

Summary
Disulfide bond formation occurs in secreted proteins in Escherichia coli when the disulfide oxidoreductase DsbA, a soluble periplasmic protein, nonspecifically transfers a disulfide to a substrate protein. The catalytic disulfide of DsbA is regenerated by the inner membrane protein DsbB. To help identify the specificity determinants in DsbB and to understand the nature of the kinetic barrier preventing direct oxidation of newly secreted proteins by DsbB, we imposed selective pressure to find novel mutations in DsbB that would function to bypass the need for the disulfide carrier DsbA. We found a series of mutations localized to a short horizontal α-helix anchored near the outer surface of the inner-membrane of DsbB that eliminated the need for DsbA. These mutations changed hydrophobic residues into nonhydrophobic residues. We hypothesize that these mutations may act by decreasing the affinity of this α-helix to the membrane. The DsbB mutants were dependent on the disulfide oxidoreductase DsbC, a soluble periplasmic thiol-disulfide isomerase, for complementation. DsbB is not normally able to oxidize DsbC, possibly due to a steric clash that occurs between DsbC and the membrane adjacent to DsbB. DsbC must be in the reduced form to function as an isomerase. In contrast, DsbA must remain oxidized to function as an oxidizing thiol-disulfide oxidoreductase. The lack of interaction that normally exists between DsbB and DsbC appears to provide a means to separate the DsbA-DsbB oxidation pathway and the DsbC-DsbD isomerization pathway. Our mutants in DsbB may act by redirecting oxidant flow to take place through the isomerization pathway.
Keywords: DsbB, disulfide bond formation, DsbA, DsbC, protein folding
Introduction
Disulfide bonds are an essential post-translational modification in many secreted proteins.1 In prokaryotes, DsbA and DsbB facilitate the introduction of disulfide bonds into newly translocated proteins in the bacterial periplasm.2; 3; 4 DsbA is a small, non-selective disulfide oxidoreductase and is a member of the thioredoxin superfamily of proteins.5 DsbA has an extremely oxidizing disulfide that acts to directly oxidize cysteines to form disulfides in secreted proteins. Null mutants in DsbA show a severe and general defect in disulfide bond formation, indicating that it is essential for protein thiol oxidation.4 For DsbA to be catalytic, it must be reoxidized. This is done by DsbB, a quinone reductase that specifically oxidizes DsbA.3 DsbB is a 20 kD inner membrane protein containing four essential cysteines, two in each of its two periplasmic domains. Cysteines 41/44 form a disulfide bond in response to quinone reduction.6; 7 This disulfide is thought to be transferred to the cysteine pair 104/130, which forms a disulfide that is donated to DsbA.8 DsbB serves as a link between the electron transport chain and oxidative protein folding.9 Quinones reduced by DsbB are reoxidized by the action of cytochrome bd and cytochrome bo oxidase. In Escherichia coli, oxygen functions as the principal electron acceptor for the electron transport system.
DsbB appears to be a very specific oxidoreductase, with DsbA serving as its only known physiological substrate. In particular, DsbB is incapable of directly oxidizing the wide variety of substrates that can be oxidized by DsbA. However, DsbB can oxidize other thioredoxin-fold proteins, including eukaryotic protein disulfide isomerase expressed in E. coli and thioredoxin, which have been artificially exported to the periplasm.10; 11 DsbA, in contrast, is a relatively nonspecific oxidoreductase; it oxidizes many E. coli substrate proteins, including OmpA, PhoA, FliC, DppA, OmpF, PhoE, HisJ, OsmY, ModA, YggN, YodA, RcsF, MdoG, YbeJ, Rna, and LivK, as well as a wide variety of eukaryotic proteins expressed in E. coli.12; 13
DsbA is the strongest disulfide oxidoreductase known, and is thought to act by sequentially oxidizing cysteines on proteins as they are secreted, forming both correct and incorrect disulfides.14; 15 Improper disulfide bonds formed by DsbA are corrected via a disulfide isomerization pathway.14; 16 DsbC, a 25 kD thioredoxin-related periplasmic protein, is thought to be the principle disulfide isomerase in E. coli. It consists of two domains: an N-terminal dimerization domain and a C-terminal thioredoxin-like domain. The thioredoxin-like domain contains an active site CXXC redox motif, which can be reversibly oxidized and reduced.17 In contrast to DsbA, which is maintained in an oxidized form in vivo, DsbC is maintained in the reduced form so that its thiol can attack disulfide bonds. The attack of a cysteine on the incorrect disulfide in the misfolded protein forms a mixed disulfide with DsbC. The mixed disulfide can then be attacked by another free thiol in the misfolded protein to form the correct disulfide and release a reduced and active DsbC. Alternatively, the free thiol in the DsbC CXXC active site may resolve the mixed disulfide between DsbC and the misfolded protein. This resolution of the mixed disulfide would result in disulfide bond formation in the DsbC active site, which may be used to oxidize the misfolded protein into a native conformation.
Dimerization appears to be important for DsbC and other protein disulfide isomerases.16 DsbC forms a V-shaped homodimer, with the two active sites facing the interior of the V. This conformation may provide a high local concentration of cysteines in the vicinity of a folding polypeptide, thereby facilitating the disulfide isomerization reaction. DsbC is maintained in the reduced state via the membrane protein DsbD,1 which gains its reducing equivalents from the cytoplasmic reductant thioredoxin and transfers them to DsbC.18
One interesting question is what keeps the DsbA-DsbB oxidation pathway separated from the DsbC-DsbD isomerization pathway. The two pathways are opposing; if components of the oxidation pathway could directly oxidize components of the isomerization pathway and vice versa, a futile cycle would occur.19; 20 To explore the nature of the kinetic barrier separating DsbB from other proteins, including both DsbA substrates and DsbC, we decided to select for DsbB mutants capable of rescuing the phenotypes of a dsbA null mutation. DsbB is capable of some oxidation of DsbC in vitro, although the reaction is >1000 fold slower than the oxidation of DsbA by DsbB and >2000 fold slower than the reduction of DsbC by DsbD.21
Selections for mutants of enzymes with altered substrate specificity have been frequently used in the past.22; 23; 24; 25 In general, these mutants make the enzymes less specific for their substrates. Examples include mutants of carotene synthase that make it able to function with alternative precursor metabolites, β-glucouronidase mutants that allow it to catalyze the breakdown of β-galactoside 500 times more efficiently than wild type, and mutants of the restriction enzyme Eco RI that allow it to recognize additional DNA targets.22; 23; 24 Identification of these mutants often provides insights into the specificity determinants of the enzymes and can help elucidate their catalytic mechanism.
Protein disulfide isomerase (PDI) is thought to serve as the direct donor of disulfides to secreted proteins in eukaryotes, and is thus comparable to DsbA in prokaryotes.26 In eukaryotes, Ero1p, reoxidizes PDI, fulfilling a role similar to that played by DsbB.26 Sevier and Kaiser recovered mutants in Ero1p that directly oxidize substrate proteins. These mutants could circumvent intrinsic regulatory mechanisms that normally act by preventing random target oxidation.27 Their success encouraged our efforts to isolate similar mutations in DsbB.
Our goal was to identify DsbB-mediated oxidation mechanisms in the environment lacking DsbB’s physiological substrate-DsbA. We found a short region in DsbB that, when mutated, can restore disulfide bond formation processes in E. coli in the absence of DsbA. The DsbB mutants are DsbC-dependent, suggesting that they act via oxidizing DsbC. Our work suggests that part of the substrate specificity of DsbB is due to a hydrophobic membrane-localized α-helix in DsbB that interferes with its ability to oxidize DsbC.
Results and Discussion
Isolating DsbB mutants that bypass the need for DsbA
In prokaryotes, DsbA oxidizes substrate proteins, whereas DsbB specifically oxidizes DsbA. We decided to isolate DsbB mutants that bypass the need for DsbA with the idea that their analysis would give us information about the determinants of substrate specificity within the DsbA-DsbB disulfide catalytic system.
To select for DsbB mutants that can bypass the need for DsbA, we introduced a randomly mutagenized dsbB-containing plasmid into JP221 (ΔdsbA ΔdsbB) and selected for cadmium resistance. ΔdsbA andΔdsbB strains are unable to grow on plates containing 15 μM cadmium, whereas wild-type strains can grow on plates containing up to 400 μM cadmium. The proposed rational for the cadmium sensitivity of dsb− strains is as follows: in the absence of DsbB or DsbA, periplasmic proteins contain multiple free thiols that can tightly bind cadmium, thus inhibiting proper folding. dsb+ strains are much more cadmium resistant (CdR), presumably because the free thiols are rapidly oxidized by the disulfide bond formation machinery.28; 29 Following targeted mutagenesis of the dsbB gene present on the plasmid, we isolated approximately 200 colonies resistant to 15 μM CdCl2 out of approximately 25,000 colonies screened.
To exclude mutants that were CdR for reasons unrelated to the restoration of disulfide bond formation, we tested CdR isolates for their ability to restore motility, a phenotype characteristic of dsb+ strains. To be motile, E. coli must properly assemble its bacterial flagella, and to do this, a critical disulfide in the flagellar component FlgI must be introduced. Thus, Δdsb strains are nonmotile.30 Of our 200 CdR clones, 170 had restored motility, suggesting that in the majority of our mutants, disulfide bond forming capabilities were at least partially restored.
Mutations map to short membrane-localized α-helix
Plasmid DNA was prepared from these clones and their DNA sequences were determined. All the plasmids contained mutations in the dsbB gene and 55% contained more than one mutation. To get an initial indication as to which regions of DsbB are associated with restoration of CdR and motility in a ΔdsbA background, we aligned all the mutant sequences and looked for common mutations. Interestingly, 90% of mutant sequences contained at least one of the following mutations: F110S, W113R, L114P, K118T, and W119G, often in the presence of other mutations. Of these, F110S, L114P, K118T, and W119G were also found in isolation, strongly suggesting that each of these dsbB mutations alone is sufficient to allow DsbB to bypass the need for DsbA. To show that plasmids containing each of these mutations are sufficient to restore CdR and motility to a ΔdsbA ΔdsbB strain, we retransformed the mutated plasmids into a freshΔdsbA ΔdsbBstrain that had not been subjected to cadmium resistance screening and showed them to be as CdR and as motile as the original isolate. This eliminates the possibility that an additional chromosomal mutation is required for the suppression phenotype. W113R was isolated twice—both times with additional mutations, once with R109P and P121Q, and once with V108D. We have not yet excluded the possibility that these mutations contribute to the ability of W113R to rescue native phenotypes. Interestingly, all of the mutations that are sufficient to restore CdR and motility map to a short membrane-localized α-helix that is horizontally oriented near the outer surface of the inner membrane. It is thought that this α-helix helps to provide an additional topological constraint to the structure of DsbB.31 This α-helix divides a large loop that was formerly thought to be periplasmically localized into two shorter periplasmic loops, each of which contain an essential, catalytically active cysteine residue.8 These two cysteines are C104 and C130.
DsbB mutants require DsbC to bypass DsbA
We considered two possible mechanisms of action of these mutants: (1) they are oxidizing newly secreted proteins indirectly via a soluble periplasmic thiol-disulfide oxidoreductase intermediate that replaces the role of DsbA, or (2) they directly interact with newly secreted proteins and oxidize them. To test these possibilities, we decided to first check if previously characterized oxidoreductases are required for the suppression. We reasoned that freely soluble and previously identified thiol-disulfide oxidoreductases may be good candidates because they are already involved in thiol-disulfide exchange and as such they could potentially serve as an intermediate allowing thiol-disulfide exchange between DsbB and oxidatively folding proteins. Identification of a protein or proteins required for the suppression would therefore argue against the possibility that the mutant DsbBs were directly oxidizing folding proteins. The most promising candidates for the DsbA substitutes are DsbC and DsbG. These proteins, like DsbA, are periplasmically located thioredoxin-like dithiol-disulfide oxidoreductases, have very similar oxidizing redox potentials, and have the same type of substrates, namely, partially folded periplasmic proteins in need of disulfide oxidation or rearrangement. Furthermore, the partial rescue of dsbA null strains by overexpression of eukaryotic protein disulfide isomerase or mutants in exported thioredoxin, both of which are members in the thioredoxin superfamily of proteins, are DsbB-dependent.10; 11 This provides evidence that DsbB can oxidize thioredoxin-related folds other than DsbA. To test if the presence of DsbC and/or DsbG is necessary for the ability of DsbB mutants to bypass DsbA, we first constructed dsbC and dsbG null mutants in JP221, the same strain background we had used to isolate the DsbA-independent DsbB mutants. We transformed these strains with our DsbB mutant plasmids and checked motility (Figure 1, black bars) and cadmium resistance (Figure 1, gray bars) of the resulting transformants. The DsbB mutants were dependent on DsbC but not on DsbG for their ability to bypass the need for DsbA.
Figure 1.

Motility and minimum inhibitory concentration (MIC) of cadmium for DsbB mutants in various dsb backgrounds. The mutation(s) of each DsbB mutant are shown on the X-axis. W113R-1 refers to a W113R mutant containing an additional R109P and P121Q mutation; W113R-2 refers to a W113R mutant containing an addition V108D mutation. Vector refers to the empty vector pKK233-2 from which each DsbB mutant is derived. wtDsbB refers to wild-type DsbB. Percent motility (black bars) and minimum inhibitory concentration (MIC in μM) of cadmium (grey bars) were calculated as described in Materials and Methods and represent independent phenotypic measures of the extent of disulfide bond formation.
Efficiency of disulfide catalysis in DsbB mutants
To directly determine the efficiency of the disulfide catalytic system in these strains, we measured the relative amounts of oxidized and reduced β-lactamase. β-lactamase serves as a good reporter for the efficiency of the disulfide catalytic system because it has a single disulfide in the mature form and the protein is stable in the absence of its disulfide. Thus, the ratio of oxidized to reduced protein present at steady state serves as a very good indicator of the redox conditions during the folding of β-lactamase. To visualize the oxidation state of β-lactamase, we alkylated whole cells with 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS), which adds 490 daltons to each free thiol, and probed for β-lactamase via western blotting. Reduced β-lactamase should run more slowly on SDS polyacrylamide gels than oxidized β-lactamase, due to the additional weight caused by AMS –thiol modification. In wild-type strains, the vast majority of β-lactamase is in the oxidized form. In dsbA or dsbB null strains, the majority of β-lactamase is in the reduced form. Our DsbB mutants that bypass the need for DsbA allow ~40–60% β-lactamase to be oxidized (Figure 2). This shows that the suppressors, although they allow a considerable amount of oxidative disulfide bond formation, do not fully restore oxidative protein folding to the level seen in wild-type strains. This is consistent with the cadmium resistance and motility phenotypes observed. In both cases, the suppressor mutations in DsbB only partially rescue the dsb− phenotype associated by the deletion of dsbA compared to wild type. We observed that deletions of dsbC virtually eliminated the oxidation of β-lactamase by these DsbB mutants, showing that DsbC is a critical component of the bypass pathway. Mutations in DsbG had no effect on rescue. The simplest interpretation of these results is that DsbC is replacing DsbA by serving as a disulfide shuttle that acts as a conduit for the transfer of oxidizing equivalents between our DsbB mutants and folding proteins.
Figure 2.
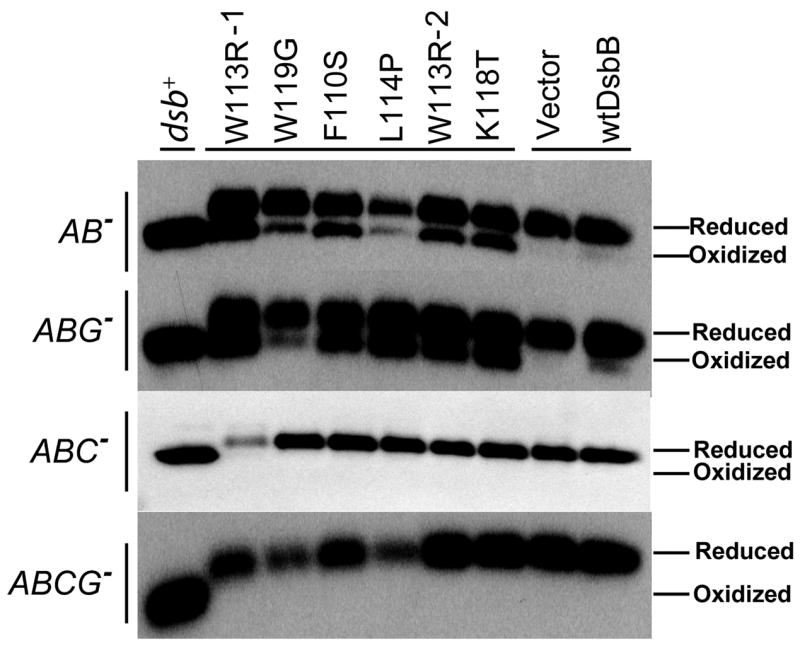
AMS trapping and western blot for β-lactamase in various Dsb mutant backgrounds. Each DsbB mutant is labeled at the top of the western blot with their corresponding mutation (labels are as described in Figure 1 legend) and the background strain is shown on the left side of the blot. β-lactamase is present in entirely the reduced form in strains lacking DsbC.
Models for DsbB mutant dependence on DsbC
One straightforward model that would explain the DsbC dependence of the bypass pathway is that the DsbB mutants have relaxed substrate specificity that allows them to oxidize DsbC. The oxidized DsbC would then be capable of oxidizing substrate proteins. A precedent for this is the observation that mutants in DsbC with disrupted dimerization interfaces allow DsbB to oxidize DsbC in vivo and in vitro and allow the rescue of motility of a ΔdsbA strain.19 One prediction of this model is that DsbB mutants will have a substantial amount of oxidized DsbC in vivo. We were unable to detect a substantial amount of oxidized DsbC in in vivo trapping experiments in ΔdsbAB strain (data not shown); however, oxidized DsbC may not need to accumulate for DsbC to be able to affect the flow of redox equivalents in the periplasm. If the combined rate of DsbC reduction by substrate proteins and by DsbD is greater than its rate of oxidation by DsbB, then DsbC will appear mostly reduced even though it is carrying out the oxidation of substrate proteins. The rate constant for DsbC reduction by DsbD was determined to be 3.9 × 106 M−1s−1, indicating a rapid reaction between DsbD and DsbC which prevents trapping of a mixed disulfides since they are kinetically unstable and quickly dissociate due to the attack of intramolecular thiols.21 A similar observation has been made in the case of eukaryotic protein disulfide isomerase. Xiao et al. observed that rat PDI had all six of its cysteines in the reduced conformation in in vivo trapping experiments. PDI is able to function as an oxidizing thiol-disulfide oxidoreductase and an isomerase in vivo and in vitro. The interpretation of these in vivo trapping results is that the presence of only a fraction of PDI in the oxidized form is sufficient to allow this protein to function as an oxidizing thiol-disulfide oxidoreductase.32 In addition, very rapid peptide oxidation occurs in in vitro experiments where the redox buffer is adjusted to allow for only 10 % of PDI to be present in the oxidized form at steady state.33
In an effort to detect oxidized state of DsbC in various DsbB mutants, we transformed the plasmids encoding the DsbB mutants into JP621, a strain derived from JP221 that also carries a dsbD deletion. In vitro oxidation of DsbC by DsbB occurs with the rate constant of 1.9 × 103 M−1s−1 21, which is >2000 fold slower than the reduction of DsbC by DsbD. Deletion of dsbD resulted in 50% oxidized and 50% reduced DsbC with wild-type DsbB, which is in an agreement with previous reports.34 However, DsbB mutants totally eliminated the reduced state of DsbC, suggesting that DsbB mutants have a better access to DsbC active sites, thereby oxidizing DsbC more efficiently than the wild-type DsbB. It should be noted that there is already some genetic evidence that DsbC contributes to the oxidation of proteins. We, in collaboration with the lab of Jean-Francois Collet, have recently found that DsbC can assist DsbA to oxidatively fold proteins. This led us to conclude that the view that DsbC’s function is limited to the disulfide isomerization pathway should be reinterpreted.35
In order to explore the alternate possibility that DsbB directly oxidizes proteins by directly replacing the function of DsbA, we performed the motility experiments in the strains lacking dsbD. If DsbB did not directly oxidize proteins and DsbC were behaving like an oxidizing thiol-disulfide oxidoreductase, then the deletion of dsbD would potentially enhance the motility; but if the DsbB were oxidizing proteins and DsbC were behaving like an isomerase, then deletion of dsbD would potentially eliminate or alleviate the motility. Deletion of dsbD resulted in an increased level of motility compared to the strain containing wild-type DsbD (Figure 3 b). Although the exact mechanism of increased motility is unknown, the result is consistent with the hypothesis in which DsbB mutants that rescue dsbA null strains oxidize DsbC, which then oxidizes folding proteins. It is less consistent with the alternative explanation that DsbB mutants are directly oxidizing newly secreted protein substrates. Oxidation of DsbC by DsbB mutants was also observed in dsbD null strain (Figure 3 a) further supporting the proposed hypothesis. The deletion of dsbD may also be acting in an additive fashion with the DsbB mutants by simply shifting the redox balance of the periplasm in the oxidizing direction. Consistent with this, it has previously been observed that deletion of dsbD leads to a partial rescue of DsbA null mutants.14
Figure 3.


(a) In vivo redox state analysis of DsbC in the presence of DsbB mutants in ΔdsbABD backgrounds. In order to establish the redox state of DsbC in various DsbB mutants (labels are described in Figure 1 legend), we used AMS trapping and western blot as described in Materials and Methods. (b) Motility of DsbB mutants in ΔdsbABD backgrounds. The mutation(s) of each DsbB mutant are shown on the X-axis (labels are as described in Figure 1 legend). In an effort to distinguish between the oxidizing thiol-disulfide oxidoreductase function and the isomerization function of DsbC in relation to the DsbB mutants, we deleted DsbD. Deletion of DsbD increased motility in the presence of the DsbB mutants..
Our results suggest that our dsbA− suppressing DsbB mutants act by oxidizing reduced DsbC in vivo. To directly demonstrate this, we sought to purify the DsbB mutant protein to determine its reactivity with wild-type DsbA and DsbC. Unfortunately, attempts to purify this protein were unsuccessful. These DsbB mutant proteins proved to be very unstable and as a result less amenable to purification than wild type. However, small amounts of approximately 60% pure mutant DsbB (K118T) were obtained, and this mutant did show ability to oxidize DsbA, and a less efficient but still significant ability to oxidize DsbC. This is in contrast to wild-type DsbB, which shows no detectable ability to oxidize DsbC (data not shown).
Glutathione not required in suppression pathway
Although we consider the direct oxidation of DsbC by our DsbB mutants the simplest interpretation of our results, our inability to measure the detailed kinetics of this direct oxidation reaction in vitro raises the possibility that our DsbB mutants are acting through some other intermediate such as a small molecule. One small redox active molecule known to be in the periplasm is glutathione.36 To eliminate the possibility that the DsbB protein mutants are functioning via a small molecule intermediate such as glutathione, we deleted gsbA, the key enzyme responsible for the synthesis of glutathione. We transformed our DsbB mutants into JP619, and showed that the deletion in gsbA did not affect the activity of our mutants, ruling out the possibility that glutathione is serving as an obligate intermediate in our suppression pathway. However, we cannot exclude the possibility that other unknown proteins or small molecules are functioning together with DsbC in our remodeled disulfide catalytic pathway.
Specificity determinants
Substrate specificity is a common theme in all biological processes because without such specificity, crosstalk between different enzymatic systems would become unmanageable. The enzymes involved in disulfide bond formation and isomerization are no exception to this rule. These enzymes need to maintain a careful balance between protein oxidation and protein reduction/isomerization in order to properly fold proteins that contain variable numbers of disulfides. Unfettered crosstalk between the DsbA-DsbB oxidation pathway and the DsbC-DsbD isomerization pathway would result in very little oxidative protein folding and the depletion of cellular resources such as NADPH. We have shown here that the oxidation of DsbC can occur in DsbB mutants that have altered residues in the major periplasmic loop. These mutations are likely to function simply by decreasing the substrate specificity of DsbB so that it can effectively oxidize DsbC either directly or through an intermediate protein or compound.
It has been shown previously that mutants in DsbC that interfere with dimerization have been able to partially restore a dsbA− phenotype. The resulting monomers can now interact with DsbB and function as an adequate replacement for DsbA.19 In addition, Segatori et al. have shown that the α-helical linker between the dimerization domain and the thioredoxin domain is critical in the regulation of the DsbC oxidizing thiol-disulfide oxidoreductase activity.20 Insertions or deletions in odd numbers in this domain appear to cause the catalytic disulfides to turn outward, which may increase exposure to the DsbB disulfides.20 Both results show there are elements in DsbC that prevent its oxidation by DsbB, and thus preclude DsbC to function as a oxidizing thiol-disulfide oxidoreductase.
Our identification of mutants in DsbB that bypass the need for DsbA has allowed us to propose the existence of an additional specificity determinant present in DsbB. When this determinant is mutated, DsbB can affect the oxidation of substrate proteins in a DsbC-dependent manner. We followed the model proposed by Inaba et al., where one of the thioredoxin domains of DsbC was superimposed with the thioredoxin domain of DsbA in DsbA-DsbB complex. The resulting structure demonstrated a severe steric clash of the second thioredoxin domain of DsbC with the membrane surface and inability of DsbC to have an access to the oxidizing power of DsbB active site cysteines (Figure 4A), as pointed by Inaba et al.31 Interestingly, the experimental evidence shows that the monomeric form of DsbC can utilize the oxidative power of DsbB in the absence of DsbA.19 The mutations that were discovered were close to the active site cysteines that catalyze DsbA reoxidation. The DsbB mutations that alleviate this restrictive nature in DsbC oxidation occur in hydrophobic amino acids between residues 110–119. These mutations result in the substitution of hydrophobic residues, namely F110, W113, L114, K118 (long-aliphatic chain), and W119, with small polar uncharged residues such as serine, threonine, large charged residues such as arginine, or helix-breaking residues such as proline and glycine. Based on the crystal structure of DsbB38, these residue changes may act by reducing the likelihood of membrane localization of the horizontal α-helix that anchors the second pair of periplasmic loops to the membrane. Mutation to nonhydrophobic residues or helix-breaking residues may create instability in the helix or decrease its affinity to the membrane. By creating this local instability in the α-helix, one can imagine that DsbB would gain more flexibility in its active site cysteine pair, C104-C130, and therefore could accommodate the DsbC (PDB access #1tjd)37 dimeric structure (Figure 4B). This would allow DsbC to be oxidized by DsbB (PDB access #2hi7)38 without a clash of the second protomer of DsbC into the inner membrane (Figure 4A). It is also possible that this enhanced flexibility of the DsbB mutants explains their relative instability in vitro and the relative difficulty in the purification of the DsbB mutants compared to wild-type DsbB.
Figure 4.

Model of DsbB mutant interaction with DsbC dimer. DsbC is shown in blue and DsbB is shown in red. The sulfurs of each molecules’ cysteines are colored in yellow. C98 of DsbC, C104 of DsbB, and the horizontal α-helix (amino acids 110–119) of DsbB is shown with spheres. It should be noted that the crystal structure of DsbB did not contain electron density in many regions due to high disorder and flexibility of the protein. The horizontal α-helix region was traced using a V120M mutation because the region contained very little electron density, and would not have been resolved without the mutation and subsequent Se-Met introduction.38 (a) Dimeric DsbC does not interact with DsbB due to steric clash between one protomer of DsbC with the membrane. Amino acids 110–119 of DsbB are shown interacting with the membrane. (b) Mutations in the α-helix (amino acids 110–119) of DsbB potentially increases its ability to interact with DsbC. Mutation of these five DsbB residues causes potential destabilization of the α-helix, which may reduce the rigidity of this region even further, allowing DsbC to access DsbB’s catalytic cysteines. Previously, these cysteines were not accessible due to spatial constraints. This restriction in part probably contributes to the ability of wild-type DsbB to specifically oxidize DsbA and not DsbC.
Careful comparison of the crystal structure of DsbB with the various hydrophobicity prediction algorithms available at http://us.expasy.org/cgi-bin/protscale.pl showed that a number of the prediction methods showed the correct localization of the various segments of DsbB quite well.39; 40 A change in the hydrophobicity of the horizontally-oriented α-helix may decrease the localization of the α-helix, which may serve to create more flexibility in that region. When we model the effects of our mutants on these hydrophobicity plots we see that most of our mutants decrease the hydrophobicity of this horizontal membrane helix with the exception of the K118T mutant, which shows an increase in hydrophobicity. (Figure 5)
Figure 5.

Comparison of hydrophobicity of DsbB mutants. Each mutant is highlighted in a unique color with wild-type heavy set in black. The region where the α-helix begins and ends is inset into the figure and is shown as a black cylinder. The heavy black line to the left of the α-helix in the inset represents a loop in DsbB, and the black dotted line to the left represents a loop in DsbB that did not contain electron density in the crystal structure. The sequence of each DsbB mutant was inputed into the Rose Algorithm33, which is available at Expasy (http://us.expasy.org/cgi-bin/protscale.pl).
Our work also may give some insight into the interesting question of how membrane protein topology evolves (recently reviewed by von Heijne41) and how this affects the function of proteins. We have selected mutants that probably change the localization of a membrane bound α-helix to the periplasm (at least partially or transiently). This change mediates a change in the specificity of the enzyme. This shows one evolutionarily straightforward mechanism of how alterations in the membrane localization of one part of a membrane protein can lead to changes in enzymatic specificity.
Conclusion
In conclusion, we have used a genetic selection to identify a key regulatory region in DsbB that prevents oxidation of DsbC. This region maps to a short α-helix that associates with the inner-membrane. This helix probably allows DsbB to maintain a highly ordered structure, which may allow DsbB to distinguish between monomeric DsbA and dimeric DsbC. This helps explain the specificity determinants of DsbB and how DsbB normally avoids oxidation of DsbC.
Materials and Methods
Strains and media
All strains are listed in Table 1. All plasmids are listed in Table 2. Strains were grown at 37°C unless otherwise noted. Antibiotics were used at the following concentrations: ampicillin (200μg/ml), kanamycin (100 μg/ml), tetracycline (12.5 μg/ml), and chloramphenicol (34 μg/ml). Luria-Bertani, terrific broth, and M9 minimal media (lacking cysteine unless otherwise stated) were made according to established protocols.42
Table 1.
Strains used in this study
| Name | Genotype | Reference |
|---|---|---|
| JP114 | ER1821: MM294 background: McrA− McrBC− EcoK r− m− Mrr− | Kelleher, J.E. and Raleigh, E.A. (1991) J.
Bacteriol., 173, 5220–5223. |
| JP118 | JP114 ΔdsbB::Tet | This study |
| JP219 | JP114 pKD46 | This study |
| JP220 | JP114 ΔdsbA::Kan (FRT) | This study |
| JP221 | JP220 ΔdsbB::Tet | This study |
| JP373 | JP114 pKK233-2 | This study |
| JP378 | JP221 pJP216 | This study |
| JP518 | JP118 ΔdsbA ΔdsbG | This study |
| JP557 | JP220 ΔdsbC::Cam (FRT) | This study |
| JP577 | JP221 ΔdsbC::Cam (FRT) | This study |
| JP579 | JP518 ΔdsbC::Cam (FRT) | This study |
| JP619 | JP221 ΔgshA::Cam(FRT) | This study |
| JP621 | JP221 ΔdsbD::Cam | This study |
| XL-1 Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F’proAB lacIqZM15 Tn10 (Tetr)] | Stratagene |
| JR6 | BL21(DE3) placIQ (TetR) | Regeimbal et al |
| BL21(DE3) | E. coli B F− dcm ompT hsdS(rB− mB−) gal λ(DE3) | Stratagene |
Table 2.
Plasmids used in this study
| Plasmid | Relevent information | Reference |
|---|---|---|
| pKK233-2 | Cloning vector (pBRori) | Lab collection |
| pKD46 | Pint-ts araC-ParaB γ β exo (from λ) | Datsenko and Wanner |
| pCP20 | pSC101 FLP+ λ cI857+, λ pR RepTS | Cherepanov et al.1 |
| pJP216 | pKK233-2 dsbB | This study |
| pAN302 | pJP216 (W113R. R109P, P121Q) | This study |
| pAN303 | pJP216 (W119R) | This study |
| pAN304 | pJP216 (F110S) | This study |
| pAN322 | pJP216 (L114P) | This study |
| pAN336 | pJP216 (W113R, V108D) | This study |
| pAN365 | pJP216 (K118T) | This study |
| pJR7 | pQE-70 dsbB | Regeimbal et al.2 |
| pJP546 | pJR7 (K118T) | This study |
(Cherepanov and Wackernagel 1995)
(Regeimbal and Bardwell 2002)
Molecular methods
Bacterial gene replacement was done by the linear transformation method described by Datsenko and Wanner.43 Briefly, primers were made flanking either the chloramphencol cassette of pKD3 43 or the kanamycin cassette of pKD4 43 that also contained ~30 nucleotides of homology to dsbA, dsbC, dsbG or gshA. Primers are listed in Table 3. The antibiotic cassette was amplified by PCR. The PCR product was transformed into electro-compentent JP219 cells that had the λ recombination system induced. Gene deletions were confirmed by PCR. All antibiotic cassettes derived from pKD3 or pKD4 contained flanking FLP recombinase recognition target sites. Transformation of pCP20 into a strain to remove the antibiotic resistance marker flanked by FLP recognition targets (FRT) was done to generate gene knock outs with conflicting antibiotic resistances.44 P1 transductions of the generated deletions were done as previously described.42
Cloning and mutagenesis of dsbB gene
The dsbB gene was cloned from pJR7, which contains mutations in two nonessential cysteines, C8V and C49A. We previously showed this protein to have the same in vivo and in vitro activities as wild-type DsbB with the exception that it is less likely to form disulfide linked aggregates. We and others refer to this variant as wild-type DsbB.45 Two primers with 5′-Nco I (Primer A) and 3′-Hind III (Primer B) restriction sites flanking the dsbB open reading frame were used to amplify the gene. The resulting PCR fragment was digested with Nco I and Hind III and ligated to Nco I and Hind III digested pKK233-2. The resulting DsbB construct, pJP216, was verified by sequencing.
Mutant plasmid generation was done using Stratagene’s EZ Clone II® kit using the recommended procedure with minor modifications. The Mutazyme II® enzyme (Stratagene) was used to amplify the dsbB gene from pJP216 using primers C and D, which are homologous to the 5′-end and the 3′-end of the dsbB gene. The cycle was: 95°C for 30 seconds, 55°C for 50 seconds, 72°C for 1 minute, repeated 30 times. Approximately 100 ng of this mutant megaprimer was used in a second PCR reaction using 5 U of PfuTurbo®®(Stratagene) and 10 ng of pJP216. The cycle for PCR was: 95°C for 50 seconds, 60°C for 50 seconds, 68°C for 12 minutes, repeated 25 times. The PCR products were visualized on a 0.8% TAE agarose gel. Approximately 50 ng of mutagenized vector from the second PCR reaction was ethanol precipitated using Pellet Paint® (Novagen). The pellet was resuspended in 1 μL of MilliQ deionized water.
Selection of DsbB mutants
Electro-competent cells of JP221 were prepared by growing a 5 mL overnight culture in terrific broth (12 g bacto-tryptone, 24 g bacto-yeast extract, 4 mL glycerol, 2.3 g NaH2PO4, 12.54 g K2HPO4) containing 100 ug/ml kanamycin to an OD600 of ~12 and then recovered by centrifugation at 3,000 × g for 10 minutes. The pelleted cells were washed twice in 2 mL ice-cold double distilled water and resuspended in 150 μL ice-cold double distilled water. The mutagenized pJP216 plasmid DNA was electroporated into JP221 using a Gene Pulser® (Bio-Rad) and plated onto LB plates containing ampicillin and allowed to grow at 37°C overnight. The resulting colonies were replica plated using Whatman paper #3 filter paper (Whatman) onto LB ampicillin plates supplemented with 15 μM cadmium chloride (Fluka) and incubated overnight at 37°C. Cadmium resistant colonies were patched onto LB Amp200 and LB Amp200 15 μM CdCl2 plates to retest their cadmium resistance. Patches that retested as cadmium resistant were restreaked from the corresponding LB ampicillin plate onto a new LB ampicillin to isolate single colonies. This procedure avoided repeated exposure to cadmium, which could result in selection of chromosomal cadmium resistant mutants. The cadmium resistant strains were stabbed into M9 minimal plates that contained 0.2% agar to test if they had the acquired motility, which is characteristic of dsb+ strains. The motility plates were incubated at 37°C overnight. Plasmid DNA was extracted from motile strains and the mutations in the dsbB gene were determined by sequencing.
Determination of in vivo redox state in mutant strains
We monitored the redox status of β-lactamase by directly measuring the redox status of the cysteines in β-lactamase. Briefly, cells were grown overnight at 30°C in LB and an aliquot of cells containing an OD600 = 1.0 was spun down. These cells were pelleted and disulfide exchange was stopped by resuspending the cells in 1 mL 10% trichloroacetic acid (TCA) in LB media. The samples were kept on ice for 1 hour, and the precipitated macromolecules were recovered by centrifugation at 16,000 × g for 30 minutes. The TCA protein pellet was resuspended in 100 mM tris(hydroxymethyl)aminomethane hydrochloride (Tris-HCl) pH 7.5, 1% sodium dodoecyl sulfate (SDS), 1 mM ethylenediaminetetraacetic acid (EDTA), and 20 mM of the alkylating agent 4-acetamido-4′-maleimidylstilbene-2′2-disulfonic acid (AMS), which adds 490 Da to each free thiol group. The samples were allowed to incubate in the dark at 37°C for 1 hour followed by the addition of SDS-nonreducing loading buffer and subsequent boiling.7 The samples were run on a 14% Novex Tris-Glycine acrylamide gel (Invitrogen). The gels were then electroblotted onto PVDF membrane (Whatman) and blocked by incubation in 5% nonfat dry milk for 1 hour. Western blotting was done with anti-β-lactamase (5-Prime 3-Prime, Inc.) at the dilution of 1:4000. The secondary antibody (Pierce), goat anti-rabbit IgG coupled to horseradish peroxidase, was used at a 1:5000 dilution. Bands were visualized by exposure to X-ray film.
In vivo trapping of the redox status of DsbC was performed as described in the above paragraph with minor modifications. Briefly, the TCA protein pellet was resuspened in the denaturing alkylation buffer composed of 200 mM Tris-HCl, pH 8.5, 6 M urea, 10mM EDTA, 0.5% (w/v) EDTA and 15 mM AMS. After 1 hour incubation in dark at 37°C, samples were processed as described above. Western blotting was done with anti-DsbC (a gift from Jean-Francois Collet) at 1:100000 dilution. Secondary antibody and visualization of the bands were performed as described above.
Quantitative motility assay
Cells were grown overnight at 30°C in 5 mL LB ampicillin. Cells were resuspended in 150 mM NaCl to OD600 = 1.0 and 5 μL of this cell suspension was used to inoculate M9 minimal plates that contained 0.2% agar, and ampicillin. The plates were incubated at 30°C for 12 hours. The diameter of the swarms was measured and expressed as a percentage of the dsb+ wild-type control JP373.
Determination of cadmium minimum inhibitory concentration
Cells were normalized to a standard OD600 = 1.0, and were diluted to 100, 10−1, 10−2, 10−3, 10−4 and 5 μL was spotted onto LBAmp200 plates containing cadmium ranging in concentrations from 0 – 50 μM. Cells were grown at 37°C overnight. Minimum inhibitory concentration was determined by lack of growth on concentration of cadmium.
Acknowledgments
We wish to thank Fabian Himstedt for his tireless efforts to attempt to trap mixed disulfides between DsbC and DsbB and his assistance in supervising Inga Sliskovic in the initial stages of her work on this project. We wish you well in your future endeavors. We would like to thank members of the Bardwell and Jakob labs for their helpful discussions.
We wish to thank Tim Tapley for assistance in making the figures. J.C.A.B is a Howard Hughes Medical Investigator. J.L.P is a Ruth L. Kirchenstein Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kadokura H, Katzen F, Beckwith J. Protein disulfide bond formation in prokaryotes. Annu Rev Biochem. 2003;72:111–35. doi: 10.1146/annurev.biochem.72.121801.161459. [DOI] [PubMed] [Google Scholar]
- 2.Akiyama Y, Kamitani S, Kusukawa N, Ito K. In vitro catalysis of oxidative folding of disulfide-bonded proteins by the Escherichia coli dsbA (ppfA) gene product. J Biol Chem. 1992;267:22440–5. [PubMed] [Google Scholar]
- 3.Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J. A pathway for disulfide bond formation in vivo. Proc Natl Acad Sci U S A. 1993;90:1038–42. doi: 10.1073/pnas.90.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bardwell JC, McGovern K, Beckwith J. Identification of a protein required for disulfide bond formation in vivo. Cell. 1991;67:581–9. doi: 10.1016/0092-8674(91)90532-4. [DOI] [PubMed] [Google Scholar]
- 5.Martin JL, Waksman G, Bardwell JC, Beckwith J, Kuriyan J. Crystallization of DsbA, an Escherichia coli protein required for disulphide bond formation in vivo. J Mol Biol. 1993;230:1097–100. doi: 10.1006/jmbi.1993.1226. [DOI] [PubMed] [Google Scholar]
- 6.Jander G, Martin NL, Beckwith J. Two cysteines in each periplasmic domain of the membrane protein DsbB are required for its function in protein disulfide bond formation. Embo J. 1994;13:5121–7. doi: 10.1002/j.1460-2075.1994.tb06841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inaba K, Ito K. Paradoxical redox properties of DsbB and DsbA in the protein disulfide-introducing reaction cascade. Embo J. 2002;21:2646–54. doi: 10.1093/emboj/21.11.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guilhot C, Jander G, Martin NL, Beckwith J. Evidence that the pathway of disulfide bond formation in Escherichia coli involves interactions between the cysteines of DsbB and DsbA. Proc Natl Acad Sci U S A. 1995;92:9895–9. doi: 10.1073/pnas.92.21.9895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bader M, Muse W, Ballou DP, Gassner C, Bardwell JC. Oxidative protein folding is driven by the electron transport system. Cell. 1999;98:217–27. doi: 10.1016/s0092-8674(00)81016-8. [DOI] [PubMed] [Google Scholar]
- 10.Debarbieux L, Beckwith J. The reductive enzyme thioredoxin 1 acts as an oxidant when it is exported to the Escherichia coli periplasm. Proc Natl Acad Sci U S A. 1998;95:10751–6. doi: 10.1073/pnas.95.18.10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jonda S, Huber-Wunderlich M, Glockshuber R, Mossner E. Complementation of DsbA deficiency with secreted thioredoxin variants reveals the crucial role of an efficient dithiol oxidant for catalyzed protein folding in the bacterial periplasm. Embo J. 1999;18:3271–81. doi: 10.1093/emboj/18.12.3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kadokura H, Tian H, Zander T, Bardwell JC, Beckwith J. Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science. 2004;303:534–7. doi: 10.1126/science.1091724. [DOI] [PubMed] [Google Scholar]
- 13.Hiniker A, Bardwell JC. In vivo substrate specificity of periplasmic disulfide oxidoreductases. J Biol Chem. 2004;279:12967–73. doi: 10.1074/jbc.M311391200. [DOI] [PubMed] [Google Scholar]
- 14.Rietsch A, Belin D, Martin N, Beckwith J. An in vivo pathway for disulfide bond isomerization in Escherichia coli. Proc Natl Acad Sci U S A. 1996;93:13048–53. doi: 10.1073/pnas.93.23.13048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sone M, Akiyama Y, Ito K. Differential in vivo roles played by DsbA and DsbC in the formation of protein disulfide bonds. J Biol Chem. 1997;272:10349–52. doi: 10.1074/jbc.272.16.10349. [DOI] [PubMed] [Google Scholar]
- 16.Hiniker A, Bardwell JC. Disulfide bond isomerization in prokaryotes. Biochemistry. 2003;42:1179–85. doi: 10.1021/bi027141t. [DOI] [PubMed] [Google Scholar]
- 17.McCarthy AA, Haebel PW, Torronen A, Rybin V, Baker EN, Metcalf P. Crystal structure of the protein disulfide bond isomerase, DsbC, from Escherichia coli. Nat Struct Biol. 2000;7:196–9. doi: 10.1038/73295. [DOI] [PubMed] [Google Scholar]
- 18.Collet JF, Riemer J, Bader MW, Bardwell JC. Reconstitution of a disulfide isomerization system. J Biol Chem. 2002;277:26886–92. doi: 10.1074/jbc.M203028200. [DOI] [PubMed] [Google Scholar]
- 19.Bader MW, Hiniker A, Regeimbal J, Goldstone D, Haebel PW, Riemer J, Metcalf P, Bardwell JC. Turning a disulfide isomerase into an oxidase: DsbC mutants that imitate DsbA. Embo J. 2001;20:1555–62. doi: 10.1093/emboj/20.7.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Segatori L, Murphy L, Arredondo S, Kadokura H, Gilbert H, Beckwith J, Georgiou G. Conserved role of the linker alpha -helix of the bacterial disulfide isomerase DsbC in the avoidance of misoxidation by DsbB. J Biol Chem. 2005;281:4911–9. doi: 10.1074/jbc.M505453200. [DOI] [PubMed] [Google Scholar]
- 21.Rozhkova A, Stirnimann CU, Frei P, Grauschopf U, Brunishilz R, Grutter MG, Capitani G, Glockshuber R. Structureal basis and kinetics of inter- and intramolecular disulfide exchange in the redox catalyst DsbD. EMBO J. 2004;23:1709–19. doi: 10.1038/sj.emboj.7600178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heitman J, Model P. Mutants of the EcoRI endonuclease with promiscuous substrate specificity implicate residues involved in substrate recognition. Embo J. 1990;9:3369–78. doi: 10.1002/j.1460-2075.1990.tb07538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumura I, Ellington AD. In vitro evolution of beta-glucuronidase into a beta-galactosidase proceeds through non-specific intermediates. J Mol Biol. 2001;305:331–9. doi: 10.1006/jmbi.2000.4259. [DOI] [PubMed] [Google Scholar]
- 24.Umeno D, Tobias AV, Arnold FH. Evolution of the C30 carotenoid synthase CrtM for function in a C40 pathway. J Bacteriol. 2002;184:6690–9. doi: 10.1128/JB.184.23.6690-6699.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Varadarajan N, Gam J, Olsen MJ, Georgiou G, Iverson BL. Engineering of protease variants exhibiting high catalytic activity and exquisite substrate selectivity. Proc Natl Acad Sci U S A. 2005;102:6855–60. doi: 10.1073/pnas.0500063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruber CW, Cemazar M, Heras B, Martin JL, Craik DJ. Protein disulfide isomerase: the structure of oxidative folding. Trends Biochem Sci. 2006;31:455–64. doi: 10.1016/j.tibs.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Sevier CS, Kaiser CA. Disulfide transfer between two conserved cysteine pairs imparts selectivity to protein oxidation by Ero1. Mol Biol Cell. 2006;17:2256–66. doi: 10.1091/mbc.E05-05-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rensing C, Mitra B, Rosen BP. Insertional inactivation of dsbA produces sensitivity to cadmium and zinc in Escherichia coli. J Bacteriol. 1997;179:2769–71. doi: 10.1128/jb.179.8.2769-2771.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stafford SJ, Humphreys DP, Lund PA. Mutations in dsbA and dsbB, but not dsbC, lead to an enhanced sensitivity of Escherichia coli to Hg2+ and Cd2+ FEMS Microbiol Lett. 1999;174:179–84. doi: 10.1111/j.1574-6968.1999.tb13566.x. [DOI] [PubMed] [Google Scholar]
- 30.Dailey FE, Berg HC. Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc Natl Acad Sci U S A. 1993;90:1043–7. doi: 10.1073/pnas.90.3.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. Crystal Structure of the DsbB-DsbA Complex Reveals a Mechanism of Disulfide Bond Generation. Cell. 2006;127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 32.Xiao R, Wilkinson B, Solovyov A, Winther JR, Holmgren A, Lundstrom-Ljung J, Gilbert HF. The contributions of protein disulfide isomerase and its homologues to oxidative protein folding in the yeast endoplasmic reticulum. J Biol Chem. 2004;279:49780–6. doi: 10.1074/jbc.M409210200. [DOI] [PubMed] [Google Scholar]
- 33.Xiao R, Lundström-Ljung J, Holmgren A, Gilbert HF. Catalysis of Thiol/Disulfide Exchange. J Biol Chem. 2005;280:21099–21106. doi: 10.1074/jbc.M411476200. [DOI] [PubMed] [Google Scholar]
- 34.Rietsch A, Bessete P, Georgiou G, Beckwith J. Reduction of the periplasmic disulfide bond isomerase, DscC, occurs by passage of electrons from cytoplasmic thioredoxin. J Bactriol. 1997;179:6602–8. doi: 10.1128/jb.179.21.6602-6608.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vertommen D, Depuydt M, Pan J, Leverrier P, Knoops L, Szikora J-P, Messens J, Bardwell JCA, Collet J-F. DsbC cooperates with DsbA in a DsbD-independent manner. Molecular Micro. doi: 10.1111/j.1365-2958.2007.06030.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pittman MS, Robinson HC, Poole RK. A bacterial glutathione transporter (Escherichia coli CydDC) exports reductant to the periplasm. J Biol Chem. 2005;280:32254–61. doi: 10.1074/jbc.M503075200. [DOI] [PubMed] [Google Scholar]
- 37.Banaszak K, Mechin I, Frost G, Rypniewski W. Structure of the reduced disulfide-bond isomerase DsbC from Escherichia coli. Acta Crystallogr, Sect D. 2004;60:1747–52. doi: 10.1107/S0907444904018359. [DOI] [PubMed] [Google Scholar]
- 38.Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. Crystal Structure of the DsbB-DsbA Complex Reveals a Mechanism of Disulfide Bond Generation. Cell. 127:789–801. doi: 10.1016/j.cell.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 39.Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science. 1985;229:834–8. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 40.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 41.von Heijne G. Membrane-protein topology. Nat Rev Mol Cell Biol. 2006;7:909–18. doi: 10.1038/nrm2063. [DOI] [PubMed] [Google Scholar]
- 42.Silhavy T, Berman M, Enquist L. Experiments in Gene Fusions. Cold Spring Harbor Laboratory Press; 1984. [Google Scholar]
- 43.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97:6640–5. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cherepanov PP, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- 45.Regeimbal J, Bardwell JC. DsbB catalyzes disulfide bond formation de novo. J Biol Chem. 2002;277:32706–13. doi: 10.1074/jbc.M205433200. [DOI] [PubMed] [Google Scholar]