetic determinants or environmental factors to explain the aetiology of complex diseases. As a result, the concept of ‘gene-environment’ interactions continues to be an effective paradigm for research [92]. However, there is a growing recognition that genetic diversity from single nucleotide polymorphisms (SNP's) and copy-number variation are primary contributors to both disease susceptibility and drug response variability [93]. For example, pharmacogenetic testing for drug-metabolizing enzymes (i.e. cytochrome P450's) may prevent adverse drug reactions [94] and one such commercial pharmacogenomic screen is currently in use [95]. More recently, genome-wide association studies have gained increasing popularity since they enable scientists to robustly associate specific genetic variants with a predisposition for complex disease, such as age-related macular degeneration, Type 2 diabetes, inflammatory bowel disease, obesity, autism and leukaemia [96].
A sense of perspective about the underlying gene-environment nature of adverse health effects is also evolving. The measurement of chemicals in the environment and the conduct of experimental chemical exposures to determine health effects can be considered in broader concept of ‘exposure biology’. An expanding view in environmental health research is taking place about the role of chemical toxicity studies in achieving better understanding of the aetiology of chronic human diseases. Much of environmental health research has been driven by intensive testing of nominated industrial chemicals and environmental contaminants in toxicity and cancer bioassays [97]. The adoption of genomic and proteomic technologies in chemical toxicity and bioassay studies attempts to better assess the molecular consequences of human exposure to chemicals, dietary and lifestyle factors, infectious agents and other stressors [98] and has been conceptualized as ‘exposure biology’ [99]. Regulatory testing and chemically-driven research continues as mainstay in environmental health but the impact of proteomic and genomic technologies will be more fully realized when new biologic sensors are eventually developed. Such sensors, which include ‘expression signatures’ would distinguish gene and protein changes between those positively linked to injury and disease from those expression changes characterized as secondary, incidental or non-specific. Discovery of such new sensors utilizing large case-control and population-based genetic studies are envisioned to be important new drivers for conducting environmental biology and human disease research [99].
The influences of nutrition and dietary intake are well recognized for their major environmental interactions with genetics in disease [100], particularly for lipids in cardiovascular disease [101] and as effectors of protein expression [102], which have gained renewed attention. Nutritional proteomics or ‘nutriproteomics’ applies proteomics methodology to nutrition-related research with a focus on interaction of bioactive food ingredients with proteins [103]. Nutrient effects on protein expression can be monitored by protein mapping and also modify post-translational modifications and small-molecule–protein interactions. Such interactions have structural and functional consequences on affected proteins, their receptors, ligand binding and protein–protein interactions [103]. The commingling of chemical and nutritional exposures upon the affected organism in affecting protein expression suggests a joint relationship of toxicoproteomics and nutriproteomics, probably accounting for a combined symposium session at a recent international proteomics conference [104]. Thus, the arrival of genomic expression technologies are rapidly changing the conceptualization of toxicity, toxicology and chemically-induced injury. Emerging disciplines like toxicoproteomics will shape the next wave of change in preclinical assessment, pharmaceutical development and environmental health research.
Key Points
Toxicoproteomics can serve to organize resources toward specific research questions suited for the unique capabilities of proteomic technologies.
Study designs in biomarker research of the plasma proteome are greatly assisted by removal of abundant proteins for toxicoproteomic profiling.
Concentration of microparticles from blood represent an untapped source of biomarkers for toxicoproteomics.
Chemical adducts represent a special type of post-translational modification of proteins that can be uniquely exploited in toxicoproteomic research.
Genetic polymorphisms that lead to functionally variant protein responses to chemicals and therapeutics may contribute to idiosyncratic reactions and disease.
Toxicology is evolving from a pathology-based to molecular-oriented discipline in which toxicoproteomics can play a significant role in the future.
Acknowledgements
This review was supported in part by the Intramural Research program of the National Institutes of Health, National Institute of Environmental Health Sciences.
References
- 1.Wetmore BA, Merrick BA. Toxicoproteomics: proteomics applied to toxicology and pathology. Toxicol Pathol. 2004;32:619–42. doi: 10.1080/01926230490518244. [DOI] [PubMed] [Google Scholar]
- 2.Merrick BA, Bruno ME. Genomic and proteomic profiling for biomarkers and signature profiles of toxicity. Curr Opin Mol Ther. 2004;6:600–7. [PubMed] [Google Scholar]
- 3.Silbergeld EK, Davis DL. Role of biomarkers in identifying and understanding environmentally induced disease. Clin Chem. 1994;40:1363–7. [PubMed] [Google Scholar]
- 4.Hood L, Heath JR, Phelps ME, et al. Systems biology and new technologies enable predictive and preventative medicine. Science. 2004;306:640–3. doi: 10.1126/science.1104635. [DOI] [PubMed] [Google Scholar]
- 5.Lin J, Qian J. Systems biology approach to integrative comparative genomics. Expert Rev Proteomics. 2007;4:107–19. doi: 10.1586/14789450.4.1.107. [DOI] [PubMed] [Google Scholar]
- 6.Merrick BA, Madenspacher JH. Complementary gene and protein expression studies and integrative approaches in toxicogenomics. Toxicol Appl Pharmacol. 2005;207:189–94. doi: 10.1016/j.taap.2005.01.040. [DOI] [PubMed] [Google Scholar]
- 7.Petricoin EF, Rajapaske V, Herman EH, et al. Toxicoproteomics: serum proteomic pattern diagnostics for early detection of drug induced cardiac toxicities and cardioprotection. Toxicol Pathol. 2004;32(Suppl 1):122–30. doi: 10.1080/01926230490426516. [DOI] [PubMed] [Google Scholar]
- 8.Sinha A, Singh C, Parmar D, et al. Proteomics in clinical interventions: achievements and limitations in biomarker development. Life Sci. 2007;80:1345–54. doi: 10.1016/j.lfs.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 9.Collins BC, Clarke A, Kitteringham NR, et al. Use of proteomics for the discovery of early markers of drug toxicity. Expert Opin Drug Metab Toxicol. 2007;3:689–704. doi: 10.1517/17425255.3.5.689. [DOI] [PubMed] [Google Scholar]
- 10.Merrick BA. Introduction to high-throughput protein expression. In: Hamadeh HK, Afshari CA, editors. Toxicogenomics: Principles and Applications. 1st edn Wiley and Sons, Inc; Hoboken, New Jersey: 2004. pp. 263–81. [Google Scholar]
- 11.Righetti PG, Castagna A, Antonucci F, et al. Critical survey of quantitative proteomics in two-dimensional electrophoretic approaches. J Chromatogr A. 2004;1051:3–17. doi: 10.1016/j.chroma.2004.05.106. [DOI] [PubMed] [Google Scholar]
- 12.Yates JR. Mass spectral analysis in proteomics. Annu Rev Biophys Biomol Struct. 2004;33:297–316. doi: 10.1146/annurev.biophys.33.111502.082538. [DOI] [PubMed] [Google Scholar]
- 13.Shen Y, Tolic N, Masselon C, et al. Nanoscale proteomics. Anal Bioanal Chem. 2004;378:1037–45. doi: 10.1007/s00216-003-2329-8. [DOI] [PubMed] [Google Scholar]
- 14.Anderson NL, Anderson NG, Haines LR, et al. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA). J Proteome Res. 2004;3:235–44. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 15.MacGregor JT. The future of regulatory toxicology: impact of the biotechnology revolution. Toxicol Sci. 2003;75:236–48. doi: 10.1093/toxsci/kfg197. [DOI] [PubMed] [Google Scholar]
- 16.Hackett JL, Gutman SI. Introduction to the food and drug administration (FDA) regulatory process. J Proteome Res. 2005;4:1110–3. doi: 10.1021/pr050059a. [DOI] [PubMed] [Google Scholar]
- 17.Bilello JA. The agony and ecstasy of “OMIC” technologies in drug development. Curr Mol Med. 2005;5:39–52. doi: 10.2174/1566524053152898. [DOI] [PubMed] [Google Scholar]
- 18.Sawada H, Takami K, Asahi S. A toxicogenomic approach to drug-induced phospholipidosis: analysis of its induction mechanism and establishment of a novel in vitro screening system. Toxicol Sci. 2005;83:282–92. doi: 10.1093/toxsci/kfh264. [DOI] [PubMed] [Google Scholar]
- 19.Meneses-Lorente G, Watt A, Salim K, et al. Identification of early proteomic markers for hepatic steatosis. Chem Res Toxicol. 2006;19:986–98. doi: 10.1021/tx060007f. [DOI] [PubMed] [Google Scholar]
- 20.Younossi ZM, Baranova A, Ziegler K, et al. A genomic and proteomic study of the spectrum of nonalcoholic fatty liver disease. Hepatology. 2005;42:665–74. doi: 10.1002/hep.20838. [DOI] [PubMed] [Google Scholar]
- 21.Kolker E, Higdon R, Hogan JM. Protein identification and expression analysis using mass spectrometry. Trends Microbiol. 2006;14:229–35. doi: 10.1016/j.tim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 22.Nesvizhskii AI. Protein identification by tandem mass spectrometry and sequence database searching. Methods Mol Biol. 2007;367:87–119. doi: 10.1385/1-59745-275-0:87. [DOI] [PubMed] [Google Scholar]
- 23.Kuster B, Schirle M, Mallick P, et al. Scoring proteomes with proteotypic peptide probes. Nat Rev Mol Cell Biol. 2005;6:577–83. doi: 10.1038/nrm1683. [DOI] [PubMed] [Google Scholar]
- 24.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–88. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.de Graauw M, Tijdens I, Cramer R, et al. Heat shock protein 27 is the major differentially phosphorylated protein involved in renal epithelial cellular stress response and controls focal adhesion organization and apoptosis. J Biol Chem. 2005;280:29885–98. doi: 10.1074/jbc.M412708200. [DOI] [PubMed] [Google Scholar]
- 26.Mascarell L, Auger R, Alcover A, et al. Characterization of a gene encoding two isoforms of a mitochondrial protein up-regulated by cyclosporin A in activated T cells. J Biol Chem. 2004;279:10556–63. doi: 10.1074/jbc.M313770200. [DOI] [PubMed] [Google Scholar]
- 27.Mascarell L, Frey JR, Michel F, et al. Increased protein synthesis after T cell activation in presence of cyclosporin A. Transplantation. 2000;70:340–8. doi: 10.1097/00007890-200007270-00019. [DOI] [PubMed] [Google Scholar]
- 28.Kawada N, Kristensen DB, Asahina K, et al. Characterization of a stellate cell activation-associated protein (STAP) with peroxidase activity found in rat hepatic stellate cells. J Biol Chem. 2001;276:25318–23. doi: 10.1074/jbc.M102630200. [DOI] [PubMed] [Google Scholar]
- 29.Rauch A, Bellew M, Eng J, et al. Computational proteomics analysis system (CPAS): an extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. J Proteome Res. 2006;5:112–21. doi: 10.1021/pr0503533. [DOI] [PubMed] [Google Scholar]
- 30.Orchard S, Hermjakob H, Julian RK, Jr, et al. Common interchange standards for proteomics data: public availability of tools and schema. Proteomics. 2004;4:490–1. doi: 10.1002/pmic.200300694. [DOI] [PubMed] [Google Scholar]
- 31.Waters MD, Fostel JM. Toxicogenomics and systems toxicology: aims and prospects. Nat Rev Genet. 2004;5:936–48. doi: 10.1038/nrg1493. [DOI] [PubMed] [Google Scholar]
- 32.Hamady M, Cheung TH, Resing K, et al. Key challenges in proteomics and proteoinformatics. Progress in proteins. IEEE Eng Med Biol Mag. 2005;24:34–40. doi: 10.1109/memb.2005.1436456. [DOI] [PubMed] [Google Scholar]
- 33.Ekins S, Nikolsky Y, Nikolskaya T. Techniques: application of systems biology to absorption, distribution, metabolism, excretion and toxicity. Trends Pharmacol Sci. 2005;26:202–9. doi: 10.1016/j.tips.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 34.Quackenbush J. Extracting meaning from functional genomics experiments. Toxicol Appl Pharmacol. 2005;207:195–9. doi: 10.1016/j.taap.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 35.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1:845–67. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 36.Swarup V, Rajeswari MR. Circulating (cell-free) nucleic acids–a promising, non-invasive tool for early detection of several human diseases. FEBS Lett. 2007;581:795–9. doi: 10.1016/j.febslet.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 37.Piccin A, Murphy WG, Smith OP. Circulating microparticles: pathophysiology and clinical implications. Blood Rev. 2007;21:157–71. doi: 10.1016/j.blre.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Pieper R, Gatlin CL, Makusky AJ, et al. The human serum proteome: display of nearly 3700 chromatographically separated protein spots on two-dimensional electrophoresis gels and identification of 325 distinct proteins. Proteomics. 2003;3:1345–64. doi: 10.1002/pmic.200300449. [DOI] [PubMed] [Google Scholar]
- 39.Anderson NL, Polanski M, Pieper R, et al. The human plasma proteome: a nonredundant list developed by combination of four separate sources. Mol Cell Proteomics. 2004;3:311–26. doi: 10.1074/mcp.M300127-MCP200. [DOI] [PubMed] [Google Scholar]
- 40.Gong Y, Li X, Yang B, et al. Different immunoaffinity fractionation strategies to characterize the human plasma proteome. J Proteome Res. 2006;5:1379–87. doi: 10.1021/pr0600024. [DOI] [PubMed] [Google Scholar]
- 41.Liu X, Valentine SJ, Plasencia MD, et al. Mapping the human plasma proteome by SCX-LC-IMS-MS. J Am Soc Mass Spectrom. 2007;18:1249–64. doi: 10.1016/j.jasms.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Follettie MT, Pinard M, Keith JC, Jr, et al. Organ messenger ribonucleic acid and plasma proteome changes in the adjuvant-induced arthritis model: responses to disease induction and therapy with the estrogen receptor-beta selective agonist ERB-041. Endocrinology. 2006;147:714–23. doi: 10.1210/en.2005-0600. [DOI] [PubMed] [Google Scholar]
- 43.Merrick BA, Bruno ME, Madenspacher JH, et al. Alterations in the rat serum proteome during liver injury from acetaminophen exposure. J Pharmacol Exp Ther. 2006;318:792–802. doi: 10.1124/jpet.106.102681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Y, Fleet JC, Adamec J, et al. Effects of hindlimb unloading and bisphosphonates on the serum proteome of rats. Bone. 2007;41:646–58. doi: 10.1016/j.bone.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 45.Santos-Gonzalez M, Gomez Diaz C, Navas P, et al. Modifications of plasma proteome in long-lived rats fed on a coenzyme Q10-supplemented diet. Exp Gerontol. 2007;42:798–806. doi: 10.1016/j.exger.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 46.Ginestra A, Monea S, Seghezzi G, et al. Urokinase plasminogen activator and gelatinases are associated with membrane vesicles shed by human HT1080 fibrosarcoma cells. J Biol Chem. 1997;272:17216–22. doi: 10.1074/jbc.272.27.17216. [DOI] [PubMed] [Google Scholar]
- 47.Gasser O, Schifferli JA. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood. 2004;104:2543–8. doi: 10.1182/blood-2004-01-0361. [DOI] [PubMed] [Google Scholar]
- 48.MacKenzie A, Wilson HL, Kiss-Toth E, et al. Rapid secretion of interleukin-1beta by microvesicle shedding. Immunity. 2001;15:825–35. doi: 10.1016/s1074-7613(01)00229-1. [DOI] [PubMed] [Google Scholar]
- 49.Ardoin SP, Shanahan JC, Pisetsky DS. The role of microparticles in inflammation and thrombosis. Scand J Immunol. 2007;66:159–65. doi: 10.1111/j.1365-3083.2007.01984.x. [DOI] [PubMed] [Google Scholar]
- 50.Pilzer D, Gasser O, Moskovich O, et al. Emission of membrane vesicles: roles in complement resistance, immunity and cancer. Springer Semin Immunopathol. 2005;27:375–87. doi: 10.1007/s00281-005-0004-1. [DOI] [PubMed] [Google Scholar]
- 51.Garcia BA, Smalley DM, Cho H, et al. The platelet microparticle proteome. J Proteome Res. 2005;4:1516–21. doi: 10.1021/pr0500760. [DOI] [PubMed] [Google Scholar]
- 52.Merrick BA, Tomer KB. Toxicoproteomics: a parallel approach to identifying biomarkers. Environ Health Perspect. 2003;111:A578–9. doi: 10.1289/ehp.111-1241639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fannin RD, Auman JT, Bruno ME, et al. Differential gene expression profiling in whole blood during acute systemic inflammation in lipopolysaccharide-treated rats. Physiol Genomics. 2005;21:92–104. doi: 10.1152/physiolgenomics.00190.2004. [DOI] [PubMed] [Google Scholar]
- 54.Talwar S, Munson PJ, Barb J, et al. Gene expression profiles of peripheral blood leukocytes after endotoxin challenge in humans. Physiol Genomics. 2006;25:203–15. doi: 10.1152/physiolgenomics.00192.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Forrest MS, Lan Q, Hubbard AE, et al. Discovery of novel biomarkers by microarray analysis of peripheral blood mononuclear cell gene expression in benzene-exposed workers. Environ Health Perspect. 2005;113:801–7. doi: 10.1289/ehp.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liew CC, Ma J, Tang HC, et al. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med. 2006;147:126–32. doi: 10.1016/j.lab.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 57.Fischer HP. Towards quantitative biology: integration of biological information to elucidate disease pathways and to guide drug discovery. Biotechnol Annu Rev. 2005;11:1–68. doi: 10.1016/S1387-2656(05)11001-1. [DOI] [PubMed] [Google Scholar]
- 58.Spickett CM, Pitt AR, Morrice N, et al. Proteomic analysis of phosphorylation, oxidation and nitrosylation in signal transduction. Biochim Biophys Acta. 2006;1764:1823–41. doi: 10.1016/j.bbapap.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 59.Kalgutkar AS, Gardner I, Obach RS, et al. A comprehensive listing of bioactivation pathways of organic functional groups. Curr Drug Metab. 2005;6:161–225. doi: 10.2174/1389200054021799. [DOI] [PubMed] [Google Scholar]
- 60.Doss GA, Baillie TA. Addressing metabolic activation as an integral component of drug design. Drug Metab Rev. 2006;38:641–9. doi: 10.1080/03602530600959466. [DOI] [PubMed] [Google Scholar]
- 61.Angerer J, Ewers U, Wilhelm M. Human biomonitoring: state of the art. Int J Hyg Environ Health. 2007;210:201–28. doi: 10.1016/j.ijheh.2007.01.024. [DOI] [PubMed] [Google Scholar]
- 62.Sturla SJ. DNA adduct profiles: chemical approaches to addressing the biological impact of DNA damage from small molecules. Curr Opin Chem Biol. 2007;11:293–9. doi: 10.1016/j.cbpa.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 63.Shin NY, Liu Q, Stamer SL, et al. Protein targets of reactive electrophiles in human liver microsomes. Chem Res Toxicol. 2007;20:859–67. doi: 10.1021/tx700031r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dennehy MK, Richards KA, Wernke GR, et al. Cytosolic and nuclear protein targets of thiol-reactive electrophiles. Chem Res Toxicol. 2006;19:20–9. doi: 10.1021/tx050312l. [DOI] [PubMed] [Google Scholar]
- 65.Koen YM, Gogichaeva NV, Alterman MA, et al. A proteomic analysis of bromobenzene reactive metabolite targets in rat liver cytosol in vivo. Chem Res Toxicol. 2007;20:511–9. doi: 10.1021/tx6003166. [DOI] [PubMed] [Google Scholar]
- 66.Hanzlik RP, Koen YM, Theertham B, et al. The reactive metabolite target protein database (TPDB) - A web accessible resource. BMC Bioinformatics. 2007;8:95. doi: 10.1186/1471-2105-8-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pumford NR, Roberts DW, Benson RW, et al. Immunochemical quantitation of 3-(cystein-S-yl)-acetaminophen protein adducts in subcellular liver fractions following a hepatotoxic dose of acetaminophen. Biochem Pharmacol. 1990;40:573–9. doi: 10.1016/0006-2952(90)90558-3. [DOI] [PubMed] [Google Scholar]
- 68.Damsten MC, Commandeur JN, Fidder A, et al. Liquid chromatography/tandem mass spectrometry detection of covalent binding of acetaminophen to human serum albumin. Drug Metab Dispos. 2007;35:1408–17. doi: 10.1124/dmd.106.014233. [DOI] [PubMed] [Google Scholar]
- 69.Dietze EC, Schafer A, Omichinski JG, et al. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by a reactive metabolite of acetaminophen and mass spectral characterization of an arylated active site peptide. Chem Res Toxicol. 1997;10:1097–103. doi: 10.1021/tx970090u. [DOI] [PubMed] [Google Scholar]
- 70.Chen W, Shockcor JP, Tonge R, et al. Protein and nonprotein cysteinyl thiol modification by N-acetyl-p-benzoquinone imine via a novel ipso adduct. Biochemistry. 1999;38:8159–66. doi: 10.1021/bi990125k. [DOI] [PubMed] [Google Scholar]
- 71.Isbell MA, Morin D, Boland B, et al. Identification of proteins adducted by reactive naphthalene metabolites in vitro. Proteomics. 2005;5:4197–204. doi: 10.1002/pmic.200401278. [DOI] [PubMed] [Google Scholar]
- 72.Lin CY, Boland BC, Lee YJ, et al. Identification of proteins adducted by reactive metabolites of naphthalene and 1-nitronaphthalene in dissected airways of rhesus macaques. Proteomics. 2006;6:972–82. doi: 10.1002/pmic.200500170. [DOI] [PubMed] [Google Scholar]
- 73.Lame MW, Jones AD, Wilson DW, et al. Monocrotaline pyrrole targets proteins with and without cysteine residues in the cytosol and membranes of human pulmonary artery endothelial cells. Proteomics. 2005;5:4398–413. doi: 10.1002/pmic.200402022. [DOI] [PubMed] [Google Scholar]
- 74.Barber DS, Stevens S, Lopachin RM. Proteomic analysis of rat striatal synaptosomes during acrylamide intoxication at a low dose rate. Toxicol Sci. 2007;100:156–67. doi: 10.1093/toxsci/kfm210. [DOI] [PubMed] [Google Scholar]
- 75.Tallman KA, Kim HY, Ji JX, et al. Phospholipid-protein adducts of lipid peroxidation: synthesis and study of new biotinylated phosphatidylcholines. Chem Res Toxicol. 2007;20:227–34. doi: 10.1021/tx600331s. [DOI] [PubMed] [Google Scholar]
- 76.Luo J, Hill BG, Gu Y, et al. Mechanisms of acrolein-induced myocardial dysfunction: implications for environmental and endogenous aldehyde exposure. Am J Physiol Heart Circ Physiol. 2007;293:H3673–84. doi: 10.1152/ajpheart.00284.2007. [DOI] [PubMed] [Google Scholar]
- 77.Parastatidis I, Thomson L, Fries DM, et al. Increased protein nitration burden in the atherosclerotic lesions and plasma of apolipoprotein A-I deficient mice. Circ Res. 2007;101:368–76. doi: 10.1161/CIRCRESAHA.107.157537. [DOI] [PubMed] [Google Scholar]
- 78.Ju C, Uetrecht JP. Mechanism of idiosyncratic drug reactions: reactive metabolite formation, protein binding and the regulation of the immune system. Curr Drug Metab. 2002;3:367–77. doi: 10.2174/1389200023337333. [DOI] [PubMed] [Google Scholar]
- 79.Uetrecht J. Idiosyncratic drug reactions: current understanding. Annu Rev Pharmacol Toxicol. 2007;47:513–39. doi: 10.1146/annurev.pharmtox.47.120505.105150. [DOI] [PubMed] [Google Scholar]
- 80.Petkov T, Pehlivanov G, Grozdev I, et al. Toxic epidermal necrolysis as a dermatological manifestation of drug hypersensitivity syndrome. Eur J Dermatol. 2007;17:422–7. doi: 10.1684/ejd.2007.0241. [DOI] [PubMed] [Google Scholar]
- 81.Hussaini SH, Farrington EA. Idiosyncratic drug-induced liver injury: an overview. Expert Opin Drug Saf. 2007;6:673–84. doi: 10.1517/14740338.6.6.673. [DOI] [PubMed] [Google Scholar]
- 82.Pieters R. Detection of autoimmunity by pharmaceuticals. Methods. 2007;41:112–7. doi: 10.1016/j.ymeth.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 83.Zaccara G, Franciotta D, Perucca E. Idiosyncratic adverse reactions to antiepileptic drugs. Epilepsia. 2007;48:1223–44. doi: 10.1111/j.1528-1167.2007.01041.x. [DOI] [PubMed] [Google Scholar]
- 84.LaRocca D, Lehmann DF, Perl A, et al. The combination of nuclear and mitochondrial mutations as a risk factor for idiosyncratic toxicity. Br J Clin Pharmacol. 2007;63:249–51. doi: 10.1111/j.1365-2125.2006.02743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ong MM, Latchoumycandane C, Boelsterli UA. Troglitazone-induced hepatic necrosis in an animal model of silent genetic mitochondrial abnormalities. Toxicol Sci. 2007;97:205–13. doi: 10.1093/toxsci/kfl180. [DOI] [PubMed] [Google Scholar]
- 86.Eason C, O'Halloran K. Biomarkers in toxicology versus ecological risk assessment. Toxicology. 2002;181−2:517–21. doi: 10.1016/s0300-483x(02)00472-9. [DOI] [PubMed] [Google Scholar]
- 87.Huby R, Tugwood JD. Gene expression profiling for pharmaceutical safety assessment. Expert Opin Drug Metab Toxicol. 2005;1:247–60. doi: 10.1517/17425255.1.2.247. [DOI] [PubMed] [Google Scholar]
- 88.Beyer RP, Fry RC, Lasarev MR, et al. Multicenter study of acetaminophen hepatotoxicity reveals the importance of biological endpoints in genomic analyses. Toxicol Sci. 2007;99:326–37. doi: 10.1093/toxsci/kfm150. [DOI] [PubMed] [Google Scholar]
- 89.Thomas RS, Allen BC, Nong A, et al. A method to integrate benchmark dose estimates with genomic data to assess the functional effects of chemical exposure. Toxicol Sci. 2007;98:240–8. doi: 10.1093/toxsci/kfm092. [DOI] [PubMed] [Google Scholar]
- 90.Korrapati MC, Chilakapati J, Witzmann FA, et al. Proteomics of S-(1, 2-dichlorovinyl)-L-cysteine-induced acute renal failure and autoprotection in mice. Am J Physiol Renal Physiol. 2007;293:F994–1006. doi: 10.1152/ajprenal.00114.2007. [DOI] [PubMed] [Google Scholar]
- 91.Holly MK, Dear JW, Hu X, et al. Biomarker and drug-target discovery using proteomics in a new rat model of sepsis-induced acute renal failure. Kidney Int. 2006;70:496–506. doi: 10.1038/sj.ki.5001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cooper RS. Gene-environment interactions and the etiology of common complex disease. Ann Intern Med. 2003;139:437–40. doi: 10.7326/0003-4819-139-5_part_2-200309021-00011. [DOI] [PubMed] [Google Scholar]
- 93.Gambaro G, Anglani F, D'Angelo A. Association studies of genetic polymorphisms and complex disease. Lancet. 2000;355:308–11. doi: 10.1016/s0140-6736(99)07202-5. [DOI] [PubMed] [Google Scholar]
- 94.Guzey C, Spigset O. Genotyping as a tool to predict adverse drug reactions. Curr Top Med Chem. 2004;4:1411–21. doi: 10.2174/1568026043387791. [DOI] [PubMed] [Google Scholar]
- 95.Jain KK. Applications of AmpliChip CYP450. Mol Diagn. 2005;9:119–27. doi: 10.2165/00066982-200509030-00002. [DOI] [PubMed] [Google Scholar]
- 96.Grant SF, Hakonarson H. Recent development in pharmacogenomics: from candidate genes to genome-wide association studies. Expert Rev Mol Diagn. 2007;7:371–93. doi: 10.1586/14737159.7.4.371. [DOI] [PubMed] [Google Scholar]
- 97.Chhabra RS, Bucher JR, Wolfe M, et al. Toxicity characterization of environmental chemicals by the US National Toxicology Program: an overview. Int J Hyg Environ Health. 2003;206:437–45. doi: 10.1078/1438-4639-00240. [DOI] [PubMed] [Google Scholar]
- 98.Weis BK, Balshaw D, Barr JR, et al. Personalized exposure assessment: promising approaches for human environmental health research. Environ Health Perspect. 2005;113:840–8. doi: 10.1289/ehp.7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schwartz D, Collins F. Medicine. Environmental biology and human disease. Science. 2007;316:695–6. doi: 10.1126/science.1141331. [DOI] [PubMed] [Google Scholar]
- 100.Mariman EC. Nutrigenomics and nutrigenetics: the ‘omics’ revolution in nutritional science. Biotechnol Appl Biochem. 2006;44:119–28. doi: 10.1042/BA20050112. [DOI] [PubMed] [Google Scholar]
- 101.Ordovas JM. Nutrigenetics, plasma lipids, and cardiovascular risk. J Am Diet Assoc. 2006;106:1074–81. doi: 10.1016/j.jada.2006.04.016. quiz 1083. [DOI] [PubMed] [Google Scholar]
- 102.Barnes S, Kim H. Nutriproteomics: identifying the molecular targets of nutritive and non-nutritive components of the diet. J Biochem Mol Biol. 2004;37:59–74. doi: 10.5483/bmbrep.2004.37.1.059. [DOI] [PubMed] [Google Scholar]
- 103.Schweigert FJ. Nutritional proteomics: methods and concepts for research in nutritional science. Ann Nutr Metab. 2007;51:99–107. doi: 10.1159/000102101. [DOI] [PubMed] [Google Scholar]
- 104.HUPO 2007 Congress. Seoul SK. [19 January 2008]. http://www.hupo2007.com/