

Abstract
In voltage-gated K+ channels (Kv), membrane depolarization promotes a structural reorganization of each of the four voltage sensor domains surrounding the conducting pore, inducing its opening. Although the crystal structure of Kv1.2 provided the first atomic resolution view of a eukaryotic Kv channel, several components of the voltage sensors remain poorly resolved. In particular, the position and orientation of the charged arginine side chains in the S4 transmembrane segments remain controversial. Here we investigate the proximity of S4 and the pore domain in functional Kv1.2 channels in a native membrane environment using electrophysiological analysis of intersubunit histidine metallic bridges formed between the first arginine of S4 (R294) and residues A351 or D352 of the pore domain. We show that histidine pairs are able to bind Zn2+ or Cd2+ with high affinity, demonstrating their close physical proximity. The results of molecular dynamics simulations, consistent with electrophysiological data, indicate that the position of the S4 helix in the functional open-activated state could be shifted by ∼7–8 Å and rotated counterclockwise by 37° along its main axis relative to its position observed in the Kv1.2 x-ray structure. A structural model is provided for this conformation. The results further highlight the dynamic and flexible nature of the voltage sensor.
INTRODUCTION
Voltage-dependent K+ (Kv) channels are formed by the assembly of four protein subunits encompassing a central aqueous pore allowing for selective K+ permeation across the lipid membrane. Each subunit is composed of six transmembrane segments (S1–S6), the first four segments (S1–S4) constitute the voltage sensor domain, while the last two segments (S5–S6) from each of the four subunits form the pore domain (Bezanilla, 2000; Hille, 2001). Upon membrane depolarization, the four voltage sensor domains undergo a conformational change promoting the opening or the closing of the ion permeation pathway, a process referred to as “voltage gating” (Sigworth, 1993; Bezanilla, 2000; Hille, 2001). Four highly conserved positively charged arginine residues within the transmembrane segment S4 have been identified as being predominantly responsible for the gating charge controlling the voltage activation of the channel (Aggarwal and MacKinnon, 1996; Seoh et al., 1996). Accessibility data show that membrane depolarization causes the S4 segment to move toward the extracellular side of the membrane (Larsson et al., 1996). However, voltage gating is obviously a very complex process involving multiple intermediate conformations of the protein (Zagotta et al., 1994). At a simple level, the process may be conveniently discussed in terms of two dominant states: a closed or resting conformation in which the four voltage sensors are resting in the “down” position and ion conduction through the central pore is blocked, and an open or active conformation in which the four voltage sensors are activated in the “up” position in which the central pore is opened to allow ion conduction.
Even though there remains significant uncertainty about the nature of the closed state (Yarov-Yarovoy et al., 2006; Campos et al., 2007; Pathak et al., 2007), there is now a broad consensus concerning the overall three-dimensional structure of Kv channels in the open-activated state. This was confirmed from the x-ray structure of the Kv1.2 channel (Long et al., 2005), which is in agreement with what had been previously deduced on the basis of a wide range of structural, functional, and biophysical experiments using the homologous Shaker K+ channel in its open-activated state (Laine et al., 2003, 2004). First, the voltage sensor domain is formed by a bundle of four anti-parallel transmembrane helices, S1–S4, packed counterclockwise with their N- and C-terminal ends exposed alternatively to the intra and extracellular solution and second, the voltage sensor makes contact with the adjacent subunit pore domain in the clockwise direction (Laine et al., 2003).
One striking feature revealed by the x-ray structure is the modular nature of the voltage sensor formed by S1–S4. In part, this explains why only a small number of specific interactions of the voltage sensor with the pore domain of Kv channels have been identified experimentally before the determination of the x-ray structure of the Kv1.2 channel (Elinder et al., 2001; Gandhi et al., 2003; Laine et al., 2003; Soler-Llavina et al., 2006). In particular, a high affinity intersubunit His–Zn2+–His metal bridge engineered in Shaker between R362 in S4 and A419 in S5 was discovered to greatly stabilize the channel in the open-activated state (Laine et al., 2003), suggesting that those two residues are in close atomic proximity. Paradoxically, the corresponding residues R294 and A351 in the x-ray structure of Kv1.2 are relatively distant from one another, with a Cβ–Cβ distance of 13.7 Å. At first view, it seems that the backbone of S4 might need to translate by at least 5–7 Å to allow the formation of the bridge.
Whether such a discrepancy is indicative of structural distortions induced by the engineered metal bridge, deviations in the crystal environment, or genuine differences between Shaker and Kv1.2, is unclear. In fact, relatively large lateral fluctuations are displayed by the voltage sensor and a tendency to shift toward a shorter distance between R294 and A351 has been observed in previous molecular dynamics (MD) simulations of the Kv1.2 channel in a membrane environment (Treptow and Tarek, 2006; Jogini and Roux, 2007). This suggests that the extracellular parts of S4 and S5 might alter their relative positions without causing costly structural distortions when the channel is in its membrane environment. The recent x-ray structure of a chimeric Kv1.2 channel in the presence of lipids, comprising the S3b–S4 segment from Kv2.1, provides an additional piece of evidence (Long et al., 2007). The distance between the Cβ–Cβ of corresponding residues in the chimera structure is 10.7 Å, shorter than the 13.7 Å in the Kv1.2 structure. However, the equivalent residue to R294 in the chimera structure is actually a neutral glutamine (Q290). In spite of the difference in sequence, the shorter distance is suggestive of the structural variability in the relative position of S4 and S5 in Kv channels.
The goal of the present study is to examine the possibility of engineering a histidine–histidine metal bridge between the voltage sensor domain and the pore domain in Kv1.2, and to use this information to refine our knowledge of the open-activated conformation of functional Kv channels in the membrane environment. Mutant Kv1.2 channels were engineered with site-directed mutagenesis and assayed electrophysiologically in Xenopus oocytes. In addition, computational models of the various mutants were constructed and simulated with MD. The experimental methods and simulation methodology are described in the next section. In the following sections, the results are first given and then discussed. The paper is concluded with a brief summary of the main results.
MATERIALS AND METHODS
Cloning and Site-directed Mutagenesis
Rat Kv1.2 (rKv1.2, accession no.: NM012970, GI:148298857) was a gift of S. Goldstein (University of Chicago). Point mutations were generated using the Quikchange Kit (Stratagene). All primers were synthesized by Integrated DNA Technologies, Inc. Plasmid DNA was isolated using the PureYield Midiprep System (Promega), and all constructs were sequenced at the University of Chicago core facility. Plasmids containing desired mutants were linearized with PacI (New England Biolabs) and purified using a Qiaquick PCR Purification Kit (QIAGEN). Capped mRNA was synthesized in vitro using the T7 mMESSAGE mMACHINE kit (Ambion). cRNA concentration was determined by spectrophotometry and visualized for qualitative analysis by gel electrophoresis.
Oocyte Preparation and RNA Injection
Xenopus laevis oocytes were prepared and isolated as follows. In brief, frogs were anesthetized in 1% tricaine (5 mM HEPES, pH 7.5) for 10 min and stage IV–V oocytes isolated and defolliculated in Ca2+-free OR-2 solution (in mM: 82.5 NaCl, 2.5 KCl, 1 MgCl2, 10 HEPES, pH 7.4 NaOH) using a combination of collagenase treatment (∼1 h in 2 mg/ml collagenase type II; Worthington) and manual defolliculation. Oocytes were injected with 23 nl (5 ng) wild-type or mutant cRNA for either Kv1.2 or Shaker using a Drummond microdispenser (Fisher Scientific) and incubated in ND-96 supplemented with 50 μg/ml gentamycin and 1% penicillin/streptomycin at 16°C. ND-96 solution contained (in mM) 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 10 HEPES (pH 7.6, NaOH).
Two-Electrode Voltage Clamp
Whole-cell currents were measured 48–60 h post-injection using a Geneclamp 500 amplifier and Axon Digidata 1322 (Axon Instruments, Inc.). Microelectrodes were filled with 3 M KCl and had resistances of 0.5–1.0 MΩ. Currents were recorded at room temperature in a bath solution containing (in mM) 96 NaCl, 4 KCl, 1 MgCl2, 0.3 CaCl2, 10 HEPES (pH 7.6) gravity perfused at ∼1–2 ml/min. Data were sampled at 5 kHz and recorded using Clampex 9.1 software (Axon Instruments, Inc.). Leak and capacitance was subtracted online using a P/4 protocol. To record voltage-activated outward K+ currents oocyte membrane potential was held at −80 mV and step depolarized to potentials ranging from −100 to +60 mV in 10-mV steps for durations of 1s. This was followed by a second voltage step to −30 mV for 500 ms to visualize tail currents before returning to the holding potential, and repeated at a frequency of 0.1 Hz. Bath solutions containing ZnCl2 or CdCl2 (concentrations between 1 nM and 10 μM) were perfused for 2 min to equilibrate before recording. To visualize inward deactivating tail currents a bath solution containing 50 mM K+ was used (replacing Na+) and oocytes subjected to the following voltage protocol: oocytes were step depolarized from a holding potential of −80 to +40 mV for 500 ms before hyperpolarizing membrane potential to voltages between −80 and −150 mV in 10-mV increments for a duration of 1s before returning to the holding potential.
Data Analysis
Data were analyzed using Clampfit 9.1 (Axon Instruments, Inc.) and tabulated in Excel (Microsoft Office). I-V curves were generated from steady-state currents after 1 s and G-V curves were derived using the normalized chord conductance, which was calculated by dividing the steady-state current by the driving force derived from the K+ equilibrium potential (internal K+ concentration was assumed to be 100 mM). Both I-V and G-V relationships were generated using Origin. G-V curves were fitted with a single Boltzmann function:
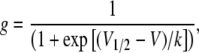 |
(1) |
where g is the conductance normalized with respect to the maximal conductance, V1/2 is the half-activation potential, V is the test voltage, and k is the slope factor. Data throughout the text and figures are shown as mean ± SEM. Data were subjected to statistical analysis were appropriate and deemed significant at P < 0.05.
Molecular Dynamics Simulations
The simulated systems represent atomic models of wild type and mutants of the Kv1.2 channel embedded into a di-palmitoyl-phosphatidyl-choline (DPPC) bilayer surrounded by an aqueous salt solution of 100 mM KCl. The procedure to construct the model of Kv1.2 was described previously in Jogini and Roux (2007). In brief, the missing coordinates of the Kv1.2 channel were constructed using the structural information from the crystal structure of the Kv1.2 channel (PDB id 2A79) and of the isolated voltage sensor of the KvAP channel (PDB id 1QRS). For the S1–S2 linker, the residues 185–214 were not included, and for the S3–S4 linker, the residues 282–286 were deleted in accord with experimental data, indicating that such deletions are not detrimental to the channel function. To complete the missing parts of the crystal structure we used structural alignments and mutagenesis functional data to assign the residues forming the S1 and S3 helices. (Jogini and Roux, 2007). The most critical parts for the present study, i.e., the S5–S6 pore domain, the S4–S5 linker and the S4 (up to residue 287), are taken directly from the x-ray structure (Long et al., 2005). The total number of atoms in the channel/membrane system is ∼77,000. The channel is oriented perpendicular to the membrane plane such that the pore is along the Z-axis, and the center of the bilayer is at Z = 0. The simulation methodology is essentially the same as that described previously in (Jogini and Roux, 2007). In brief, all the simulations were performed at constant temperature and constant pressure with fixed cross-sectional area and no truncation of electrostatic interactions using the program CHARMM version c32a2 (Brooks et al., 1983; MacKerell et al., 1998a). Periodic boundary conditions were applied to mimic a multilayer membrane system extending in the XY plane. The all-atom potential energy function PARAM27 for protein (MacKerell et al., 1998b), phospholipids (Schlenkrich et al., 1996; Lee et al., 2005; Klauda et al., 2006), and ions (Beglov and Roux, 1994) were used with the TIP3P water model (Jorgensen et al., 1983). The histidine side chain was modeled as a deprotonated imidazole carrying a net charge of −1, yielding a neutral complex with the divalent Zn2+. Though the protonation state of histidine depends on its environment (Lin and Lim, 2004), the details of the charge distribution are not critical because the geometry of the bridge is imposed by harmonic distance restraints in the present simulations.
The final configurations from the two previous MD simulations of 20 ns were used to construct the various mutants simulated for the present study. Two systems were generated and simulated for each of the three mutants: R294H-A351H, R294H-A351H-D352G-E353S, and R294H-D352H. A flat-bottom harmonic energy restraint with maximum distance of 3.0 Å and force constant of 10 kcal/mol/Å2 was imposed between the Zn2+ and the NE2 atom of each of the two histidine side chains in S4 and S5 of adjacent subunits (respectively at positions 294 and 351 or 352). Because the optimized distance between Zn2+ and a NE2 atom is on the order of 2.1 Å, the energy restraint only maintains the existence of the metal bridge but does not interfere with its local geometry. The distance restraints are rapidly satisfied during the early stage of the trajectory without distorting the local structure. Taking the end-point of previous simulations as starting point (Jogini and Roux, 2007), a total of two MD simulations per mutant, each of 2.5 ns, were generated (six MD simulations in total). The trajectories were stored every 2 ps for analysis. To provide a unique reference frame for analyzing the orientation of the voltage sensor, the channel configurations were reoriented with respect to the pore domain (S5–S6) accounting for the fourfold symmetry of the channel; see Fig. 3 in Jogini and Roux (2007). Coordinates of the model corresponding to open-activated Kv1.2-R294H-A351H-D352G-E353S channel can be accessed as online supplemental material.
Figure 3.

Cd2+ can form a metal bridge between R294H and A351H. (A) Representative current traces from an oocyte expressing Kv1.2-R294H-A351H channels recorded in the absence (left) and presence (right) of 1 μM Cd2+. Black arrowheads indicate where steady-state current was measured to generate I-V and G-V relationships. (B) Mean current–voltage relationships from oocytes expressing Kv1.2-R294H-A351H channels recorded in the absence (solid squares, n = 6) and presence (open circles, n = 6) of 1 μM Cd2+. (C) Normalized G-V relationships from oocytes expressing Kv1.2-R294H-A351H channels recorded in the absence (solid squares, n = 6) and presence (open circles, n = 6) of 1 μM Cd2+.
Online Supplemental Material
The online supplemental material (available at http://www.jgp.org/cgi/content/full/jgp.200809962/DC1) includes the coordinate of the model corresponding to open-activated Kv1.2-R294H-A351H-D352G-E353S channel. Also, it includes Fig. S1, which displays an analysis of the rate of activation with and without Zn2+ on the quadruple Kv1.2 mutant.
RESULTS
R294H-A351H Metal Bridge Inhibits Channel Activation
Previously it was demonstrated that there was a direct interaction between residues R362 and A419 in the Shaker K+ channel via engineering a cadmium (Cd2+) and a zinc (Zn2+) binding site by replacing these residues with cysteine and histidine, respectively (Laine et al., 2003). A spontaneous intersubunit disulphide bond was formed between residues 362 and 419, suggesting that S4 interacts directly with the pore domain in the Shaker K+ channel. The determination of the structure of the mammalian voltage-gated K+ channel Kv1.2 by x-ray crystallography (Long et al., 2005) prompted us to assess the S4-pore interaction in the context of a channel of known 3D structure. Exploiting the similarity between Kv1.2 and Shaker to implement the methodology previously used successfully with Shaker, the equivalent R294 and A351 residues in Kv1.2 (see Fig. 1) were mutated to histidine, and their ability to form a high-affinity Zn2+ or Cd2+ binding site was assayed.
Figure 1.

Comparison of amino acid sequence of Shaker and Kv1.2 in the region of the S4 and S5 segments. (A) A model describing the membrane topology of a Kv1.2 subunit showing transmembrane segments S1–S6 and the reentrant P loop. The approximate positions of residues R294 of S4 segment and A351 of the S5 pore domain are indicated. (B) Alignment of S4–S5 regions of Kv1.2, Shaker, KvAP, and the recently crystallized chimera of Kv1.2-Kv2.1. The black line indicates the S4 and S5 transmembrane segments and the asterisks denote the position of residues R294 and A351 of Kv1.2. Highlighted in blue and red are basic and acidic residues, respectively.
Fig. 2 (A and B) shows that the double mutant R294H-A351H of Kv1.2 generates robust membrane currents when expressed in Xenopus oocytes and that upon addition of 1 μM Zn2+ this current is significantly reduced by ∼5-fold at +60 mV (P < 0.05). This was an unexpected result and one that differs markedly from those obtained with Shaker, where the equivalent residues form a strong disulphide bridge shifting the V1/2 by ∼35 mV in the hyperpolarized direction (Laine et al., 2003), stabilizing the open channel conformation without reducing current magnitude. The loss of current in the Kv1.2 R294H-A351H mutant was also apparent in the normalized G-V relationships (Fig. 2 C) but was not observed in either of the single mutants (Figs. 2, D and E). However, addition of lower concentrations of Zn2+ (10 nM) did lead to a small, but unambiguous, hyperpolarizing shift in V1/2 of −15.8 mV, without any significant loss of current magnitude (Fig. 2 C, P < 0.05). This suggests that Zn2+ is able to bind both R294H and A351H in Kv1.2, confirming these residues are in close proximity in the open-activated state. The strong effect seen by the addition of small concentrations of Zn2+ also implies that the engineered binding site has high affinity. The loss of current magnitude in the R294H-A351H mutant Kv1.2 channel at concentrations of Zn2+ >10 nM infers that Zn2+ is able to bind to a nonspecific binding site with lower affinity that likely involves the histidine introduced at position R294 and one or more acidic residues that are present in the S4–S5 loop adjacent to A351 (D352 and E353), residues that are absent at the equivalent positions in Shaker. Furthermore, 1 μM Zn2+ is able to induce a small but significant shift (P < 0.05) in the V1/2 of the single mutant R294H but not A351H (Fig. 2, D and E, and Table I), suggesting that the introduced histidine at R294 and acidic residues at D352 and E353 probably form an interaction site for Zn2+. We were able to overcome the problem of Zn2+-induced loss of current by using Cd2+, which also has the ability to bind histidine but with lower affinity (Rulisek and Vondrasek, 1998). The addition of 1 μM Cd2+ to Kv1.2-R294H-A351H significantly shifted the voltage dependence of activation in the hyperpolarized direction by −17.2 mV from −26.7 ± 0.9 mV to −43.9 ± 0.6 mV, without loss of current magnitude (Fig. 3, A–C, P < 0.05). No significant shift in V1/2 was observed in either of the single mutants with Cd2+ (unpublished data).
Figure 2.

R294H-A351H forms a high affinity Zn2+ binding site that reduces current magnitude. (A) Representative current traces from an oocyte expressing Kv1.2-R294H-A351H channels recorded in the absence (left) and presence (right) of 1 μM Zn2+, using the protocol outlined in the Materials and methods. Black arrowheads indicate where steady-state current was measured to generate I-V and G-V relationships. (B) Mean current–voltage relationships from oocytes expressing Kv1.2-R294H-A351H channels recorded in the absence (solid squares, n = 8) and presence (open circles, n = 8) of 1 μM Zn2+. (C) Normalized G-V curves for Kv1.2-R294H-A351H obtained in the absence (black squares, n = 8) and presence of various concentrations of Zn2+; 10 nM (gray triangles, n = 7), 100 nM (open diamonds, n = 7), and 1 μM (open circles, n = 8) showing dose-dependent reduction in conductance with increasing concentrations of extracellular Zn2+. To demonstrate loss of current with Zn2+ application, conductance (G) values obtained in either the absence or presence of Zn2+ were normalized to the maximal G value obtained in the absence of Zn2+. Data for control (no Zn2+) and 10 nM Zn2+ were fitted with a single Boltzmann function, shown as solid black and gray curves respectively. (D and E) Normalized G-V curves obtained in the absence (solid squares) and presence (open circles) of 1 μM Zn2+ for (D) Kv1.2-R294H (n = 9) and (E) Kv1.2-A351H (n = 8). Single mutants showed no loss of current magnitude with application of 1 μM Zn2+; hence G values were normalized to the maximal G in each experimental condition (absence or presence of Zn2+).
TABLE I.
Summary of V1/2 Changes Associated with Zn2+ Application to Kv1.2 Mutations
| n | Control V1/2 | 1 μM Zn2+ V1/2 | ΔV1/2 | |
|---|---|---|---|---|
| mV | mV | mV | ||
| R294H | 9 | −4.7 ± 0.5 | −13.6 ± 0.6 | −8.9 |
| A351H | 8 | −7.9 ± 0.5 | −11.3 ± 0.5 | −3.4 |
| D352H | 7 | −0.9 ± 0.6 | −0.3 ± 0.6 | +0.6 |
| R294H-D352H | 7 | −10.8 ± 0.4 | −25.7 ± 0.6 | −14.9 |
| D352G-E353S | 7 | −17.3 ± 0.5 | −15.9 ± 0.6 | +1.4 |
| R294H-D352G-E353S | 7 | −1.7 ± 0.5 | −1.6 ± 0.5 | +0.1 |
| A351H-D352G-E353S | 7 | −12.8 ± 0.5 | −16.5 ± 0.5 | −3.7 |
| R294H-A351H-D352G-E353S | 9 | −2.2 ± 0.8 | −29.6 ± 0.6 | −27.4 |
Taken together these results suggest that a metal bridge can be formed between histidine residues introduced at positions 294 of S4 and 351 of the S5 pore domain. However, the introduction of these histidine residues allows additional interactions in the presence of Zn2+ that are not present in Shaker and that appear to promote a nonconducting channel conformational state.
Neutralization of D352 and E353 and the R294H-A351H Metal Bridge
Alignment of Shaker and Kv1.2 S4-S5 region (Fig. 1) indicates the presence of two acidic residues in Kv1.2 between the transmembrane segment S5 and the pore loop (D352 and E353) that are absent in the corresponding positions in Shaker (G420 and S421). The fact that Zn2+ can bind acidic groups such as aspartate and glutamate, and that D352 and E353 lie adjacent to the metal binding site A351H, strongly suggests that these two residues could compete for Zn2+ binding to the introduced histidine residues. Therefore both residues D352 and E353 were neutralized to the Shaker-like glycine (D352G) and serine (E353S) respectively (the functional effects of charge-modifying mutations in Shaker and Kv1.2 are described below). The effect of 1 μM Zn2+ on the quadruple Kv1.2 mutant (R294H-A351H-D352G-E353S) was assessed by two-electrode voltage clamp. In contrast to the double mutant R294H-A351H, there was no loss of current magnitude with the addition of 1 μM Zn2+ for the quadruple mutant (Fig. 4 A). In this case, 1 μM Zn2+ significantly shifted the voltage dependence of activation in the hyperpolarized direction by −27.4 mV from −2.2 ± 0.8 mV to −29.6 ± 0.6 mV (P < 0.05, Fig. 4 B and Table I). Assuming Eq. 1, a hyperpolarizing shift of ∼25 mV corresponds roughly to a shift of ∼2 kBT in the relative stability of the open and closed states. Furthermore, 1 μM Zn2+ had no effect on any of the control mutants: the double Kv1.2-D352G-E353S mutant or the triple mutants Kv1.2-A351H-D352G-E353S and Kv1.2-R294H-D352G-E353S (Fig. 4, D and E). In fact, with the acidic residues neutralized, the Zn2+-induced shift observed previously in the single mutant Kv1.2-R294H (Fig. 2 D) was now completely removed (Table I; ΔV1/2 of Kv1.2-R294H = −8.9 mV versus ΔV1/2 of Kv1.2-R294H-D352G-E353S = 0.1 mV).
Figure 4.

Metal ion binding to R294H-A351H stabilizes the activated state after neutralization of acidic residues D352 and E353. (A) Representative current traces from an oocyte expressing Kv1.2-R294H-A351H-D352G-E353S channels recorded in the absence (left) and presence (right) of 1 μM Zn2+, using the protocol outlined in the Materials and methods. (B) Normalized G-V relationships from oocytes expressing Kv1.2-R294H-A351H-D352G-E353S channels recorded in the absence (solid squares, n = 9) and presence (open circles, n = 9) of 1 μM Zn2+. (C) A plot of ΔV1/2 versus Zn2+ concentration in oocytes expressing Kv1.2-R294H-A351H-D352G-E353S channels, where ΔV1/2 is the difference between the fitted V1/2 values obtained in the absence and presence of varying concentrations of Zn2+. The data were fitted with a hyperbolic function (solid curve) with a half-maximal concentration of 0.36 ± 0.18 μM (n = 4–8). (D and E) Normalized G-V curves obtained in the absence (solid squares) and presence (open circles) of 1 μM Zn2+ for (D) Kv1.2-R294H-D352G-E353S (n = 7) and (E) Kv1.2-A351H-D352G-E353S (n = 7).
To estimate the Zn2+ affinity of the quadruple mutant Kv1.2-R294H-A351H-D352G-E353S the half-maximal voltage of activation was determined with varying Zn2+ concentrations. Fig. 4 C shows the calculated ΔV1/2 values plotted against Zn2+ concentrations ranging between 1 nM and 10 μM. The data were fitted with a rectangular hyperbola, yielding a half-maximal effective Zn2+ concentration of ∼360 nM. This value is very similar to 400 nM obtained previously for the corresponding metal bridge engineered between R362H-A419H in Shaker (Laine et al., 2003). The high apparent affinity is consistent with the conclusion that the Zn2+ ion binds simultaneously to the two histidine side chains (Kiefer and Fierke, 1994; Loland et al., 1999; Norregaard et al., 2000), confirming that these positions are within atomic distance of each other in the open-activated state. For comparison, the apparent affinities of Zn2+ binding sites formed by two histidine residues in the active site of the enzyme carbonic anhydrase II range from 100 nM to 1 μM (Kiefer and Fierke, 1994).
The strong apparent Zn2+ affinity detected in the electrophysiological experiments confirms that residues R294 and A351 in Kv1.2 are closer in an open-activated state than the 13.7 Å observed in the Kv1.2 x-ray crystal structure (Long et al., 2005). Furthermore, the observations indicate that residues D352 and E353 are also in close proximity to R294, as suggested by previous MD results (Jogini and Roux, 2007).
Metal Bridge Formation Stabilizes the Open-Activated State
Our data suggest that the generation of a metal bridge between R294H and A351H with either Zn2+ or Cd2+ promotes a stabilization of the open-activated conformation of Kv1.2 relative to the closed state. This view is supported by simple considerations based on the available structural (Long et al., 2005) and functional data (Gandhi et al., 2003; Ruta et al., 2005; Tombola et al., 2006; Campos et al., 2007; Pathak et al., 2007) and is also in accord with recently proposed models of the closed state of Kv1.2, which all have in common an increase in the distance separating R294 and A351 relative to the open-activated state (Campos et al., 2007; Long et al., 2007; Pathak et al., 2007), suggesting that metal bridge formation between S4 and S5 should become unfeasible in the closed state. Nevertheless, one might be tempted to argue the converse, namely that the formation of the metal bridge causes a destabilization of the closed state of Kv1.2. To draw clear structural inferences from the experimental observations requires that one be able to distinguish between these two possibilities. To address this issue, we studied deactivating tail currents from the quadruple mutant Kv1.2-R294H-A351H-D352G-E353S in the absence and presence of 1 μM Zn2+. In Fig. 5 we show that metal bridge formation induces a slowing of tail current deactivation kinetics at all voltages between −150 and −80 mV. This data shows that in the presence of Zn2+ it is harder for the channel to return to the closed state, suggesting it is indeed the open-activated channel conformation that sits in a deeper free energy well under conditions were the metal bridge is formed. In fact, the kinetic data in Fig. 5 are in semi-quantitative agreement with the shift in channel activation shown in Fig. 4. The presence of Zn2+ slows the deactivation rate by a factor of ∼8, corresponding to an increased stabilization of the open state by ∼2 kBT, which manifests itself by a 27.4 mV hyperpolarizing shift in V1/2. It is essentially as if the shift in the open-activated (O) state versus the closed (C) state was due to a slowing of the O→C rate with no change in the O←C rate.
Figure 5.

Metal bridge formation slows channel closure signifying stabilization of the open-activated channel conformation. (A) Representative current traces from an oocyte expressing Kv1.2-R294H-A351H-D352G-E353S channels recorded in the absence (left) and presence (right) of 1 μM Zn2+. Bath solution contained 50 mM K+ in order to visualize inward tail currents. (B) Plot of time constant for deactivation versus voltage for channels recorded in the absence (solid squares) or presence (open circles) of 1 μM Zn2+ (n = 7 each). Tail currents were fitted with a single exponential and the time constant of deactivation (τ, ms) plotted on a log scale against voltage.
Residue R294 Is Also in Close Proximity to D352 in the Open-Activated State
Residue D352 was mutated to histidine and the double mutant Kv1.2-R294H-D352H assayed by two-electrode voltage clamp for metal binding with Zn2+. As illustrated in Fig. 6, 1 μM Zn2+ significantly shifted the half maximal voltage of activation in the hyperpolarized direction by −15 mV from −10.8 ± 0.4 to −25.7 ± 0.6 mV (P < 0.05). However, 1 μM Zn2+ had no effect on the single Kv1.2-D352H mutant (Fig. 6 B and Table I). As performed previously with the Kv1.2 quadruple mutant, the Zn2+ affinity of the Kv1.2-R294H-D352H mutant was assessed by calculating the change in the voltage dependence of activation as a function of Zn2+ concentration. Fig. 6 C shows the calculated ΔV1/2 values plotted against Zn2+ concentrations ranging between 1 nM and 10 μM. The data were fitted with a rectangular hyperbola, which indicated that the half-maximal effective Zn2+ concentration was ∼470 nM. This high affinity suggests that the Zn2+ ion binds simultaneously to R294H and D352H in the open-activated channel conformation.
Figure 6.

R294H-D352H forms a lower affinity Zn2+ site but is still able to promote the activated conformational state. (A and B) Normalized G-V relationships from oocytes expressing (A) Kv1.2-R294H-D352H (n = 7) and (B) Kv1.2-D352H (n = 7) channels recorded in the absence (solid squares) and presence (open circles) of 1 μM Zn2+. (C) A plot of ΔV1/2 versus Zn2+ concentration in oocytes expressing Kv1.2-R294H-D352H channels, where ΔV1/2 is the difference between the fitted V1/2 values obtained in the absence and presence of varying concentrations of Zn2+. The data were fitted with a hyperbolic function (solid curve) with a half-maximal concentration of 470 nM (n = 5–10).
Charge Modified Mutations in Shaker and Kv1.2
It was inferred from previous MD simulations (Jogini and Roux, 2007) that residue R294 of Kv1.2 may form a salt bridge with acidic residues D352 and E353 adjacent to residue A351 in the pore domain (see Fig. 1). Removal of the acidic residues in Kv1.2 should induce a rightward shift in the voltage of half-maximal activation by eliminating the salt bridge formed between R294 of S4 and D352 and/or E353 of S5 in the activated state, thereby making it harder to open the channel. To ascertain this hypothesis, we mutated the acidic residues D352 and E353 of Kv1.2 to the equivalent neutral glycine and serine residues of Shaker. As shown in Table II, neutralization of these two residues did indeed induce a depolarizing shift in V1/2 by +9.4 mV (P < 0.05). Conversely, substituting the normally neutral G420 and S421 residues of Shaker by the negatively charged aspartic and glutamic acid residues of Kv1.2, we would expect to see the opposite effect, making it easier to open the channel by promoting salt bridge formation between Shaker residues R362 of S4 and 420D and/or 421E of S5. As predicted, when the equivalently positioned acidic residues were introduced into Shaker, we observed a significant hyperpolarizing shift in V1/2 by −8.5 mV (P < 0.05) in Shaker-G420D-S421E (Table II). Collectively, these data present further evidence to suggest that the N-terminal region of S4 in Kv1.2 reorients during membrane depolarization to within close proximity of those residues in the pore domain.
TABLE II.
Summary of V1/2 Changes Associated with Various Kv1.2 and Shaker Mutations
| n | V1/2 | ΔV1/2 from WT | |
|---|---|---|---|
| mV | mV | ||
| Shaker-WT | 10 | −17.2 ± 0.6 | |
| Shaker-G420D | 10 | −23.7 ± 0.8 | −6.5 |
| Shaker-S421E | 10 | −17.8 ± 0.7 | −0.6 |
| Shaker-G420D-S421E | 10 | −25.7 ± 0.9 | −8.5 |
| Kv1.2-WT | 10 | −14.8 ± 0.5 | |
| Kv1.2-D352G | 10 | −9.9 ± 0.6 | +4.9 |
| Kv1.2-E353S | 10 | −13.2 ± 0.7 | +1.6 |
| Kv1.2-D352G-E353S | 10 | −5.4 ± 0.6 | +9.4 |
Open-Activated Functional Conformation of Kv1.2 from MD
To predict the conformation of the activated channel that can satisfy the metal bridge formation, we have performed all-atom MD simulations. During all the MD simulations the channel structure fluctuates around a well-defined conformation, and as in previous simulations, the pore domain remains very stable (Jogini and Roux, 2007). The imposed harmonic distance restraints to satisfy the metal bridge between the position corresponding to the first charge in S4 and S5 from the adjacent subunit are rapidly satisfied in the early stage of the trajectory. Representative configurations of the channel from the MD simulations of the various mutants are shown in Fig. 7. The interaction of D352 with the metal is eliminated in the Kv1.2 quadruple mutant R294H-A351H-D352G-E353S (Fig. 7 A). In the Kv1.2 double mutant R294H-A351H, the carboxylate group of the side chain of D352 reaches over to coordinate the Zn2+ (Fig. 7 B). No significant coordination of the Zn2+ by E353 was observed in the MD simulations. The S4 helix with the metal bridge formed in the double mutant Kv1.2-R294H-D352H channel adopts a position similar to that of the quadruple mutant (Fig. 7 C).
Figure 7.

Comparison of the Kv1.2 crystal structure (Long et al., 2005) with instantaneous configurations taken from the MD simulations of the wild-type channel and mutant channels. (A) R294H-A351H-D352G-E353S (side and top view). (B) R294H-A351H (top view). (C) R294H-D352H (top view). Crystal structure is shown in gray. Mutant channel is represented as following: S1-cyan, S2-yellow, S3-red, S4-blue, pore-green, and Zn2+-violet. All the mutant structures were taken from the last snapshot of the MD trajectories. (See online supplemental material for the coordinates of the open activated Kv1.2 model obtained from R294H-A351H-D352G-E353S simulation in the presence of metal bridge).
For the sake of comparison, the x-ray structure of Kv1.2 (Long et al., 2005) is also shown in Fig. 7 (gray). To form the metal bridge with S5 of the adjacent subunit, the S4 helix must shift slightly in a counterclockwise direction and adopt a position that is more vertical than in the x-ray structure. Overall, a displacement of ∼7–8 Å relative to the x-ray structure is required at the level of the Cβ of R294. The voltage sensor fluctuates significantly, even in the presence of the metal bridge. To better appreciate the systematic shift in S4 position, the Cβ of residue 294 is shown in Fig. 8 for the MD simulation of wild-type channel (Jogini and Roux, 2007) (orange mesh), the x-ray structure of Kv1.2 (Long et al., 2005) (red sphere), and of the chimeric channel (Long et al., 2007) (green sphere), as well as the various mutants studied here. The multiple instantaneous configurations from the MD simulations of the various systems cover a range of positions for S4, which form a large cluster that has a width of ∼4–5 Å. The position corresponding to R294 in the x-ray structure of Kv1.2 sits at one extreme (bottom left) below the center of the cluster. The positions corresponding to the MD of the metal bridges (R294H-A351H, R294H-A351H-D352G-E353S, and R294H-D352H) overlap significantly (top right). The instantaneous snapshots taken from the MD of the wild-type Kv1.2 channel (Jogini and Roux, 2007) are distributed in between, ranging from the x-ray structure (2A79) up to the configurations corresponding to channels with engineered S4–S5 metal bridge. It is of particular significance that the position of Q290, the residue corresponding to the first charge in S4 in the x-ray structure of the chimera channel (2R9R), is also shifted away by 5.5 Å from the position of R294 in the Kv1.2 structure, in a direction similar to the instantaneous snapshots obtained from MD simulations of wild-type Kv1.2 (Jogini and Roux, 2007).
Figure 8.

Position of the first gating charge along the S4 segment (R294) in the open-activated conformation of Kv1.2 channel deduced from MD. The mesh plots encompass the volume occupied 60% of the time by the Cβ of residue 294 during the different MD simulations. The data shown includes the wild-type Kv1.2 channel (A, orange mesh), the Kv1.2 channel with R294H-A351H Zn2+ bridge (B, gray mesh), the Kv1.2 channel with the R294H-D352H Zn2+ bridge (C, cyan mesh), and the Kv1.2 channel with the R294H-D352H Zn2+ bridge and the substitution D352G-E353S (D, blue mesh). On the left is a side view from membrane and on the right is a side view from the pore. As a reference, the S4 segment (sky blue ribbon) and the pore domain (pale green ribbon) from the crystal structure of the Kv1.2 channel (PDB id 2A79) are shown. Three K+ ions are shown in the pore as orange spheres (left). The Cβ of residue 294 and the Cβ of residue 351 from the Kv1.2 x-ray structure are shown as red and magenta spheres, respectively. The Cβ of the residue at the corresponding position in the x-ray structure of the Kv1.2-Kv2.1 chimera PDB id 2R9R (Q290) is shown as a green sphere (assuming the same position for the pore domain). To record the probability density of the Cβ of residue 294 from the MD, the pore domain (S5–S6) was oriented with respect to the x-ray structure for every snapshot, followed by increments of 90° rotations to superimpose the voltage sensor modules of the four subunits (Jogini and Roux, 2007), yielding four data points per snapshot. For the wild-type channel, one snapshot every 5 ps is included (total 20,000 points), for each mutant one snapshot every 2 ps from two trajectories is included (total 10,000 points).
DISCUSSION
The first atomic view of a voltage-gated K+ channel was provided by the x-ray structure of the KvAP channel (Jiang et al., 2003). Although inconsistencies of this structure with other data were noted very early on (Cohen et al., 2003; Gandhi et al., 2003; Laine et al., 2003, 2004), the view that the KvAP x-ray structure was distorted into a nonnative conformation was recognized only after the determination of the x-ray structure of Kv1.2 (Lee et al., 2005; Long et al., 2005). As a consequence, the recent history with KvAP and Kv1.2 might lead to the view that crystal structures are either detectably distorted (like KvAP), or are otherwise representative of the native conformation in all respects (like Kv1.2). The reality is probably more complex. In fact, assessing the functional significance of any protein conformation captured in a crystal environment is always a fundamental issue that must be addressed carefully.
The four highly conserved arginine residues along S4 are largely responsible for the gating charge controlling the voltage activation of the channel (Aggarwal and MacKinnon, 1996; Seoh et al., 1996). Accessibility data show that membrane depolarization causes the S4 segment to move toward the extracellular side of the membrane (Larsson et al., 1996). In particular, the position of the first gating charge near the N terminus of S4 is an important marker of the conformational change that underlies voltage gating. This first charge is known to alternate between two locations, exposed to the extracellular side of the membrane in the open-activated state of the channel, and displaced toward the intracellular side of the membrane in the closed resting channel state (Starace and Bezanilla, 2001, 2004; Ruta et al., 2005). To refine our knowledge of the open-activated state of Kv1.2, three different sources of information were taken into consideration: (1) the crystallographic x-ray structure (2A79), (2) electrophysiological measurements of Kv1.2 mutant channels, and (3) MD simulations of all the systems studied experimentally.
A first step is to obtain electrophysiological data leading to unambiguous interpretation. In Fig. 2, the results for the double mutant R294H-A351H are shown. In the absence of Zn2+, the mutant channels behave essentially as wild type. However, in the presence of Zn2+ the ionic currents of the mutant channels are significantly reduced, an effect that is not observed in the wild-type Kv1.2 channel. In the presence of Cd2+ the G-V relationship is shifted to a more negative voltage without any reduction of the current. This phenotype is qualitatively similar to the R362H-A419H bridge engineered in Shaker (Laine et al., 2003). Differences between the sequence of Shaker and Kv1.2 in this region could be responsible for the markedly different behavior. As shown in Fig. 1, two acidic residues near A351 at the extracellular end of S5 in Kv1.2 (D352 and E353) are neutral residues in Shaker (G420 and S421). The Zn2+ cation can be effectively coordinated by the nitrogen atom of histidine as well as by carboxylate (Alberts et al., 1998; Jain and Jayaram, 2007), but not so for Cd2+, which prefers to bind softer ligands such as sulfur (Rulisek and Vondrasek, 1998). A reasonable hypothesis is that the negatively charged side chains, somehow, interfere with the activated position of S4 in the presence of the His-Zn2+-His metal bridge, perhaps promoting some inactivation of the pore. In fact, the double mutant R294H-A351H displays a slight shift of the G-V at very low Zn2+ concentration before the current starts to collapse (see Fig. 2 C). This suggests that activation of the voltage sensor and inactivation of the pore involve two different processes, which are not affected in the same way by the formation of the bridge. Although more work would be required to ascertain and characterize this phenomenon in wild-type Kv1.2, this is not our main interest here. Rather, we try to recover the physical environment of metal coordination engineered previously in Shaker by substituting the neutral glycine and serine residues of Shaker back into Kv1.2. As shown in Fig. 4, the His-His metal bridge behaves essentially as the corresponding bridge in Shaker once the two acidic residues have been neutralized. In particular, there is a shift of −27.4 mV in the G-V, indicating that the open-activated state of functional channels is stabilized in the presence of Zn2+. In Shaker, the corresponding metal bridge caused a shift of −40 mV (Laine et al., 2003). The dissociation constant for Zn2+ is 360 nM, which is quantitatively similar to the value previously reported for Shaker (400 nM) (Laine et al., 2003). The high apparent affinity is consistent with the conclusion that the Zn2+ ion binds simultaneously to the two histidine side chains (Kiefer and Fierke, 1994; Loland et al., 1999; Norregaard et al., 2000), confirming that these positions are within atomic distance of each other in the open-activated state. Furthermore, analysis of the deactivation rate from tail currents demonstrates that the observed effect clearly arises from a stabilization of the open-activated state promoted by the formation of the metal bridge, as opposed to a destabilization of the closed state. The hyperpolarizing shift in the activation (Fig. 4) and the slowed deactivation rate (Fig. 5) are both consistent with an open-activated conformation that sits in a free energy well deepened by ∼2 kBT upon formation of the His-His metal bridge. By comparison, the activation rate of the quadruple Kv1.2 mutant (R294H-A351H-D352G-E353S) slows down by less than a factor of 2 in the presence of Zn2+ (see Fig. S1, available at http://www.jgp.org/cgi/content/full/jgp.200809962/DC1), again indicating that the closed state of the channel is not destabilized by the formation of the metal bridge. The structural interpretation of the effect of Zn2+ as stabilizing a functional open-activated state is therefore unambiguous.
From a functional point of view, the open-activated state of the Kv1.2 channel is structurally stabilized by the formation of a His-His metal bridge engineered between position 294 in S4 and position 351 in S5. The metal bridge promoting the activated open state is not unique, as it is also possible to engineer a bridge between R294H and D352H. The voltage shift in the G-V is slightly smaller, −15 mV, which suggests that this metal bridge might be promoting a conformation that does not match the open-activated state as well as the R294H-A351H bridge. In the x-ray structure of Kv1.2, the distance between those residues is on the order of 13.7 Å. While this distance seems large, the implications for the formation of a His-His metal bridge must be ascertained carefully. In crystal structures of metal binding proteins the Cα-Cα distance between residues involved in a His-His metal bridge can be as short as 5–6 Å (Alberts et al., 1998). However, in a fully extended position of the histidine side chain, it is also possible to form a bridge while keeping the backbone Cα separated by a distance of 9–10 Å. Therefore, whether the formation of a R294H-A351H metal bridge stabilizing the open-activated state is truly inconsistent with the relative position of S4, with respect to the pore domain observed in the x-ray structure, needs to be clarified. Simple arguments based on backbone-to-backbone distances are actually too simplistic, as one must also consider the relative orientation of the S4 and S5 helices, as well as steric clashes with neighboring residues. Without further information, it is nearly impossible to assess the relative significance of the x-ray structure and of the functional data, and further advance our knowledge of the open-activated state.
Atomistic MD simulations of the Kv1.2 channel and the various mutants studied experimentally can help shed some light on this issue. In a previous study of the Kv1.2 channel embedded in a lipid membrane (Jogini and Roux, 2007), it was reported that residue R294 of S4 was shifting toward residue A351 of S5. Furthermore, intersubunit salt bridges between the side chains of R294/R297 and D352 near the extracellular end of S5 from the adjacent subunit formed spontaneously during the simulation (Jogini and Roux, 2007). Similar observations were made in an independent MD simulation of Kv1.2 based on a different model (Treptow and Tarek, 2006). Following the shift of the Cβ corresponding to the first side chain atom at position 294 is most informative. As shown in Fig. 8, the position of 294 in the simulation of the wild-type Kv1.2 form a wide cluster that is displaced roughly by 7.5 Å compared with the x-ray structure. Although there is significant overlap with the configurations generated with the wild-type Kv1.2 channels, the simulations in which the R294H-Zn2+-A351H bridge was imposed via a harmonic restraint form a more well-defined cluster. A slightly more compact cluster of positions is obtained when the metal bridge is simulated in the background of the Shaker-like neutralized D352G-E353S mutations (Fig. 7 D, blue mesh). Systematically, the shift in the position of the Cβ of 294 in S4 relative to the x-ray structure is observed for all simulations. Interestingly, the x-ray structure of the Kv1.2-Kv2.1 chimera channel (PDB id 2R9R) in the presence of a lipid environment displays a shift of ∼5.5 Å in a similar direction (the Cβ of Q290 is indicated by a green sphere). The latter observation highlights the importance of a dynamic lipid membrane environment on the conformation of Kv channels, a point previously emphasized in crystallographic studies of the KvAP and Kv1.2 channels (Lee et al., 2005).
Before concluding this discussion, we wish to summarize the issue one more time for the sake of clarity. The residues R294 and A351 are too far apart in the Kv1.2 crystal structure to allow the formation of the metal bridge that was previously engineered in Shaker. Only four possible reasons could explain this observation: (1) the Kv1.2 channel is slightly different from Shaker; (2) the formation of the Zn2+ bridge destabilizes the closed state of Kv1.2; (3) the metal bridge stabilizes a somewhat distorted open-activated state; (4) the conformation seen in the Kv1.2 crystal structure does not correspond precisely to that of the open-activated state of a Kv channel. Several factors argue in favor of the latter explanation.
The possibility that Shaker and Kv1.2 are different (1) can be readily eliminated. We were able to engineer the same S4–S5 high affinity metal bridge in a charge-neutralized mutant of Kv1.2 that worked previously in Shaker, thus ruling out the possibility that those channels display any fundamental differences. The possibility that the metal bridge stabilizes the closed state (2) is not consistent with the electrophysiological data. As demonstrated by the rate of activation and the analysis of tail currents, the bridge is clearly stabilizing the open-activated state, as opposed to destabilizing the closed state. The possibility that the metal bridge actually stabilizes a somewhat distorted open-activated state (3) deserves to be addressed carefully. First, it is important to establish the magnitude of the distortion. MD simulations of the mutant Kv1.2 channels indicate that a shift of ∼7–8 Å is needed to allow the formation of the His-His metal bridge, yielding an unambiguous structural constraint. Second, it is important to recall that high affinity metal bridges (nM) are typically occurring in undistorted structures like active site of enzymes (Kiefer and Fierke, 1994; Loland et al., 1999; Norregaard et al., 2000). To retain the high Zn2+ affinity, the flexibility of the voltage sensor would need to be such that very little energy cost would be incurred by the structural distortion. Furthermore, even in the case of a very freely flexible sensor with no energy cost, the loss of conformational entropy upon formation of the metal bridge would act to decrease the high Zn2+ affinity. In this context, the possibility that the metal bridge might actually stabilize some open channel conformation distorted by 7–8 Å at nM Zn2+ concentration seems very unlikely.
This leaves the only possible alternative explanation (4): the crystal structure of Kv1.2 does not correspond precisely to that of the open-activated state. According to the structural constraint deduced from MD, a 7–8 Å shift is needed to allow the formation of the His-His metal bridge. It is particularly noteworthy that a similar shift—in the same direction—was already observed to occur spontaneously in unbiased MD simulations of the WT channel, without any biasing constraints between R294 and A351 (Jogini and Roux, 2007), and that a similar shift of about 4–5 Å—in the same direction—is also seen in the recent x-ray structure of the Kv1.2-Kv2.1 chimera in which R294 is a Q (Long et al., 2007). This analysis gives us the confidence to conclude that the position and orientation of R294 in S4 in the activated and open functional state of the Kv1.2 channel in a fluid lipid membrane is shifted by ∼7–8 Å and rotated by ∼37° counterclockwise along its main axis relative to the x-ray structure 2A79. As a consequence, the first two arginines of S4 are exposed to the water molecules near the bulk-membrane interface, or interact with the lipid headgroups as shown in MD simulations (Treptow and Tarek, 2006; Jogini and Roux, 2007). Consequently, the picture emerging from the present results contrasts with the conclusion initially drawn from the x-ray structure that those side chains pointed directly into the lipid hydrocarbon (Long et al., 2005).
The position of S4 in the open-activated state deduced here on the basis of functional data and MD simulations can be put within the context of a proposed atomic model of the voltage sensor in the resting-closed state (Yarov-Yarovoy et al., 2006; Pathak et al., 2007), which is consistent with a wide range of experimental constraints (Starace and Bezanilla, 2004; Ahern and Horn, 2005; Chanda et al., 2005; Posson et al., 2005; Tombola et al., 2006; Campos et al., 2007). According to the resting state model, the S4 segment is configured such that the first charge R294 points toward S1 and S2 (Campos et al., 2007; Pathak et al., 2007). In this conformation, a salt bridge is formed between R294 and E226 in S2, previously identified experimentally using second site charge reversal mutations in the homologous Shaker channel where it corresponds to E283 (Tiwari-Woodruff et al., 1997; Tiwari-Woodruff et al., 2000). To reach this proposed resting state starting from the Kv1.2 crystal structure, the helix S4 must undergo a counterclockwise helical screw rotation of ∼180° along its main axis. However, a more modest displacement and rotation of S4 would be required if the conformational transition started from the functional open-activated conformation proposed here. In fact, when starting from the x-ray structure the charges R294 and R297 must initially move up toward the extracellular side before coming down in between S1 and S2, a process that presumably would not be accelerated by membrane hyperpolarization. On a speculative note, this raises the interesting possibility that the crystal structure might have captured the channel in a conformation related to a zero-voltage inactivated state of the voltage sensor domain. This functional state, which differs from the resting (down) state and the activated (up) state, manifests its existence as a history-dependent hysteresis in measured gating currents (Bezanilla et al., 1982; Olcese et al., 1997; Kuzmenkin et al., 2004).
Conclusion
The present analysis provides evidence that, in the “functional” open-activated state of Kv1.2, the S4 helix is actually shifted by ∼7–8 Å and rotated counterclockwise by ∼37° along its main axis relative to its position in the crystal structure. As a result, the helix sits in a more upright orientation relative to the membrane. While the overall topology of Kv channels in the open-activated state is not in question (Laine et al., 2003; Long et al., 2005), the magnitude of the conformational shift deduced here is certainly not negligible and should be taken into account in efforts aimed at relating channel structure to function. The present findings highlight the difficulties in trying to grasp the genuine molecular realities of Kv channels. Details from any experiments and computations have to be interpreted with caution, in large part because the voltage sensor is dynamic, flexible, and highly sensitive to its environment. The wide range of dynamical fluctuations also partly explains the contrasting results concerning the magnitude of the conformational change underlying voltage gating (Chanda et al., 2005; Posson et al., 2005; Ruta et al., 2005). The complexity of Kv channels and voltage sensors argues in favor of a perspective built from a broad consensus of all available information from structural, spectroscopic, biophysical, functional, and computational approaches.
Supplementary Material
Acknowledgments
We thank Prof. Steve Goldstein for his gracious support of A. Lewis through grant GM051851 from the National Institutes of Health (NIH), and Prof. Francisco Bezanilla for valuable discussions.
This work was supported by grant GM062342 from the NIH.
Olaf S. Andersen served as editor.
References
- Aggarwal, S.K., and R. MacKinnon. 1996. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 16:1169–1177. [DOI] [PubMed] [Google Scholar]
- Ahern, C.A., and R. Horn. 2005. Focused electric field across the voltage sensor of potassium channels. Neuron. 48:25–29. [DOI] [PubMed] [Google Scholar]
- Alberts, I.L., K. Nadassy, and S.J. Wodak. 1998. Analysis of zinc binding sites in protein crystal structures. Protein Sci. 7:1700–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beglov, D., and B. Roux. 1994. Finite representation of an infinite bulk system: solvent boundary potential for computer simulations. J. Chem. Phys. 100:9050–9063. [Google Scholar]
- Bezanilla, F. 2000. The voltage sensor in voltage-dependent ion channels. Physiol. Rev. 80:555–592. [DOI] [PubMed] [Google Scholar]
- Bezanilla, F., R.E. Taylor, and J.M. Fernandez. 1982. Distribution and kinetics of membrane dielectric polarization. 1. Long-term inactivation of gating currents. J. Gen. Physiol. 79:21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks, B.R., R.E. Bruccoleri, B.D. Olafson, D.J. States, S. Swaminathan, and M. Karplus. 1983. CHARMM: a program for macromolecular energy minimization and dynamics calculations. J. Comput. Chem. 4:187–217. [Google Scholar]
- Campos, F.V., B. Chanda, B. Roux, and F. Bezanilla. 2007. Two atomic constraints unambiguously position the S4 segment relative to S1 and S2 segments in the closed state of Shaker K channel. Proc. Natl. Acad. Sci. USA. 104:7904–7909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda, B., O.K. Asamoah, R. Blunck, B. Roux, and F. Bezanilla. 2005. Gating charge displacement in voltage-gated ion channels involves limited transmembrane movement. Nature. 436:852–856. [DOI] [PubMed] [Google Scholar]
- Cohen, B.E., M. Grabe, and L.Y. Jan. 2003. Answers and questions from the KvAP Structures. Neuron. 39:395–400. [DOI] [PubMed] [Google Scholar]
- Elinder, F., P. Arhem, and H.P. Larsson. 2001. Localization of the extracellular end of the voltage sensor S4 in a potassium channel. Biophys. J. 80:1802–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, C.S., E. Clark, E. Loots, A. Pralle, and E. Isacoff. 2003. The orientation and molecular movement of a K+ channel voltage-sensing domain. Neuron. 40:515–525. [DOI] [PubMed] [Google Scholar]
- Hille, B. 2001. Ion Channels of Excitable Membranes. Third edition. Sinauer, Sunderland, MA. 814 pp.
- Jain, T., and B. Jayaram. 2007. Computational protocol for predicting the binding affinities of zinc containing metalloprotein-ligand complexes. Proteins. 67:1167–1178. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B. Chait, and R. MacKinnon. 2003. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Jogini, V., and B. Roux. 2007. Dynamics of the Kv1.2 voltage-gated K+ channel in a membrane environment. Biophys. J. 93:3070–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen, W.L., J. Chandrasekhar, J.D. Madura, R.W. Impey, and M.L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- Kiefer, L.L., and C.A. Fierke. 1994. Functional characterization of human carbonic anhydrase II variants with altered zinc binding sites. Biochemistry. 33:15233–15240. [DOI] [PubMed] [Google Scholar]
- Klauda, J.B., B.R. Brooks, and R.W. Pastor. 2006. Dynamical motions of lipids and a finite size effect in simulations of bilayers. J. Chem. Phys. 125:144710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmenkin, A., F. Bezanilla, and A.M. Correa. 2004. Gating of the bacterial sodium channel, NaChBac: voltage-dependent charge movement and gating currents. J. Gen. Physiol. 124:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine, M., M.C.A. Lin, J.P.A. Bannister, W.R. Silverman, A.F. Mock, B. Roux, and D.M. Papazian. 2003. Atomic proximity between S4 segment and pore domain in Shaker potassium channels. Neuron. 39:467–481. [DOI] [PubMed] [Google Scholar]
- Laine, M., D.M. Papazian, and B. Roux. 2004. Critical assessment of a proposed model of Shaker. FEBS Lett. 564:257–263. [DOI] [PubMed] [Google Scholar]
- Larsson, H.P., O.S. Baker, D.S. Dhillon, and E.Y. Isacoff. 1996. Transmembrane movement of the shaker K+ channel S4. Neuron. 16:387–397. [DOI] [PubMed] [Google Scholar]
- Lee, S.Y., A. Lee, J. Chen, and R. MacKinnon. 2005. Structure of the KvAP voltage-dependent K+ channel and its dependence on the lipid membrane. Proc. Natl. Acad. Sci. USA. 102:15441–15446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, Y.L., and C. Lim. 2004. Factors governing the protonation state of Zn-bound histidine in proteins: a DFT/CDM study. J. Am. Chem. Soc. 126:2602–2612. [DOI] [PubMed] [Google Scholar]
- Loland, C.J., L. Norregaard, and U. Gether. 1999. Defining proximity relationships in the tertiary structure of the dopamine transporter. Identification of a conserved glutamic acid as a third coordinate in the endogenous Zn2+-binding site. J. Biol. Chem. 274:36928–36934. [DOI] [PubMed] [Google Scholar]
- Long, S.B., E.B. Campbell, and R. Mackinnon. 2005. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 309:897–903. [DOI] [PubMed] [Google Scholar]
- Long, S.B., X. Tao, E.B. Campbell, and R. MacKinnon. 2007. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 450:376–382. [DOI] [PubMed] [Google Scholar]
- MacKerell, A.D., Jr., B. Brooks. C.L. Brooks III, L. Nilsson, Y. Won, B. Roux, and M. Karplus. 1998. a. CHARMM: the energy function and the program. In The Encyclopedia of Computational Chemistry. P.v.R. Schleter, N.L. Allinger, T. Clark, J. Gasteiger, P.A. Kollman, H.F. Schaefer III, and P.R. Schreiner, editors. John Wiley & Sons, Chichester, UK. 271–277.
- MacKerell, A.D.J., D. Bashford, M. Bellot, R.L. Dunbrack, J.D. Evanseck, M.J. Field, S. Fischer, J. Gao, H. Guo, D.J.-M.S. Ha, L. Kuchnir, K. Kuczera, F.T.K. Lau, C. Mattos, S. Michnick, T. Ngo, D.T. Nguyen, B. Prodhom, W.E. Reiher III, B. Roux, M. Schlenkrich, J. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, and M. Karplus. 1998. b. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Norregaard, L., I. Visiers, C.J. Loland, J. Ballesteros, H. Weinstein, and U. Gether. 2000. Structural probing of a microdomain in the dopamine transporter by engineering of artificial Zn2+ binding sites. Biochemistry. 39:15836–15846. [DOI] [PubMed] [Google Scholar]
- Olcese, R., R. Latorre, L. Toro, F. Bezanilla, and E. Stefani. 1997. Correlation between charge movement and ionic current during slow inactivation in Shaker K+ channels. J. Gen. Physiol. 110:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak, M.M., V. Yarov-Yarovoy, G. Agarwal, B. Roux, P. Barth, S. Kohout, F. Tombola, and E.Y. Isacoff. 2007. Closing in on the resting state of the Shaker K+ channel. Neuron. 56:124–140. [DOI] [PubMed] [Google Scholar]
- Posson, D.J., P. Ge, C. Miller, F. Bezanilla, and P.R. Selvin. 2005. Small vertical movement of a K+ channel voltage sensor measured with luminescence energy transfer. Nature. 436:848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rulisek, L., and J. Vondrasek. 1998. Coordination geometries of selected transition metal ions (Co2+, Ni2+, Cu2+, Zn2+, Cd2+, and Hg2+) in metalloproteins. J. Inorg. Biochem. 71:115–127. [DOI] [PubMed] [Google Scholar]
- Ruta, V., J. Chen, and R. MacKinnon. 2005. Calibrated measurement of gating-charge arginine displacement in the KvAP voltage-dependent K+ channel. Cell. 123:463–475. [DOI] [PubMed] [Google Scholar]
- Schlenkrich, M.J., J. Brickmann, A.D.J. MacKerell, and M. Karplus. 1996. An empirical potential energy function for phospholipids: criteria for parameters optimization and applications. In Biological Membranes. A Molecular Perspective from Computation and Experiment. Birkhauser, Boston. 31–81.
- Seoh, S., D. Sigg, D. Papazian, and F. Bezanilla. 1996. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 16:1159–1167. [DOI] [PubMed] [Google Scholar]
- Sigworth, F.J. 1993. Voltage gating of ion channels. Q. Rev. Biophys. 27:1–40. [DOI] [PubMed] [Google Scholar]
- Soler-Llavina, G.J., T.H. Chang, and K.J. Swartz. 2006. Functional interactions at the interface between voltage-sensing and pore domains in the Shaker K(v) channel. Neuron. 52:623–634. [DOI] [PubMed] [Google Scholar]
- Starace, D.M., and F. Bezanilla. 2001. Histidine scanning mutagenesis of basic residues of the S4 segment of the shaker K+ channel. J. Gen. Physiol. 117:469–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starace, D.M., and F. Bezanilla. 2004. A proton pore in a potassium channel voltage sensor reveals a focused electric field. Nature. 427:548–553. [DOI] [PubMed] [Google Scholar]
- Tiwari-Woodruff, S.K., C.T. Schulteis, A.F. Mock, and D.M. Papazian. 1997. Electrostatic interactions between transmembrane segments mediate folding of Shaker K+ channel subunits. Biophys. J. 72:1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari-Woodruff, S.K., M.A. Lin, C.T. Schulteis, and D.M. Papazian. 2000. Voltage-dependent structural interactions in the Shaker K+ channel. J. Gen. Physiol. 115:123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombola, F., M.M. Pathak, P. Gorostiza, and E.Y. Isacoff. 2006. The twisted ion-permeation pathway of a resting voltage-sensing domain. Nature. 445:546–549. [DOI] [PubMed]
- Treptow, W., and M. Tarek. 2006. Environment of the gating charges in the Kv1.2 Shaker potassium channel. Biophys. J. 90:L64–L66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy, V., D. Baker, and W.A. Catterall. 2006. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K+ channels. Proc. Natl. Acad. Sci. USA. 103:7292–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta, W.N., T. Hoshi, and R.W. Aldrich. 1994. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J. Gen. Physiol. 103:321–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}