

Abstract
The cytochrome P450 3A (CYP3A) enzymes represent one of the most important drug-metabolizing systems in human. Recently, our group has generated cytochrome P450 3A knockout mice to study this drug-handling system in vivo. Here, we have characterized the Cyp3a knockout mice by studying the metabolism of midazolam, one of the most widely used probes to assess CYP3A activity. We expected that the midazolam metabolism would be severely reduced in the absence of CYP3A enzymes. We used hepatic and intestinal microsomal preparations from Cyp3a knockout and wild-type mice to assess the midazolam metabolism in vitro. In addition, in vivo metabolite formation was determined after intravenous administration of midazolam. Surprisingly, our results demonstrated that there is still marked midazolam metabolism in hepatic (but not intestinal) microsomes from Cyp3a knockout mice. Accordingly, we found comparable amounts of midazolam as well as its major metabolites in plasma after intravenous administration in Cyp3a knockout mice when compared to wild-type mice. These data suggested that other hepatic cytochrome P450 enzymes could take over the midazolam metabolism in Cyp3a knockout mice. We provide evidence that CYP2C enzymes, which were found to be upregulated in Cyp3a knockout mice, are primarily responsible for this metabolism and that several but not all murine CYP2C enzymes are capable of metabolizing midazolam to its 1’-OH and/or 4-OH derivatives. These data illustrate interesting compensatory changes that may occur in Cyp3a knockout mice. Such flexible compensatory interplay between functionally related detoxifying systems is probably essential to their biological role in xenobiotic protection.
Introduction
The cytochrome P450 enzymes (CYPs) play a pivotal role in the phase I metabolism of drugs and other xenobiotics. In addition, CYPs are involved in the synthesis and metabolism of a broad range of endogenous substrates, including steroids, bile acids and arachidonic acids. Members of the cytochrome P450 3A (CYP3A) subfamily are of particular interest because of their broad substrate specificity and their high inter- and intra-individual variation in expression and activity levels. In humans, four CYP3A enzymes have been identified, but only CYP3A4 and CYP3A5 are considered to be relevant for drug metabolism in adults. In general, CYP3A4 and CYP3A5 have similar substrate specificities, although they may have distinct affinities and turnovers for some substrates (Williams et al., 2002).
It is estimated that CYP3A enzymes contribute to the metabolism of approximately half of the currently marketed drugs (Guengerich, 1999). Since the CYP3A enzymes are strategically located in the liver and intestinal wall, they have a strong effect on the first-pass metabolism, oral bioavailability, and elimination of administered drugs. Furthermore, the induction and inhibition of CYP3A enzymes are considered to be important determinants in the therapeutic efficacy and toxicity of numerous drugs (Dresser et al., 2000; Lamba et al., 2002). Accordingly, interactions at the CYP3A level are often the cause of pronounced drug-drug interactions (Thummel and Wilkinson, 1998).
Given the importance of CYP3A, the in vitro screening of novel drug candidates as potential substrates of CYP3A has become routine in the pre-clinical drug development stage. Yet, these in vitro studies are not always indicative for the in vivo situation. To allow a more systematic in vivo evaluation of CYP3A-mediated metabolism, we have recently generated mice lacking all Cyp3a genes (Cyp3a−/−) as well as CYP3A4 transgenic mice in a Cyp3a knockout background (van Herwaarden et al., 2007). Interestingly, Cyp3a−/− mice are viable, fertile and do not show obvious physiological abnormalities. These observations suggest that the CYP3A enzymes do not have an essential endogenous physiological function and could be primarily dedicated to the detoxification of xenobiotics.
A classical probe for CYP3A activity in humans is midazolam. This drug is considered highly specific since no other human P450s contribute significantly to its metabolism. The biotransformation of midazolam by CYP3A enzymes yields 1’-OH and 4-OH midazolam as the principal metabolites (Kronbach et al., 1989). Also minor quantities of the secondary metabolite 1’,4-OH midazolam are formed. In this study, we aimed to further evaluate our Cyp3a knockout model by studying the metabolism of midazolam in vitro and in vivo. We hypothesized that the midazolam metabolism would be severely reduced in the absence of CYP3A enzymes. Interestingly, however, we still observed significant midazolam 1’- and 4-hydroxylation in Cyp3a−/− mice. Results of the present study indicate that this can be attributed to upregulated CYP2C enzymes in the Cyp3a−/− mice.
Materials and Methods
Materials
Midazolam was obtained from Roche Diagnostics (Almere, The Netherlands). NADPH-generation system, pooled human liver and intestinal microsomes as well as microsomes from baculovirus cells expressing human CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP3A4 and CYP3A5 were obtained from BD Bioscience (Alphen aan den Rijn, The Netherlands). RT-PCR primers were from Qiagen (Venlo, The Netherlands). 1’-OH and 4-OH midazolam were purchased from Sigma (St. Louis, MO, USA). Methoxyflurane (Metofane) was obtained from Medical Developments Australia Pty. Ltd., (Springvale, Australia). All other chemicals were of analytical grade and were obtained from commercial sources.
Animals
The generation and characterization of Cyp3a−/− mice is described elsewhere (van Herwaarden et al., 2007). Cyp3a+/+ or Cyp3a−/− mice (on a FVB/N genetic background) used for pharmacokinetic experiments or for the preparation of microsomes were male and 8−12 weeks of age. Mice were housed and handled according to institutional guidelines complying with Dutch legislation. Animals were kept in a temperature-controlled environment with a 12-h light/dark cycle and received a standard diet (AM-II; Hope Farms, Woerden, The Netherlands) and acidified water ad libitum.
Preparation of microsomes
Mouse liver and small intestinal microsomes were prepared by a procedure analogous to that of Emoto et al. (2000). Briefly, a cardiac puncture under anesthesia with methoxyflurane was performed after which the mice were sacrificed by cervical dislocation. Subsequently, organs from Cyp3a+/+ or Cyp3a−/− mice (each n=5) were collected and immediately washed with an ice-cold buffer solution A (50 mM Tris–HCl pH 7.4 containing 250 mM sucrose, 1 mM EDTA and 1 tablet Complete® (Protease inhibitor cocktail) per 45 ml). The whole small intestine, including duodenum, jejunum and ileum was isolated. The whole tissues were subsequently homogenized in ice-cold buffer solution B (50 mM Tris–HCl pH 7.4 supplemented with 150 mM KCl, 1 mM EDTA, 1 tablet Complete® per 45 ml and 20% v/v glycerol) and differential centrifugation was performed for 20 min at 9,000 g after which the supernatant was subjected to a 1-hour spin at 105,000 g. The resulting pellet was resuspended in ice-cold solution B and stored at −80°C until use. Protein concentrations were determined using the Bio-Rad protein assay based on the method of Bradford.
Microsomal incubations
Incubations were carried out in a total volume of 200 μl containing 100 mM KPi buffer pH 7.4 and either 0.5 mg/ml liver microsomes or 1 mg/ml intestinal microsomes. Protein concentrations and incubation times were chosen within the linear range of product formation. Control experiments without cofactor were performed to ascertain CYP-dependent metabolism. To determine kinetic parameters, midazolam was added in 10−12 different concentrations ranging from 0 to 400 μM. Final concentration of methanol was 0.5% in all incubations. After 5 min of pre-incubation at 37 °C, the reactions were initiated with a NADPH-regenerating system (final concentrations 1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 0.4 U/ml glucose-6-phosphate dehydrogenase and 3.3 mM MgCl2). The reactions were allowed to proceed for 5 min before they were terminated by adding 100 μl of ice-cold acetonitrile and the mixture was subsequently cooled on ice for 5 minutes before it was centrifuged (10 min at 6,800 g). 100 μl of the supernatant was subjected to HPLC analysis.
Kinetic parameters for 1’-OH- and 4-OH midazolam were determined using GraphPad Prism 4.0. 4-OH midazolam data were analyzed using the standard Michaelis-Menten equation: V = Vmax*[S] / Km+[S]. Data for 1’-OH midazolam formation were fitted in a Michaelis-Menten kinetics model with noncompetitive substrate inhibition as described previously (von Moltke et al., 1996): V = Vmax*[S] / (Km + [S]*(1+[S]/Ks)).
Chemical- and immunoinhibition
Two different antibodies against rat CYP2C11 were used and were obtained from Daiichi Pure Chemicals (Tokyo, Japan) and Invitrogen (Breda, The Netherlands), respectively. After a preincubation of 15 min at 37 °C with ketoconazole (2.5 μM final concentration, considered to be specific for CYP3A (Newton et al., 1995)) and/or anti-CYP2C11 antibody, the mixture was incubated for 6 minutes. The final concentration of midazolam in the incubations was 50 μM. All other conditions were as described above.
Preparation and reconstitution of recombinant murine CYP2C enzymes
The heterologous expression of the recombinant murine CYP2C enzymes in E. coli as well as their partial purification and reconstitution has been previously reported (Luo et al., 1998; Tsao et al., 2001; DeLozier et al., 2004; Wang et al., 2004). CYP2C65 CYPC65 and CYP2C70 were prepared in E. coli using similar methods (J.A. Bradbury and D.C. Zeldin, personal communication). The final concentration of midazolam in the incubations was 25 μM and 25 pmol of CYP2C enzyme was used. After a preincubation of 5 min the mixture was incubated for 15 minutes. All other conditions were as described for the microsomal incubations. In case of the recombinant human CYP2C and CYP3A enzymes, 20 pmol of enzyme was used and the reactions were allowed to proceed for 10 min.
Plasma pharmacokinetics
Midazolam was dissolved in 0.9% NaCl and was injected i.v. into the tail vein of mice at 0.5 or 10 mg/kg bodyweight. At t = 7.5, 15, 30, 60 and 90 min, blood samples were taken by cardiac puncture under anesthesia with methoxyflurane after which mice were sacrificed by cervical dislocation (n = 3−5 for each time point). Blood samples were centrifuged at 2,100 g for 10 minutes at 4°C, and the plasma fraction was collected and stored at −20°C until analysis. Samples were processed and measured by LC-MS/MS as described below.
HPLC
HPLC analyses were performed using a Symmetry C18 column; 3.0 × 150 mm, 3.5 μm (Waters, Etten-Leur, The Netherlands). Isocratic analyses were carried out at a flow rate of 0.4 ml/min. The mobile phase consisted of 33% acetonitrile / 23% methanol / 44% 10 mM phosphate buffer (pH 7.4; 0.2% triethylamine). The identities of 1’-OH and 4-OH midazolam were verified by comparing the retention times with authentic standards. Metabolites were detected at 230 nm and quantitated by using standard curves for 1’-OH and 4-OH midazolam.
LC-MS/MS
Mouse plasma samples were measured by LC-MS/MS. To a 20-μl plasma sample, 100 μl water, 20 μl of 0.5 μg/ml clonazepam (internal standard) in 50% (v/v) methanol and 200 μl of 5 mM sodium hydroxide were added. The analytes were extracted with 2 ml diethylether and the organic phase was evaporated under a stream of nitrogen. The residue was reconstituted in 100 μl of 0.1% (v/v) acetic acid in 5% (v/v) acetonitrile before injection in the chromatographic system. The LC-MS/MS equipment consisted of a DGU-14A degasser, a Sil-HTc autosampler, two LC10-ADvp-μ pumps and a CTO10-Avp column oven (all from Shimadzu, Kyoto, Japan) and a Finnigan TSQ Quantum Discovery Max triple quadrupole mass spectrometer with electrospray ionization (Thermo Electron, Waltham, MA, USA). Data were processed with the Finnigan Xcalibur software (version 1.4, Thermo Electron). 30 μl injections were made on a Polaris 3 C18-A column (50 × 2 mm, dp = 3 μm, average pore diameter = 10 nm, Varian, Middelburg, The Netherlands) with a Polaris 3 C18-A pre-column (10 × 2 mm, dp = 3 μm, Varian). The column temperature was maintained at 35°C and the autosampler was maintained at 4°C. The flow rate was 0.3 ml/min and the eluent comprised 45% (v/v) of 0.01% (v/v) formic acid in water and 55% (v/v) methanol. Mass transitions (collision energies (V)) were 326.1→291.1(26) for midazolam, 342.1→324.1(20), 203.1(25) and 168.0(33) for 1’-OH midazolam, 342.1→325.1(20) for 4-OH midazolam and 316.0→269.9(25) for clonazepam. The mass resolutions were set at 0.2 full with at half height for the first quadrupole and at 0.7 full with at half height (unit resolution) for the third quadrupole for all compounds.
RNA isolation and cDNA synthesis
Mouse livers were excised and immediately placed in an appropriate volume of RNAlater (Qiagen). They were stored at 4°C for several days until RNA was extracted using the RNeasy mini kit (Qiagen) according to the manufacturers protocol for the purification of total RNA from animal tissues. Subsequently, cDNA was generated using 5 μg of total RNA in a synthesis reaction using random hexamers (Applied Biosystems) and superscript II reverse transcriptase (Invitrogen, Carlsbad, CA) according to the supplier's protocols. The reverse transcription reaction was performed for 60 min at 42°C with a deactivation step of 15 min at 70°C. cDNA was stored at −20°C until use.
RT-PCR analysis
Real-time PCR (RT-PCR) was performed using specific primers (Qiagen) for the individual mouse CYP2C subfamily members on an Applied Biosystems 7500 real-time cycler system according to the manufacturers protocol. Briefly, in a MicroAmp Fast Optical 96-well reaction plate (Applied Biosystems), 25 μl reaction mixtures containing 5 μl cDNA (0.1 ng/μl), 12.5 μl SyBr Green PCR master mix, 2.5 μl sample primer mix (QuantiTect Primer Assays (Qiagen)) and 5 μl aqua Braun were pipetted. After sealing the plates with optical adhesive film (Applied Biosystems), the plates were briefly centrifuged. The cycling conditions were initiated at 50°C for 2 min with an enzyme activation step of 95°C for 10 min, followed by 45 PCR cycles of denaturation at 95°C for 15 s, and annealing/extension at 60°C for 1 min.
Analysis of the results was done by the comparative Ct method as described (Livak and Schmittgen, 2001). Briefly, quantitation of the target cDNAs in all samples was normalized to GAPDH cDNA (CtCYP2CX – CtGAPDH = ΔCt), and the difference in expression for each target cDNA in the Cyp3a−/− mice was expressed relative to the amount in the wild-type mice (ΔCtwild-type–ΔCtCyp3a−/− = ΔΔCt). Subsequently, fold changes in target gene expression were determined by taking 2 to the power of this number (2–ΔΔCt). Statistics was performed on ΔCt values (Yuan et al., 2006).
Western blot analysis
Immunohistochemical staining of CYP2C55 in mouse liver tissues was performed as described previously (Wang et al., 2004; van Herwaarden et al., 2005). Blots were probed with rabbit anti-CYP2C55pep1 IgG antibody (1:1,000). After extensive washing, the secondary HRP-labeled antibody (Goat anti Rabbit Igg HRP (1:10,000)) was added. Bands were visualized by ECL. Equal loading across the lanes was confirmed with total protein staining (ponceau S and India ink).
Data analysis
The two-sided unpaired Student's t test was used throughout the study to assess the statistical significance of differences between two sets of data. Differences were considered to be statistically significant when P < 0.05.
Results
Midazolam metabolism in wild-type and Cyp3a−/− mouse liver microsomes
In wild-type mouse liver microsomes midazolam was metabolized to its 1’-OH and 4-OH metabolites with kinetic parameters as reported in Table 1. Whereas normal Michaelis-Menten kinetics was observed for the 4-hydroxylation, substrate inhibition was seen for the 1’-hydroxylation reaction (Ks = 204 ± 27 μM) (Figure 1). These results are consistent with those observed in other mouse strains (Perloff et al., 2000; Granvil et al., 2003). Surprisingly, when using liver microsomes from Cyp3a−/− mice, we still found substantial 1’-OH- and 4-OH midazolam formation in an NADPH-dependent manner, indicating that other Cytochrome P450 enzymes can take over these reactions from CYP3A (Table 1). The Km for the midazolam 1’-hydroxylation was increased about 7-fold with a concomitant modest increase in Vmax, resulting in an intrinsic clearance that was roughly 6-fold lower compared to wild-type. Interestingly, analogous to wild-type microsomes, data for the 1’-hydroxylation by Cyp3a−/− microsomes could also be fitted in a substrate inhibition model (Ks = 354 ± 56 μM) (Figure 1). Analysis of the 4-hydroxylation reaction in Cyp3a−/− liver microsomes revealed that the Km was increased 2-fold with a concomitant 4.5-fold decrease in Vmax when compared to wild-type microsomes (Table 1). Overall, these data indicate that there is still significant midazolam metabolism in liver microsomes from Cyp3a−/− mice.
Table 1.
Kinetic parameters for midazolam metabolism by liver and intestinal microsomes. All values are the means of three independent experiments ± S.D. Incubations were performed as described in Materials and Methods.
| |
|
1′-OH Midazolam |
4-OH Midazolam |
||||
|---|---|---|---|---|---|---|---|
| Microsomes | Strain | Kma | Vmaxb | Vmax/Kmc | Kma | Vmaxb | Vmax/Kmc |
| Liver | Cyp3a+/+ | 0.95 ± 0.18 | 630 ± 29 | 662 | 8.43 ± 0.27 | 224 ± 12.7 | 26.6 |
| Cyp3a−/− | 6.38 ± 1.2 | 735 ± 11 | 115 | 17.5 ± 6.9 | 49.8 ± 5.0 | 2.36 | |
| Human | 2.93 ± 0.33 | 2097 ± 155 | 715 | 38.7 ± 6.3 | 1226 ± 87 | 27.8 | |
| Intestine | Cyp3a+/+ | 10.4 ± 1.8 | 62.3 ± 3.2 | 6.02 | 55.9 ± 2.0 | 38.0 ± 5.2 | 0.68 |
| Cyp3a−/− | -d | -d | -d | -d | -d | -d | |
| Human | 1.61 ± 0.04 | 596 ± 18 | 369 | 24.5 ± 3.3 | 330 ± 11 | 13.5 | |
Km expressed in μM
Vmax expressed in pmol/min/mg protein
Vmax/Km expressed in μL/min/mg protein
No metabolite detected (detection limit <5 pmol/min/mg protein)
Figure 1.

Representative plots of 1’-OH midazolam (A) and 4-OH midazolam (B) formation by liver microsomes of Cyp3a+/+ and Cyp3a−/− mice. Incubations were performed as described in Materials and Methods. The corresponding kinetic parameters are summarized in Table 1.
Midazolam metabolism in Cyp3a+/+ and Cyp3a−/− mouse intestinal microsomes
In addition to liver, we also investigated the midazolam metabolism in small intestinal microsomes from wild-type and Cyp3a−/− mice. In intestinal microsomes from wild-type mice significant 1’-OH and 4-OH midazolam formation was observed (Table 1). Consistent with the kinetic profiles in mouse liver microsomes, substrate inhibition was observed for the 1’-OH midazolam formation and normal Michaelis-Menten kinetics was seen for the 4-OH midazolam formation (not shown).
In contrast to Cyp3a−/− mouse liver microsomes, no midazolam metabolism was observed in Cyp3a−/− intestinal microsomes, indicating that in wild-type intestine the CYP3A enzymes are primarily responsible for the midazolam metabolism. In agreement with this, co-incubation with the CYP3A inhibitor ketoconazole (2.5 μM) showed a virtually complete inhibition of both 1’-OH and 4-OH midazolam formation in intestinal microsomes from wild-type mice (data not shown).
Chemical and immunochemical inhibition
Inhibition experiments with ketoconazole in liver microsomes from wild-type mice supported that both midazolam 1’- and 4-hydroxylation reactions are mostly CYP3A mediated. However, ketoconazole (2.5 μM) was not able to completely inhibit the 1’-OH midazolam formation in wild-type mice (Figure 2A). In contrast, complete inhibition of the 4-OH midazolam formation with ketoconazole was observed, suggesting that in wild-type liver microsomes this reaction is entirely CYP3A dependent (Figure 2B). As expected, ketoconazole had no inhibitory effect on the 1’- and 4-OH metabolite formation in liver microsomes from Cyp3a−/− mice.
Figure 2.

Inhibition of 1’OH midazolam (A) and 4-OH (B) midazolam formation by ketoconazole and/or two different antibodies against rat CYP2C11 from Invitrogen (monoclonal, anti-CYP2C-A) or Daiichi (polyclonal, anti-CYP2C-B), respectively, in mouse liver microsomes. After a preincubation of 15 min at 37 °C with vehicle, ketoconazole (2.5 μM) and/or the anti-CYP2C11 antibodies, the reaction was started by adding a NADPH-regenerating system and the mixture was subsequently incubated for 6 minutes. The final concentration of midazolam in the incubations was 50 μM. All values are the means of n = 2−5 determinations.
Previous studies have indicated that murine CYP2C enzymes may contribute to the 1’-OH midazolam formation in the mouse (Perloff et al., 2000; Perloff et al., 2003). In the absence of established inhibitors for the murine CYP2C enzymes, we used 2 different antibodies raised against rat-CYP2C11 to evaluate the involvement of CYP2C enzymes. Both antibodies were able to partially inhibit the 1’-OH midazolam formation in liver microsomes from wild-type mice (Figure 2A). The combined use of either of the two antibodies with ketoconazole resulted in a complete inhibition of the 1’-OH midazolam formation in wild-type microsomes, indicating that primarily CYP3A and CYP2C enzymes are responsible for this reaction, with no significant contribution of other CYPs. Consistent with these observations, we found that in Cyp3a−/− mouse liver microsomes the anti-CYP2C11 antibodies were able to significantly inhibit the 1’-OH metabolite formation (down to ∼5% of control values), although clear differences in the degree of inhibition were observed between the 2 antibodies (Figure 2A). The differences in inhibition potential between these two anti-CYP2C11 antibodies were even more pronounced for the 4-OH midazolam formation and, interestingly, reversal compared to the inhibition of 1’-OH midazolam formation: Whereas antibody A inhibited the 4-OH midazolam formation efficiently, antibody B was not able to inhibit this reaction in Cyp3a−/− microsomes at all (Figure 2B). It should be noted that these antibodies have not been evaluated against the murine CYP2C enzymes but our results strongly suggest that they differ markedly in their capability to inhibit different mouse CYP2C enzymes. These data do suggest, however, that in Cyp3a−/− liver microsomes distinct CYP2C enzymes are responsible for 1’-OH and 4-OH midazolam formation, respectively.
Expression levels of CYP2C enzymes in Cyp3a−/−
Based on the findings above, we hypothesized that one or more CYP2C enzymes would be upregulated in the Cyp3a−/− mice. We performed RT-PCR analyses for a set of Cyp2c genes to see whether alterations in mRNA expression levels in the liver of Cyp3a−/− compared to wild-type mice could be detected. Indeed, we found that CYP2C29, CYP2C38, CYP2C39, CYP2C50 and CYP2C66 were significantly upregulated with roughly 1.5- to 3-fold differences (Figure 3). None of the Cyp2c genes tested was downregulated in livers from Cyp3a−/− mice. Most notable was the more than 30-fold upregulation of CYP2C55, although its RNA levels remained low even after induction (Supplementary data 1). Also at the protein level, CYP2C55 appeared to be highly upregulated in the livers of Cyp3a−/− mice compared to wild-type mice (Figure 4).
Figure 3.

CYP2C mRNA expression measured by RT-PCR. Results are expressed as the fold change in liver of Cyp3a−/− mice (n=4) compared with wild-type mice (n=4). Data were normalized against the endogenous control GAPDH. Each sample was assayed in duplicate in at least two independent experiments. * P < 0.05, ** P < 0.01 and *** P < 0.001, indicate that the ΔCt value for Cyp3a−/− mice is significantly different from wild-type mice. ΔCt values are given in Supplementary data 1.
Figure 4.
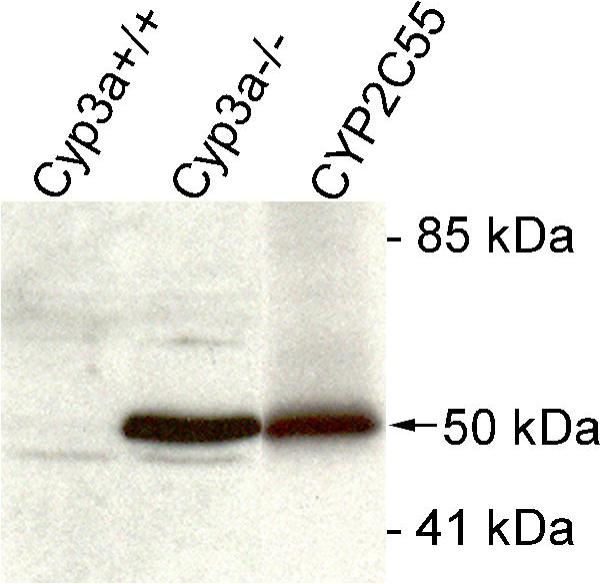
Expression of mouse CYP2C55 in liver of Cyp3a+/+ and Cyp3a−/− mice as detected by Western blotting. 30 μg of microsomal protein was loaded per lane. Recombinant CYP2C55 enzyme, migrating at 50 kDa (arrow), was used as positive control (0.5 pmol/lane). Total protein staining (Ponceau S and India Ink) confirmed equal loading across the lanes (not shown).
Midazolam metabolism by recombinant mouse CYP2C enzymes
The murine CYP2C subfamily consists of many closely related enzymes that can - unlike CYP3A enzymes - differ strikingly in substrate specificity (Goldstein and de Morais, 1994). We screened a panel of currently available recombinant expressed mouse CYP2C enzymes to identify which enzymes are capable of metabolizing midazolam (Table 2). Among these enzymes, it appeared that CYP2C29 and CYP2C65 were capable of hydroxylating midazolam at both its 1’ and 4 positions, whereas CYP2C39 and CYP2C55 solely catalyzed the 1’-OH midazolam reaction and CYP2C70 primarily did the 4-hydroxylation. For several CYP2C enzymes, however, we did not detect any 1’- or 4-OH metabolites. Unfortunately, it is not possible to compare these activities with the individual murine CYP3A enzymes, as these enzymes are not readily available.
Table 2.
Turnover rates for midazolam metabolite formation of a panel of 10 mouse CYP2C enzymes. The final concentration of midazolam in the incubations was 25 μM and 25 pmol of enzyme was used. After a preincubation of 5 min the reaction was started by adding a NADPH-regenerating system and the mixture was subsequently incubated for 15 minutes. All values are the means of three independent experiments ± S.D.
| v (min−1) | |||||
|---|---|---|---|---|---|
| Enzyme | 1′-OH MDZ | 4-OH MDZ | Enzyme | 1′-OH MDZ | 4-OH MDZ |
| CYP2C29 | 0.91 ± 0.04 | 0.019 ± 0.001 | CYP2C54 | -a | -a |
| CYP2C37 | -a | -a | CYP2C55 | 0.016 ± 0.002 | -a |
| CYP2C38 | -a | -a | CYP2C65 | 0.021 ± 0.006 | 0.008 ± 0.003 |
| CYP2C39 | 0.10 ± 0.1 | -a | CYP2C66 | -a | -a |
| CYP2C44 | -a | -a | CYP2C70 | -a | 0.28 ± 0.06 |
No metabolite detected (detection limit < 0.005 min−1)
We also tested whether human CYP2C enzymes are capable of metabolizing midazolam and compared this with the activities of CYP3A4 and CYP3A5 (Figure 5). For all the known human CYP2C enzymes we found modest amounts of the 1’-OH metabolite formed but could not detect any 4-OH midazolam. However, compared to CYP3A4 and CYP3A5 the formation of 1’-OH midazolam by human CYP2C enzymes was very low.
Figure 5.
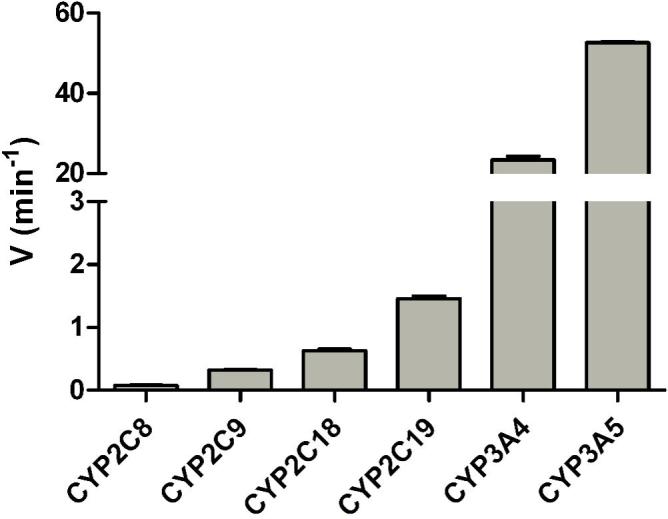
1’-OH midazolam formation by human CYP2C and CYP3A enzymes. The final concentration of midazolam in the incubations was 25 μM and 20 pmol of enzyme was used. After a preincubation of 5 min the reaction was started by adding a NADPH-regenerating system and the mixture was subsequently incubated for 10 minutes. All values are the means of duplicate determinations.
In vivo midazolam metabolism
Most of the in vivo metabolism and clearance of midazolam in wild-type mice occurs through CYP3A, as previously demonstrated by ketoconazole inhibition experiments (e.g., Granvil et al., 2003). Given the upregulation of CYP2C enzymes and their marked contribution to midazolam metabolism in Cyp3a−/− microsomes, we wanted to know the consequences of the Cyp3a knockout for the in vivo metabolism of midazolam. We therefore administered 0.5 mg/kg midazolam intravenously and subsequently determined the plasma levels of midazolam and its 1’- and 4-OH metabolites at several time points. Interestingly, as indicated in Figure 6A and Table 3, the plasma concentration curves and AUCs of midazolam were not significantly different between wild-type and Cyp3a−/− mice. Accordingly, the clearance did not differ between the two strains. Also the plasma levels of the 1’- and 4-OH metabolites were comparable (Figure 6B and C). Similar results were obtained when administering a much higher dose of 10 mg/kg midazolam (Supplementary data 2 and Table 3). Taken together, these data clearly demonstrate that also in vivo there is still marked midazolam metabolite formation in Cyp3a−/− mice and hence that midazolam is very efficiently cleared despite the absence of CYP3A.
Figure 6.
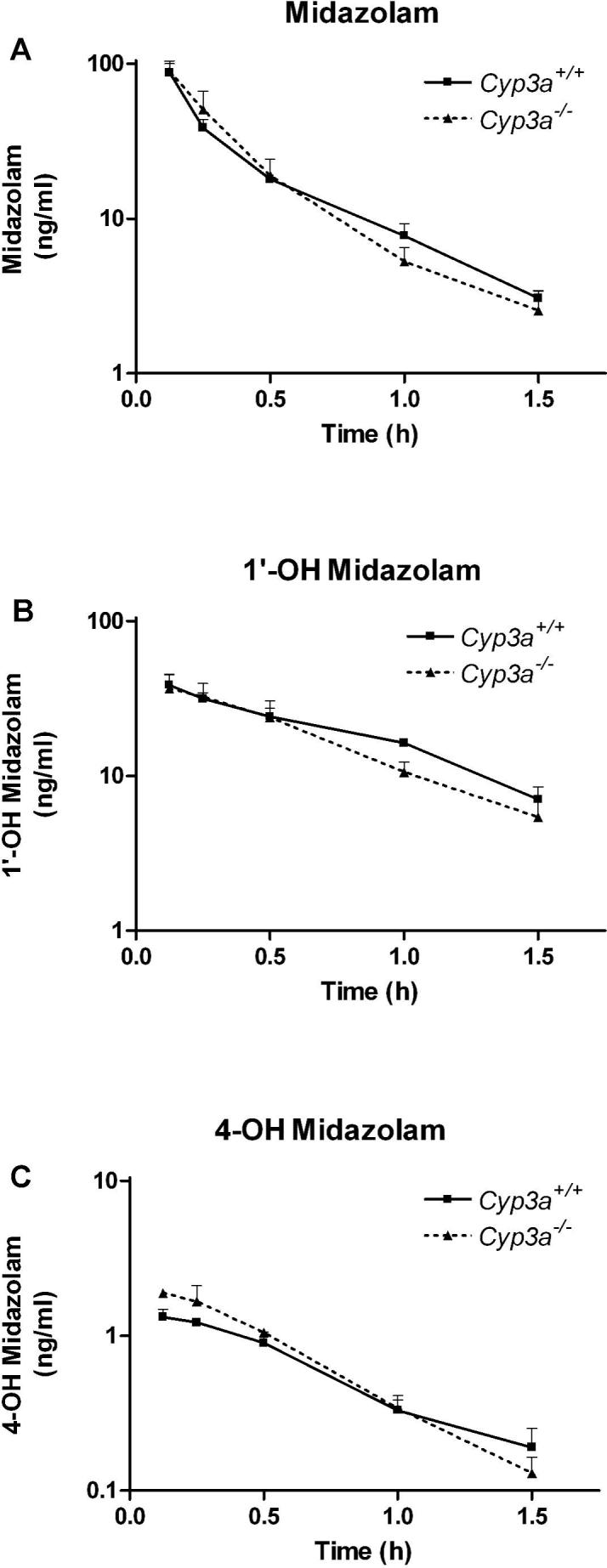
Plasma concentration versus time curves of midazolam (A), 1’-OH midazolam (B) and 4-OH midazolam (C) after intravenous midazolam administration (0.5 mg/kg) are shown for Cyp3a+/+ and Cyp3a−/− mice. Note the different axis scale for 4-OH midazolam. n = 3 ± S.D. for each time point.
Table 3.
Plasma pharmacokinetic parameters after i.v. administration of 0.5 or 10 mg/kg midazolam to Cyp3a+/+ and Cyp3a−/− mice.
| |
Midazolam |
1′-OH Midazolam |
4-OH Midazolam |
|||
|---|---|---|---|---|---|---|
| Cyp3a+/+ | Cyp3a−/− | Cyp3a+/+ | Cyp3a−/− | Cyp3a+/+ | Cyp3a−/− | |
| 0.5 mg/kg | ||||||
| AUC(0−1.5h), hr.μg/L | 29.5 ± 2.07 | 31.1 ± 4.03 | 29.7 ± 1.62 | 26.2 ± 3.25 | 0.95 ± 0.08 | 1.14 ±0.0 |
| Cl, 1/hr.kg | 0.017 ± 0.001 | 0.016 ± 0.002 | ||||
| 10 mg/kg | ||||||
| AUC(0−1.5h), hr.μg/L | 842 ± 168 | 833 ± 111 | 1849 ± 234 | 1147 ± 199 * | 23.6 ± 5.1 | 15.8 ± 4 |
| Cl, 1/hr.kg | 0.012 ± 0.002 | 0.012 ± 0.002 | ||||
AUC(0−1.5h), area under plasma concentration-time curve up to 1.5 hr; Cl, plasma clearance. Data are means ± SD, n = 3−5. * P < 0.05.
Discussion
Midazolam is one of the most widely used probes to assess CYP3A activity in vitro and in vivo and in this study we have used this drug to further characterize our recently generated Cyp3a knockout mouse model (van Herwaarden et al., 2007). We here report that CYP2C enzymes able to metabolize midazolam are upregulated in Cyp3a−/− mice and that consequently midazolam is still very efficiently metabolized and cleared in Cyp3a−/− mice.
The biotransformation of midazolam in both human and mouse is comparable, yielding 1’-OH midazolam as the major metabolite and 4-OH midazolam as a minor metabolite. Consistent with previous in vitro studies (Perloff et al., 2000; Perloff et al., 2003), our data suggest that in wild-type mice 1’-OH midazolam formation is not only dependent on CYP3A but also has a significant CYP2C component. Accordingly, we had anticipated to still observe some 1’-OH midazolam formation in Cyp3a−/− mice as a result of CYP2C activity, but we were surprised by the extent of this formation as it was only marginally reduced compared to wild-type mice. Even more surprising was that we also observed significant 4-OH midazolam formation in Cyp3a−/− mouse liver microsomes, a reaction that was also in mice considered to be CYP3A specific (Perloff et al., 2000). Consistent with this latter report, our own ketoconazole studies demonstrated complete inhibition of the 4-OH metabolite formation in wild-type microsomes. As we could not detect midazolam metabolism in small intestinal microsomes from Cyp3a−/− mice, it is likely that the small intestinal expression of CYP2C enzymes involved in midazolam metabolism is too low to significantly contribute to the intestinal metabolism of midazolam. This is consistent with studies that were not able to detect small intestinal CYP2C expression at the protein level (Emoto et al., 2000).
We demonstrated that several but not all CYP2C enzymes are upregulated in Cyp3a−/− mice. Notably, almost all CYP2C enzymes that were able to metabolize midazolam also had upregulated mRNA levels. Most prominent was the ∼35-fold upregulation of CYP2C55, which was also capable of performing the 1’-OH midazolam formation. This RNA upregulation was also reflected in a large increase in CYP2C55 protein levels as demonstrated by immunoblotting. However, the upregulated RNA expression level of CYP2C55 as judged from RT-PCR would still be lower than for most other CYP2C genes (e.g. CYP2C29). The contribution of CYP2C55 to the total 1’-OH midazolam formation in Cyp3a−/− mice might therefore still be limited.
Given the high upregulation of CYP2C55, it might be interesting to investigate whether this enzyme is partly under a different regulatory mechanism from the other Cyp2c genes. It is noteworthy that also in mice with a liver-specific deletion of the NADPH-Cytochrome P450 reductase gene, a 17-fold induction in CYP2C55 mRNA levels was observed (Weng et al., 2005). The mechanism of regulation of the different mouse CYP2C enzymes is still subject of investigation. The mouse Cyp2c locus is complex and contains 15 genes whereas in humans only 4 CYP2C genes have been identified (Nelson et al., 2004). It has recently been shown that the induction of CYP2C29 and CYP2C37 can be mediated by the constitutive androstane receptor (CAR) (Jackson et al., 2004; Jackson et al., 2006). However, CYP2C44 could not be upregulated by either CAR or PXR activators (DeLozier et al., 2004). Although little is yet known about the regulation of the other CYP2C enzymes, there clearly is accumulating evidence that various members of the CYP2C subfamily are differently regulated.
The CAR-mediated induction of various CYP2C family members indicates that these genes can be regulated by a diverse range of xenobiotic inducers. This could provide a clue to the possible mechanism behind the CYP2C upregulation in the Cyp3a knockout mice. It is likely that CYP3A normally metabolizes one or more inducers of the various Cyp2c genes, and that levels of these inducers are much higher in Cyp3a−/− mice. These inducers might in part be endogenous (e.g. steroids, bile acids), but an obvious source of these inducers would be xenobiotics (phytoestrogens, etc.) that occur in the normal food. Indeed, we have recently tested the effect of replacing normal food with a semisynthetic diet, and found much lower induction levels of CYP2C55 RNA in the livers of Cyp3a−/− mice compared to wild-type (5-fold instead of ∼35-fold on normal food). This suggests that a major factor in the CYP2C upregulation in the Cyp3a−/− mice is induction by food-derived xenobiotics that are normally metabolized by CYP3A.
During the preparation of this manuscript Emoto and Iwasaki (2007) reported that in addition to CYP3A enzymes, human CYP2C19 is also capable of catalyzing the midazolam 1’-hydroxylation. We found that not only CYP2C19 but also the 3 other human CYP2C enzymes could catalyze this reaction, although with much lower turnover rates than CYP3A enzymes. Based on the turnover rates as well as the lower expression levels of CYP2C versus CYP3A enzymes, the contribution of CYP2C enzymes to the total midazolam metabolism in humans will most likely be negligible.
Initially we used docetaxel for the in vitro and in vivo pharmacokinetic characterization of our Cyp3a−/− mice (van Herwaarden et al., 2007). Docetaxel is a widely used anti-cancer drug and is known to be primarily metabolized by members of the CYP3A subfamily (Shou et al., 1998). It appeared that the formation of docetaxel metabolites was completely absent in hepatic and intestinal microsomes from Cyp3a−/− mice, indicating that no other mouse CYPs could take over the docetaxel metabolism. In vivo, the lack of CYP3A mediated metabolism resulted in a 18-fold and 7-fold higher AUC for docetaxel in Cyp3a−/− mice after oral and i.v. administration, respectively (van Herwaarden et al., 2007). Consistent with the in vitro data from the present study, but in strong contrast to the earlier docetaxel data, our in vivo experiments demonstrated that after i.v. administration of midazolam there were only marginal differences in plasma levels of midazolam and of its 1’- and 4-OH metabolites between Cyp3a−/− and wild-type mice.
Combined with our recently generated CYP3A4 transgenic mouse models (van Herwaarden et al., 2007), we consider the Cyp3a−/− mouse model as an appropriate tool to study the impact of CYP3A on drug levels in an in vivo situation. However, other upregulated CYP enzymes can for some drugs affect the results obtained. For example, CYP3A and CYP2C enzymes have overlapping substrate specificities and this overlap may be different between species. Clearly, proper in vitro evaluation of the background metabolism of drugs of interest in Cyp3a−/− mice would be recommended to optimize application of this novel mouse model.
In summary, we investigated the metabolism of midazolam in Cyp3a−/− mice. Both our in vitro and in vivo data show that in the absence of CYP3A the metabolism of midazolam is only marginally reduced. We provided evidence that CYP2C enzymes are primarily responsible for this compensatory metabolism and we demonstrated that several but not all CYP2C enzymes are capable of catalyzing the 1’- and/or 4-hydroxylation reactions. Moreover, the Cyp3a knockout apparently results in a significant upregulation of some of the CYP2C enzymes. From a biological point of view, this study demonstrates that in the absence of an important xenobiotic metabolizing enzyme subfamily, organisms can still deal with some xenobiotics as a result of the overlapping substrate specificity of CYPs and the potential upregulation of these enzymes. Such flexibility is likely essential to the important biological function of detoxification of these enzyme systems.
Acknowledgments
We thank Dr. Conchita Vens (Division of Experimental Therapy, the Netherlands Cancer Institute) for assistance with RT-PCR analysis. This work was primarily supported by NWO / STW and, in part, by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Abbreviations
- CYP
cytochrome P450
- Cyp3a+/+
wild-type mice
- Cyp3a−/−
cytochrome P450 3A knockout mice
- MDZ
midazolam
- 1’-OH MDZ
1’-hydroxy midazolam
- 4-OH MDZ
4-hydroxy midazolam
- keto
ketoconazole
- CAR
constitutive androstane receptor (CAR)
- PXR
pregnane × receptor
References
- DeLozier TC, Tsao C-C, Coulter SJ, Foley J, Bradbury JA, Zeldin DC, Goldstein JA. CYP2C44, a New Murine CYP2C That Metabolizes Arachidonic Acid to Unique Stereospecific Products. J Pharmacol Exp Ther. 2004;310:845–854. doi: 10.1124/jpet.104.067819. [DOI] [PubMed] [Google Scholar]
- Dresser GK, Spence JD, Bailey DG. Pharmacokinetic-pharmacodynamic consequences and clinical relevance of cytochrome P450 3A4 inhibition. Clin Pharmacokinet. 2000;38:41–57. doi: 10.2165/00003088-200038010-00003. [DOI] [PubMed] [Google Scholar]
- Emoto C, Iwasaki K. Relative roles of CYP2C19 and CYP3A4/5 in midazolam 1'-hydroxylation. Xenobiotica. 2007;37:592–603. doi: 10.1080/00498250701358749. [DOI] [PubMed] [Google Scholar]
- Emoto C, Yamazaki H, Yamasaki S, Shimada N, Nakajima M, Yokoi T. Characterization of cytochrome P450 enzymes involved in drug oxidations in mouse intestinal microsomes. Xenobiotica. 2000;30:943–953. doi: 10.1080/00498250050200104. [DOI] [PubMed] [Google Scholar]
- Goldstein JA, de Morais SM. Biochemistry and molecular biology of the human CYP2C subfamily. Pharmacogenetics. 1994;4:285–299. doi: 10.1097/00008571-199412000-00001. [DOI] [PubMed] [Google Scholar]
- Granvil CP, Yu AM, Elizondo G, Akiyama TE, Cheung C, Feigenbaum L, Krausz KW, Gonzalez FJ. Expression of the human CYP3A4 gene in the small intestine of transgenic mice: in vitro metabolism and pharmacokinetics of midazolam. Drug Metab Dispos. 2003;31:548–558. doi: 10.1124/dmd.31.5.548. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. Cytochrome P-450 3A4: regulation and role in drug metabolism. Annu Rev Pharmacol Toxicol. 1999;39:1–17. doi: 10.1146/annurev.pharmtox.39.1.1. [DOI] [PubMed] [Google Scholar]
- Jackson JP, Ferguson SS, Moore R, Negishi M, Goldstein JA. The constitutive active/androstane receptor regulates phenytoin induction of Cyp2c29. Mol Pharmacol. 2004;65:1397–1404. doi: 10.1124/mol.65.6.1397. [DOI] [PubMed] [Google Scholar]
- Jackson JP, Ferguson SS, Negishi M, Goldstein JA. Phenytoin induction of the cyp2c37 gene is mediated by the constitutive androstane receptor. Drug Metab Dispos. 2006;34:2003–2010. doi: 10.1124/dmd.106.012005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronbach T, Mathys D, Umeno M, Gonzalez FJ, Meyer UA. Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol Pharmacol. 1989;36:89–96. [PubMed] [Google Scholar]
- Lamba JK, Lin YS, Schuetz EG, Thummel KE. Genetic contribution to variable human CYP3A-mediated metabolism. Adv Drug Deliv Rev. 2002;54:1271–1294. doi: 10.1016/s0169-409x(02)00066-2. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Luo G, Zeldin DC, Blaisdell JA, Hodgson E, Goldstein JA. Cloning and expression of murine CYP2Cs and their ability to metabolize arachidonic acid. Arch Biochem Biophys. 1998;357:45–57. doi: 10.1006/abbi.1998.0806. [DOI] [PubMed] [Google Scholar]
- Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics. 2004;14:1–18. doi: 10.1097/00008571-200401000-00001. [DOI] [PubMed] [Google Scholar]
- Newton DJ, Wang RW, Lu AY. Cytochrome P450 inhibitors. Evaluation of specificities in the in vitrometabolism of therapeutic agents by human liver microsomes. Drug Metab Dispos. 1995;23:154–158. [PubMed] [Google Scholar]
- Perloff MD, von Moltke LL, Court MH, Kotegawa T, Shader RI, Greenblatt DJ. Midazolam and Triazolam Biotransformation in Mouse and Human Liver Microsomes: Relative Contribution of CYP3A and CYP2C Isoforms. J Pharmacol Exp Ther. 2000;292:618–628. [PubMed] [Google Scholar]
- Perloff MD, Von Moltke LL, Greenblatt DJ. Differential metabolism of midazolam in mouse liver and intestine microsomes: a comparison of cytochrome P450 activity and expression. Xenobiotica. 2003;33:365–377. doi: 10.1080/0049825031000066259. [DOI] [PubMed] [Google Scholar]
- Shou M, Martinet M, Korzekwa KR, Krausz KW, Gonzalez FJ, Gelboin HV. Role of human cytochrome P450 3A4 and 3A5 in the metabolism of taxotere and its derivatives: enzyme specificity, interindividual distribution and metabolic contribution in human liver. Pharmacogenetics. 1998;8:391–401. doi: 10.1097/00008571-199810000-00004. [DOI] [PubMed] [Google Scholar]
- Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- Tsao C-C, Coulter SJ, Chien A, Luo G, Clayton NP, Maronpot R, Goldstein JA, Zeldin DC. Identification and Localization of Five CYP2Cs in Murine Extrahepatic Tissues and Their Metabolism of Arachidonic Acid to Regio- and Stereoselective Products. J Pharmacol Exp Ther. 2001;299:39–47. [PubMed] [Google Scholar]
- van Herwaarden AE, Smit JW, Sparidans RW, Wagenaar E, van der Kruijssen CM, Schellens JH, Beijnen JH, Schinkel AH. Midazolam and cyclosporin a metabolism in transgenic mice with liver-specific expression of human CYP3A4. Drug Metab Dispos. 2005;33:892–895. doi: 10.1124/dmd.105.004721. [DOI] [PubMed] [Google Scholar]
- van Herwaarden AE, Wagenaar E, van der Kruijssen CM, van Waterschoot RA, Smit JW, Song JY, van der Valk MA, van Tellingen O, van der Hoorn JW, Rosing H, Beijnen JH, Schinkel AH. Knockout of cytochrome P450 3A yields new mouse models for understanding xenobiotic metabolism. J Clin Invest. 2007;117:3583–3592. doi: 10.1172/JCI33435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Moltke LL, Greenblatt DJ, Schmider J, Duan SX, Wright CE, Harmatz JS, Shader RI. Midazolam hydroxylation by human liver microsomes in vitro: inhibition by fluoxetine, norfluoxetine, and by azole antifungal agents. J Clin Pharmacol. 1996;36:783–791. doi: 10.1002/j.1552-4604.1996.tb04251.x. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhao Y, Bradbury JA, Graves JP, Foley J, Blaisdell JA, Goldstein JA, Zeldin DC. Cloning, expression, and characterization of three new mouse cytochrome p450 enzymes and partial characterization of their fatty acid oxidation activities. Mol Pharmacol. 2004;65:1148–1158. doi: 10.1124/mol.65.5.1148. [DOI] [PubMed] [Google Scholar]
- Weng Y, DiRusso CC, Reilly AA, Black PN, Ding X. Hepatic gene expression changes in mouse models with liver-specific deletion or global suppression of the NADPH-cytochrome P450 reductase gene. Mechanistic implications for the regulation of microsomal cytochrome P450 and the fatty liver phenotype. J Biol Chem. 2005;280:31686–31698. doi: 10.1074/jbc.M504447200. [DOI] [PubMed] [Google Scholar]
- Williams JA, Ring BJ, Cantrell VE, Jones DR, Eckstein J, Ruterbories K, Hamman MA, Hall SD, Wrighton SA. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos. 2002;30:883–891. doi: 10.1124/dmd.30.8.883. [DOI] [PubMed] [Google Scholar]
- Yuan JS, Reed A, Chen F, Stewart CN., Jr. Statistical analysis of real-time PCR data. BMC Bioinformatics. 2006;7:85. doi: 10.1186/1471-2105-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]