

Abstract
Sulfiredoxin and sestrin are cysteine sulfinic acid reductases that selectively reduce or repair the hyperoxidized forms of typical 2-Cys peroxiredoxins within eukaryotes. As such these enzymes play key roles in the modulation of peroxide-mediated cell signaling and cellular defense mechanisms. The unique structure of sulfiredoxin facilitates access to the peroxiredoxin active site and novel sulfur chemistry.
Keywords: cysteine sulfinic acid, retroreduction, sulfiredoxin, sestrin
1. INTRODUCTION
The typical 2-Cys subclass of peroxiredoxins (Prxs) is a homodimer in which the peroxidatic cysteine residue (Cys-SpH) from one monomer attacks the O-O bond of the ROOH substrate, producing the first product (ROH) and the oxidized, sulfenic acid (Cys-SpOH) of the peroxidatic cysteine (Hofmann et al., 2002; Wood et al., 2003). During normal catalysis the resolving Cys residue (Cys-SRH), located on the C-terminus of the adjacent monomer, reacts with the sulfenic acid intermediate to form a disulfide bond. Eukaryotic, typical 2-Cys Prxs are unique in that the sulfenic acid intermediate can react with a second molecule of peroxide to form a sulfinic acid (Cys-SPO -2). This hyperoxidized Cys modification can occur under oxidative stress conditions and leads to a loss in peroxidase activity. It has long been thought that this type of modification was irreversible (Claiborne et al., 1999). With the discovery of the sulfinic acid reductases, sulfiredoxin (Srx) and sestrin, not only is this modification reversible, but may play a key role in regulating peroxide-mediated cell signaling by acting as a sulfinic acid switch (Jacob et al., 2004). Sulfiredoxin employs many novel structural and catalytic strategies to specifically repair or retroreduce typical 2-Cys Prxs. Currently little is known about the sestrins, but these proteins appear to utilize a similar catalytic mechanism despite no known similarity to Srx.
2. THE HYPEROXIDATION OF PEROXIREDOXINS
Early studies on oxidative stress in yeast revealed that a 25 kDa protein called thiol specific antigen (TSA) was able to protect glutamate synthetase from inactivation in the presence of Fe3+, O2 and dithiothreitol (DTT) (Kim et al., 1985). TSA was later shown to belong to the ubiquitous peroxiredoxin family that uses redox-sensitive cysteine residues to detoxify hydrogen peroxide, lipid hydroperoxides and peroxynitrite (Chae et al., 1994). As a result of kinetic studies that showed TSA receives reducing equivalents from the NADPH/thioredoxin reductase/thioredoxin system to break down hydrogen peroxide, tert-butyl peroxide and cumene peroxide, TSA was renamed thioredoxin peroxidase (TPx) (Chae et al., 1999). The rate of peroxide removal was initially fast and decreased gradually when 1 mM H2O2 was used as the substrate (Chae et al., 1994). However, 5 mM H2O2 caused a marked decrease in the rate of peroxide consumption. Substrate inactivation was also more rapid with t-butyl peroxide. Peroxide consumption was restored by replenishing the reaction with TPx. The decrease in rate was not attributed to the exhaustion of substrate or product inhibition by NADP+, but rather to the inactivation of TPx by peroxide. Substrate inactivation was also shown to occur in the peroxiredoxins (Prxs) from Kinetoplastida, including Leishmania major, L. donovani and Trypanosoma brucei, but not in Crithidia fasciculate (Nogoceke et al., 1997; Castro et al., 2002; Flohe et al., 2002; Budde et al., 2003). In contrast, the homologous bacterial peroxiredoxin AhpC efficiently detoxifies mM levels of hydrogen peroxide and cumene peroxide without any indication of peroxide-dependent inactivation (Niimura et al., 1995; Poole, 1996). Furthermore, in vivo peroxide stress studies in Salmonella typhimurium showed no indication of post-translational modification in AhpC (Christman et al., 1985; Morgan et al., 1986).
A clearer understanding of substrate inhibition within Prxs came from the determination of the crystal structure of human PrxII, a typical 2-Cys Prx, purified from red blood cells (Schröder et al., 2000). Surprisingly, the peroxidatic cysteine, Cys51, was found in the sulfinic acid form, suggesting that excess peroxide had post-translationally modified the cysteine via the sulfenic acid intermediate routinely using in catalysis. It was unclear at the time whether this unique derivative among Prx structures was a crystallographic artifact or could be formed in vivo. Studies on oxidative stress in human cell lines using 2D-PAGE analysis demonstrated that most Prxs are converted into variants with a lower isoelectric point (pI) upon exposure to hydroperoxides (Mitsumoto et al., 2001). The acidic shift was contributed to phosphorylation or sulfinic acid formation, modifications that were later proven to be possible (Chang et al., 2002; Chevallet et al., 2003). These findings led the Rhee laboratory to investigate the nature of substrate inactivation in human Prxs (Yang et al., 2002). Initial studies on recombinant PrxI-III showed similar substrate inhibition previously observed with yeast TPx (Chae, 1999). Further studies showed that inactivation of PrxI activity was coincident with the conversion of PrxI to a more acidic species (Yang et al., 2002). Mass spectral analysis and studies with cysteine mutants determined that the shift in pI was due to selective oxidation of the catalytic site Cys51-SH to Cys51-SO2 when in the presence of a thiol reductant and excess hydrogen peroxide. Kinetic analysis of PrxI inactivation in the presence of a low, steady-state level (<1 mM) of H2O2 indicated that PrxI was hyperoxidized at a rate of 0.072% per turnover at 30 °C.
Concurrent with the studies on human PrxI, the Chae laboratory purified a truncated form of TPx from Schizosaccharomyces pombe that showed resistance to inactivation by H2O2 when compared to the full-length form of the enzyme. This observation was further examined by generating a series of C-terminal truncations in the recombinant protein. Only the C-terminal truncation that removed residues 176-191 lead to a loss in peroxide sensitivity, suggesting that these residues play an important role in inactivation. Moreover, transformation of wild-type S. pombe with a construct bearing the oxidation-resistant, C-terminally truncated TPx provided protection against peroxide stress (Koo et al., 2002).
A rationale for why only some Prxs are hyperoxidized to the sulfinic acid form came from comparing the structure of S. typhimurium AhpC (C46S mutant) with human PrxII (Woodet al., 2003). AhpC lacks an YF motif-containing, C-terminal helix which in PrxII interacts with a conserved GGLG-motif in close proximity to the peroxidatic cysteine. It is thought that these additional structural motifs hinder or slow the ability of the resolving cysteine to access the peroxidatic cysteine located ∼13 Å away. As a result this kinetic “pause” leaves the Cys-SpOH intermediate increasingly exposed to attack by another peroxide molecule resulting in hyperoxidation to the sulfinic acid form. This model is consistent with the findings presented above for the truncated form of S. pombe TPx. Removal of the C-terminal helix in TPx most likely allows for a faster attack of the resolving cysteine on the sulfenic acid intermediate and thereby prevents hyperoxidation.
3. RETROREDUCTION OF TYPICAL 2-CYS PEROXIREDOXINS
Protein cysteine sulfinic acid formation is not unusual given that 1-2% of the cysteine residues of soluble proteins from rat liver were detected in this oxidation state (Hamann et al., 2002). Until recently these cysteine derivatives were viewed as irreversible; although sulfinic acids can be reduced in vitro by reductants such as 2-mercaptoethanol under very acidic (< pH 4) conditions (Finlayson, 1979; Claiborne et al., 1999). Interestingly, bacteria have enzymatic systems, such as the E. coli SsuE and SsuD, that during sulfate starvation scavenge sulfur from alkane sulfonates to generate the corresponding aldehyde and sulfite (Eichhorn et al., 1999).
The first indication of cysteine sulfinic acid reduction in vivo came when Woo et al. monitored the response of human cells to H2O2 treatment using metabolic 35S-labeling of proteins and 2D-PAGE analysis (Woo et al., 2003). Peroxide treatment led to the immediate hyperoxidation of Prx I and PrxII and an acidic shift of the protein spot. Other early experiments detected the overoxidation of five of the six Prx isoforms in humans (Prxs I-IV and VI) (Mitsumoto et al., 2001). The spot shift was reversed with similar rates in several cell types even in the presence of cycloheximide which prevents new protein synthesis (Woo et al., 2003). Chevallet et al., in contrast, found that the repair of PrxII in HeLa cells was more efficient than PrxI (Chevallet et al., 2003). In HepG2 liver cells PrxI and PrxII were repaired at the same rate (Cesaratto et al., 2005).
3.1 DISCOVERY AND INITIAL CHARACTERIZATION OF SULFIREDOXIN
The critical breakthrough in the field of cysteine oxidation and reversal came with the identification of the protein in Saccharomyces cerevisisae designated “sulfiredoxin” (abbreviated Srx1). Srx1 catalyzes the reversal of the hyperoxidized forms of two yeast Prxs (Tsa1 and Tsa2; also know as TPxI and TPxII) (Biteau et al., 2003). Srx1 was identified through a screen for hydrogen peroxide-induced genes. Srx1 mRNA levels were tremendously increased (>200-fold) in the first 15 minutes after peroxide treatment. Moreover, deletion of the Srx1 gene caused a decreased tolerance to hydrogen peroxide treatment. Several lines of evidence suggested that Srx1 might have some involvement in modulating the redox state of Prxs: the identification of peroxide-induced complexes in vivo between HA-tagged or 6H is-tagged Srx1 and Tsa1. The former complexes were not formed with the putative catalytic cysteine (C84S) Srx1 mutant or the Δtsa1 strain. The latter complexes were DTT-sensitive suggesting that a disulfide bond had formed between the two proteins. Moreover, wild-type Srx1, but not the C84S mutant, was able to restore the repair of Tsa1 in Δsrx1 cells as monitored by 2D-PAGE analyses. The addition of cycloheximide to cells also caused a delay in the repair of Tsa1 suggesting that de novo protein synthesis is required for efficient Cys sulfinic acid reduction.
In in vitro studies, recombinant Srx1 was able to reduce hyperoxidized Tsa1 (either generated in peroxide-treated cells or in purified form) in the presence of ATP and Mg2+ or Mn2+, but not other metals. In these studies the reduction of the sulfinic acid moiety was monitored using 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) labeling and SDS-PAGE analysis. AMS covalently modifies free thiol groups yielding a ∼500 Da increase in mass. Thus upon reduction of the sulfinic acid group to the thiol form, an increase in the mass of the Prx can be readily visualized. Unexpectedly, ADP supported Srx1-dependent catalysis, although less efficiently, and neither GTP nor AMP-PNP, a non-hydrolyzable ATP homologue, acted as inhibitors of catalysis. A reductant, either DTT or thioredoxin, was also required for the return of the Tsa1-SO2- to the Tsa1-SH form. As shown in the in vivo studies, the recombinant C84S mutant of Srx1 was inactive.
Given the requirement for ATP hydrolysis, generation of a phosphorylated intermediate, a sulfinic phosphoryl ester, was hypothesized (Figure 1) (Biteau et al., 2003). The investigators were not able to detect this intermediate, however, presumably due to its highly unstable nature. The apparent involvement of Cys84 in the covalent linkage to Tsa1 also led Toledano and coworkers to hypothesize the nucleophilic attack of Cys84-SH on the phosphorylated intermediate, resulting in formation of a thiosulfinate bond, i.e. a disulfide mono-oxide. Resolution of this complex with excess reductant would then return both enzymes to their reduced states through hypothesized Prx-SOH and Srx1-S-S-R intermediates.
Figure 1.
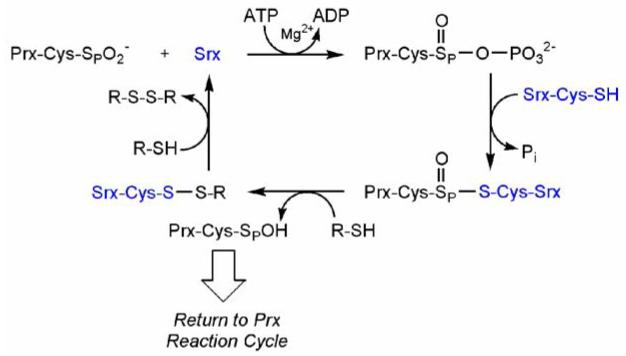
Proposed reaction mechanism for the reduction of the cysteine-sulfinic acid of Tsa1 by yeast Srx1. The reaction proceeds from a sulfinic acid through sulfinic phosphoryl ester and thiosulfinate intermediates (Biteau et al., 2003). The position of ATP hydrolysis and the identity of the exogenous thiol (R-SH) were proposed to be between the β-γ phosphate and thioredoxin, respectively.
3.2 DISCOVERY AND INITIAL CHARACTERIZATION OF SESTRIN
The sestrin family of proteins has also been shown to repair Prxs (Budanov et al., 2004). The sestrins are ∼48-60 kDa in size and show no sequence homology to the Srx family. The sestrins were originally discovered through subtractive cDNA cloning experiments designed to identify targets of the p53 protein (Velasco-Miguel et al., 1999). One of the predominant genes identified by the Kley laboratory was PA26 (p53-activated protein #26, also known as sestrin 1). Treatment of cells with genotoxic agents (e.g. the anti-cancer agent doxorubicin, UV- and γ-radiation) induced the mRNA levels of PA26 in a p53-dependent manner. These results are consistent with p53 induction and activation as a result of DNA damage and oxidative stress. The characterization of the PA26 gene and its transcripts revealed the presence of an intragenic p53 response element and three transcripts (T1, T2 and T3) of different size. The transcripts result in the production of three proteins that differ in sequence and length at their N-termini in in vitro transcription/translation experiments. In vivo data, however, suggests that the predominant transcript/protein is the PA26-T2 variant. Moreover, the T2 variant is also upregulated by serum deprivation (Velasco-Miguel et al., 1999). This latter characteristic has been used to further classify PA26 as a Growth Arrest and DNA Damage (GADD) inducible gene.
Subsequently, the Hi95 protein (sestrin 2) and sestrin 3 were identified through cDNA microarray experiments designed to discover genes upregulated in the hypoxia response. The induction by hypoxia treatment is p53 independent while the induction by oxidative stress and DNA damage is p53 dependent and modulated by serum levels as seen for PA26 (Budanov et al., 2002). Moreover, the overexpression of Hi95 protects cells from hydrogen peroxide treatment. While these studies and those for PA26 support a role for these proteins in the p53-regulated response to environmental stressors, the biological function of the sestrins was not proposed until it was recognized that the sestrins repair oxidized Prxs (Budanov et al., 2004). Based on the low sequence homology (less than 20% for only the first 200/551 residues) to proteins that are known to interact with bacterial Prxs, Chumakov’s group suspected that Hi95 may interact with human Prxs. In a series of experiments they showed that: (i) Hi95 repairs both human PrxI and PrxII; (ii) inhibition of Hi95 by siRNAs leads to higher intracellular levels of ROS while overexpression leads to lower ROS levels; and (iii) enzymatic activity in vitro requires a critical cysteine residue, ATP, Mg+2 and the reductant DTT. These characteristics suggest that the sestrin-catalyzed repair of Prxs may be similar to that proposed for Srx despite the lack of any sequence similarity and the disparity in size. The identification of two reductase families supports the cellular importance of the reduction of the sulfinic acid moiety. Moreover, it is intriguing to postulate that Srx and the sestrins modulate the activity of proteins/enzymes other than the Prxs in humans (Budanov et al., 2004). At this time no further characterization of the sulfinic acid reductase activity of sestrins has been reported. Data presented from the Rhee laboratory in section 6.2 has shown that Srx is specific for typical 2-Cys Prxs (Woo et al., 2005).
4. DISTRIBUTION OF PRX REPAIR ENZYMES
The hyperoxidation of typical 2-Cys Prxs and their repair by Srx and sestrins are highly correlated. Tissue and organellar localization may play a key role in some oxidiative stress responses. Srx exhibits a much broader organismal distribution than sestrin while maintaining strong sequence conservation.
4.1 PRX OXIDATION SUSCEPTIBILITY AND REPAIR CORRELATIONS
Typical 2-Cys Prxs have conserved features across all kingdoms with 30% or higher sequence identity. The most conserved regions include the peroxidative cysteine within the DFTFVCPTEI motif and the resolving cysteine within the VCP-motif. The YF helix and GGLG structural motifs, as described earlier, promote hyperoxidation and have been identified in most eukaryotic organisms and cyanbacteria (Wood et al., 2003). Many bacteria contain a GG(L/I)G motif, but the critical YF-motif is substituted with a YL motif resulting in a Prx that is insensitive to hyperoxidation (Baker et al., 2001). Interestingly, these bacteria do not contain the genes for sulfiredoxin or sestrin. In summary, Srxs are only found in organisms that contain peroxide-sensitive Prxs. This observation suggests that higher organisms have evolved to incorporate the phenylalanine instead of the leucine in the YF motif, and subsequently introduced the sulfiredoxin or sestrin gene.
Sestrins, in contrast to Srx, are only present in multicellular organisms ranging from nematodes to mammals. Vertebrates generally have only one Srx gene and multiple isoforms of sestrins. There are, however, organisms that contain hyperoxidizable, typical 2-Cys Prxs, but do not contain a Prx repair protein. The tryparedoxin-dependent peroxidases in kinetoplastida are one example. This apparent anomaly may be due to the high concentration of Prxs, in Crithidia fasciculata at least, amounting to more than 5% of the soluble protein (Nogoceke et al., 1997). Caenorhabditis elegans represents an organism that does not contain Srx but has a sestrin gene. C. elegans has two typical 2-Cys Prxs, CePrxI and CePrxII, where CePrxI is mitochondrial. Since mitochondrial Prx repair has yet to be reported, CeSestrin would only repair CePrxII. However, CePrxII expression is limited to two distinct neurons, I2 and I4, suggesting a very specific role for hyperoxidation and repair (Isermann et al., 2004).
4.2 TISSUE DISTRIBUTION
Typical 2-Cys Prxs in rat and human are abundant proteins constituting approximately 0.1-1% of soluble proteins in most tissue and cell lines (Chae et al., 1999). For example, the highest concentration of PrxII is found in red blood cells (5 mg ml-1) (Moore et al., 1991). Srx appears to be a ubiquitous protein with large differences in concentration among tissues, suggesting that the level of Srx may be an indicator of the oxidative stress response. A higher abundance of Srx is observed in brain, colon, liver, spleen and spinal cord (Chang et al., 2004). In Arabidopsis both the typical 2-Cys Prx and Srx are located in the chloroplast (Dietz et al., 2006; Liu et al., 2006). Northern blot analysis suggests that sestrin 1 is widely expressed in human tissues (Velasco-Miguel et al., 1999). In this study the highest mRNA levels were seen in skeletal muscles. The substrate specificity and subcellular distribution of the sestrins are, however, not known.
4.3 CONSERVATION OF SRX
A recent BLAST search using the human Srx (hSrx) sequence revealed the presence of Srx in organisms ranging from cyanobacterium and baker’s yeast to human (representative alignment in Figure 2) with at least 30% sequence identity. The active site motif GCHR containing the catalytic Cys99 (human numbering scheme) is strictly conserved. The alignment suggests that the Srxs vary predominantly at their N-termini. The plant Arabidopsis thaliana contains a N-terminal targeting sequence for the chloroplast (Liu et al., 2006). Human Srx contains a glycine-rich sequence (35%; 13 of 37 residues) that is readily removed by limited tryptic digest and leaves the remainder of the protein intact and active (Jönsson et al., 2005). This truncated form of hSrx approaches the size of Nostoc Srx (species PCC7120) that contains only 87 amino acids and represents the minimal catalytic core needed for sulfinic acid reductase activity. It is at the present moment unclear if the extended N-termini of human, rat and Drosophila Srx have a particular function. Interestingly, S. cerevisiae, which lacks most of the N-terminal extension, has numerous inserts between structurally conserved regions. A recent report suggests that Srx has sequence and structural similarity to a functionally unrelated protein ParB, a DNA binding protein involved in chromosome partitioning in bacteria (Basu et al., 2005). Human Srx, however, shows no structural similarity to ParB (Jönsson et al., 2005).
Figure 2.
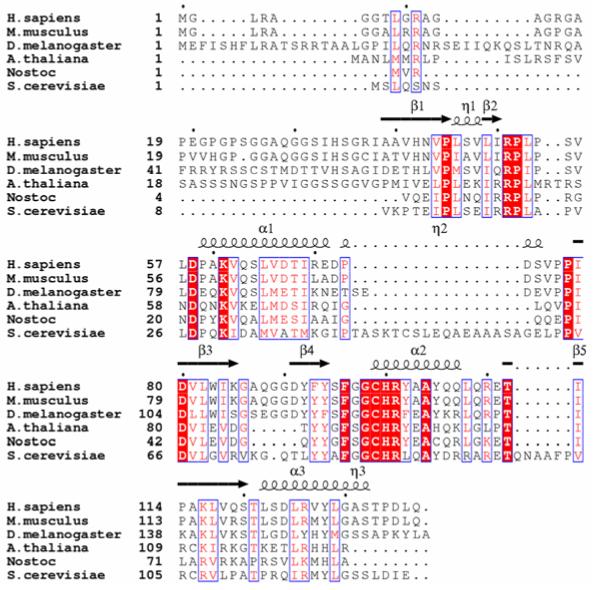
Sequence alignment of representative sulfiredoxins. The homology of the proteins to human Srx decreases down the alignment. The Srxs from mouse, Drosophila, Arabidopsis, Nostoc species PCC7120 (a cyano-bacterium), and S. cerevisiae show 91%, 60%, 43%, 41%, 33% sequence identity to hSrx, respectively. The secondary structural elements for human Srx are shown above the alignment: α, α-helices; β, β-strands; η, 310 helices (See Figure 3 and section 5.1). The residues highlighted by the red background and white lettering are strictly conserved. Residues that are either conserved in the majority of the proteins or have conservative substitutions are boxed in blue and colored red. The black dots above the alignment indicate every tenth residue of human Srx.
5. STRUCTURE OF SULFIREDOXIN AND PUTATIVE INTERACTIONS WITH PRXS
In an effort to understand the molecular basis for the novel sulfur chemistry mediated by Srx and the Srx:Prx interaction, the structure of human Srx has been determined by X-ray crystallography and NMR (PDB codes 1XW3, 1XW4, and 1YZS) (Jönsson et al., 2005; Lee et al., 2005). These structures have revealed a unique protein fold and nucleotide-binding motif. These studies have also laid the foundation for future structural work and the biochemical characterization of site-directed mutants to investigate the role of Srx residues in Prx recognition and the retroreduction process. It is clear that significant structural rearrangements are required within the Prx molecule in order for Srx to access the Cys sulfinic acid moiety. Given the symmetry relationships of the Prx molecule and an analysis of the Srx surface features, a binding mode for Srx has been proposed.
5.1 NOVEL PROTEIN FOLD AND NUCLEOTIDE-BINDING MOTIF
The peptide backbone which represents the overall fold of hSrx (Figure 3A) has considerable agreement between the crystallographic and NMR structures (RMSD of Cα carbon superposition of residues 38-137 is 1.1 Å). This “core” of Srx corresponds to the conserved region of Srx among different species (Figure 2). The conformational differences in the N-terminal portion of the models are consistent with lower sequence homology across species and how these experimental techniques observe structural plasticity. For example, residues 29-37 in the 1.65 Å X-ray structure are most likely only visible as a consequence of the formation of a novel crystal contact involving the same region from two other Srx molecules (Jönsson et al., 2005).
Figure 3.
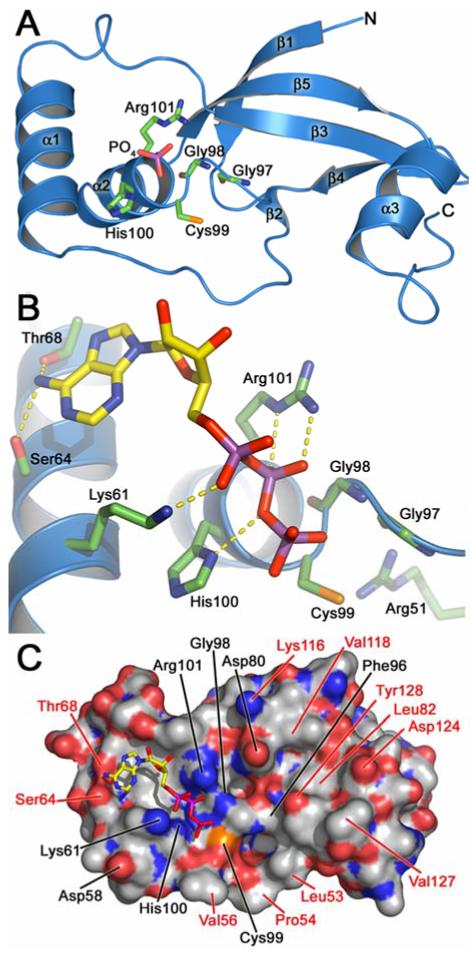
Crystal structure of human Srx. (A) Native enzyme in complex with phosphate. The α-helices and β-strands within the novel fold of Srx are numbered consecutively based on the primary sequence. Residues of the signature sequence of Srx are highlighted. For clarity residues 29-36 and the 310-helicies are not shown. Atom color scheme: green, carbon atoms for Srx; yellow, carbon atoms for ATP; red, oxygen; blue, nitrogen; magenta, phosphorous; orange, sulfur. (B) Model of ATP bound to Srx. The ATP:Srx complex is based on the crystal structure of the ADP complex. Putative hydrogen bonding interactions are indicated by dashed yellow lines. (C) Surface representation of Srx in complex with ATP. Strictly and semi-conserved residues are indicated in black and red, respectively. Carbon atoms for the Srx surface are shown in gray.
The N-terminus of Srx leads into a five-stranded, anti-parallel β-sheet (Figure 3A) (Jönsson et al., 2005). Three α-helices generate a curved surface with helix α2 and α3 located on opposite sides of the β-sheet. Helix α2 and the surrounding coil structures contain the signature sequence for Srxs, Gly98-Cys99-His100-Arg101 (Figure 2). This motif in the native structure of human Srx coordinates through multiple hydrogen bonding interactions a phosphate ion present in the crystallization buffer. Arg51 is located adjacent to Cys99 (Figure 3B) and most likely functions to activate Cys99 as the nucleophile of the reaction as originally proposed (Figure 1). Several water molecules were also found in the vicinity interacting only with the phosphate ion.
In order to understand how ATP binds to the human Srx surface, crystals of the native enzyme were equilibrated with a solution containing 200 mM ADP. The 2.0 Å structure revealed that the β-phosphate group of ADP replaced the phosphate ion seen in the native structure. Similar interactions to His100 and Arg101 were observed. Lys61, also a conserved residue, was found to hydrogen bond to an oxygen atom of the α-phosphate group. The only other hydrogen bonds to ADP were from Ser64 and Thr68 to the adenine ring which fits into a shallow surface depression. As a whole these interactions represent a novel nucleotide-binding motif (Traut, 1994). Somewhat surprisingly, no hydrogen bonds were observed to the oxygen atoms of the ribose ring.
A model of the ATP:Srx complex (Figure 3B) was generated using the ADP structure. In this model the γ-phosphate hydrogen bonds to the amide nitrogen atom of Gly98 and is positioned just above Cys99. Adjacent to the nucleotide binding site are two other pockets (Figure 3C) that presumably provide contacts to the Prx decameric structure. These pockets contain many conserved and semi-conserved residues including Leu53, Pro54, Val56, Asp80, Leu82, Phe96, Lys116, Asp124, and Tyr128. As described in more detail in section 6.2, site-directed mutants have been analyzed for some of these residues further supporting their role in mediating contacts with the Prx molecule. The ATP:Srx model and the biochemical data from yeast and human Srx available at that time suggested that the Srx reaction begins with the attack of the Prx-Cys-SPO2- group onto the γ-phosphate of ATP (Figure 4). Lys61, Gly98, His100, and Arg101 play key roles in holding the ATP molecule in the correct register for catalysis. His100 may also function to stabilize the developing negative charge on the oxygen atom that bridges the β- and γ-phosphates (Jönsson et al., 2005).
Figure 4.
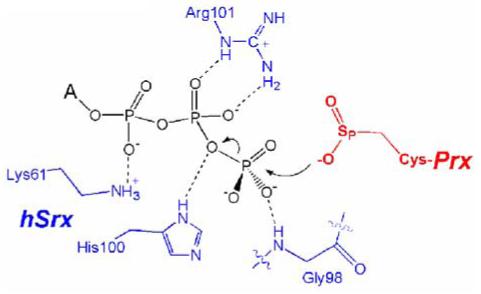
Proposed binding mode of ATP and the first step in the Srx reaction. Putative hydrogen bonding interactions are indicated by dashed lines. The nucleotide and sugar structures of the ATP molecule are abbreviated by the letter A.
A survey of the literature and other nucleotide- and phosphate-binding proteins, however, suggests some potential similarities to DNA ligases and protein tyrosine phosphatases despite little structural similarity between the folds of the proteins. For example, ATP- and NAD-dependent DNA ligases utilize a conserved Lys residue that becomes adenylated (Timson et al., 2000; Martin et al., 2002). The presence of the Lys61-α-phosphate hydrogen bond in Srx is reminiscent of this type of interaction. However, HPLC analysis of the reactions products has confirmed that ATP hydrolysis occurs at the β-γ phosphodiester bond thus excluding the potential adenylated reaction intermediate. Moreover, the Cys99Ser mutant and the alkylated, wild-type enzyme are not able to facilitate ATP hydrolysis or the repair of the Prx molecule (Jönsson et al., 2005). This latter finding coupled with the structural similarity to protein tyrosine phosphatases (e.g. PTP1B) (Pannifer et al., 1998) suggests yet another putative intermediate, the phosphorylated form of Srx. In this comparison both Srx and PTP1B contain a conserved Arg residue within a phosphate-binding motif adjacent to an essential Cys residue. An analysis of the geometry of the ATP molecule and its relationship to Cys99 in the ATP:Srx model, however, suggests that an inline attack of the γ-phosphate, like that seen in PTP1B, would require significant movement of the ATP molecule, Srx or both. Nonetheless, as described in section 6.3, the analysis of site-directed mutants provides some evidence for this intermediate and suggests that the originally proposed reaction scheme may require some modification.
5.2 MODEL FOR THE INTERACTION OF SRX WITH PRX
A comparison of the crystal structure of human PrxII in the hyperoxidized state to the structures of other Prxs in a variety of oxidation states has revealed several structural motifs (Schröder et al., 2000; Wood et al., 2003). These include the juxtaposition of the GGLG and YF motifs and the establishment of hydrogen bonds between the Cys51-SPO2- moiety (Csd51 in Figure 5) and Arg127, a conserved residue. These structural features occlude the active site for repair and therefore must be displaced or rearranged in order for Srx to perform its function. These conformational changes are quite reasonable given the known flexibility of the C-terminal amino acids and the local unfolding of the helix containing the Cys-SPH residue required for normal Prx peroxidase activity (Wood et al., 2003). As described previously, it is the presence of the GGLG and YF motifs that make the eukaryotic, 2-Cys Prxs susceptible to hyperoxidation.
Figure 5.
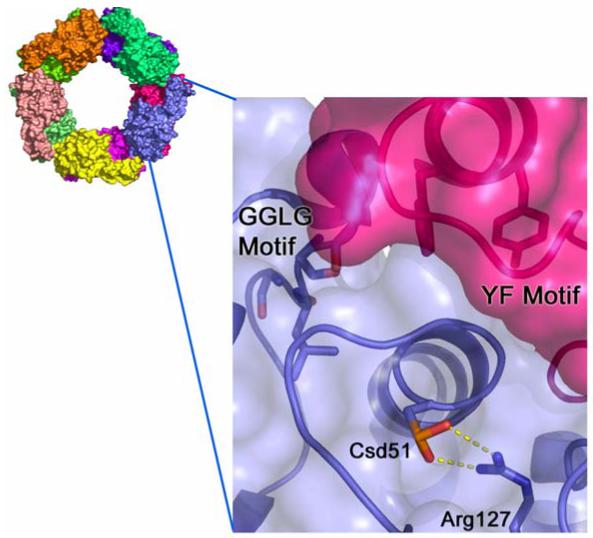
Inaccessibility of the hyperoxidized active site of human PrxII. Hyperoxidized 2-Cys Prxs form stabilized decamers. Each monomer is colored differently. A close-up view of one PrxII active sites (blue surface and ribbon) illustrates the difficulty Srx has in gaining access to the Cys51-SPO2- moiety (Csd51) which is involved in a hydrogen bonding interaction with Arg127. The Prx active site is occluded primarily by residues of the YF motif within the C-terminal α-helix of the adjacent Prx molecule (magenta surface and ribbon). This latter structural feature also interacts with the GGLG motif, another conserved region found primarily in Prxs sensitive to hyperoxidation.
Based on the above discussion and the symmetry and proportions of the PrxII decamer and Srx, it is evident that one Srx molecule could bind to each Prx molecule once the active site has been exposed. Srx most likely binds such that its narrow dimensions are adjacent to each other with the α1 and α3 helices acting as molecular calipers. The top panel in Figure 6 shows the “edge on” view of four subunits of the Prx decamer. The active sites of the middle Prx dimer are located on the periphery of the molecule and related by a two-fold symmetry axis. Srx could bind or dock to the circumference of the Prx decamer (Figure 6, bottom panel). As described above, in order for Srx to access the Cys sulfinic acid moiety of one monomer of a Prx dimer, the C-terminal YF motif and associated helix from the adjacent monomer must move out of the way. It is unclear if Srx actively facilitates this process. It has been suggested that the conserved Asp80 residue, predominately located on the Srx surface (Figure 3C), could disrupt the Csd51-Arg127 interaction of PrxII (Jönsson et al., 2005). Additional surface residues of Srx undoubtedly play a role as indicated by the mutagenesis studies described in section 6.2.
Figure 6.
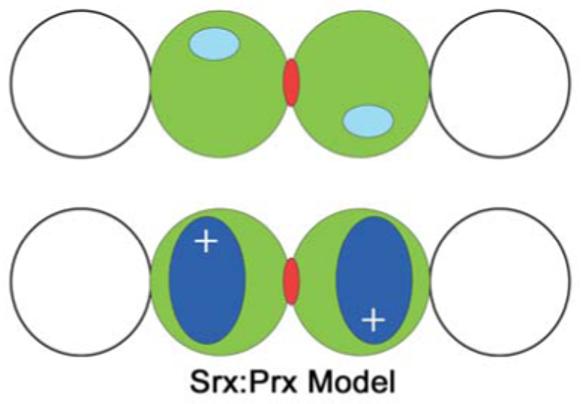
Model for the Srx:Prx interaction. The top panel indicates the edge-on view of four subunits of a Prx decamer. The active sites of the middle Prx dimer (green) are indicated in cyan. The two-fold axis is indicated by the red dyad symbol. Adjacent subunits in the Prx decamer are shown as the non-filled circles. The bottom panel illustrates the putative binding mode of Srx (blue) to the circumference of the decamer and the central Prx dimer. The Srx molecules most likely also adopt a two-fold relationship indicated by the plus signs. It is possible that the Srx molecules dock more to the side of the Prx decamer maintaining the symmetry relationship.
6. CATALYTIC PROPERTIES OF SRX
Since the initial characterization of yeast Srx1 and the determination of the human Srx structure, several studies have provided further insight into the catalytic mechanism and substrate specificity of human and rat Srx. In these studies three activity assays have been utilized: (i) sulfinic acid reductase activity based on the decrease in the immuno-reactive band to Prx-SO2/3-, (ii) ATPase activity corresponding to the increase in the amount of inorganic phosphate released, (iii) binding of Srx to Prx using the Srx-GST fusion and GSH-sepharose (Woo et al., 2003; Chang et al., 2004; Woo et al., 2005; Jeong et al., 2006). The results from these studies suggest that the initial steps in the reaction may need to be reconsidered and evaluated by further experimentation.
6.1 COFACTOR REQUIREMENTS AND CATALYTIC EFFICIENCY
Woo et al. set out to improve upon the AMS method for monitoring the sulfininc acid reductase activity of Srx by generating an antibody specific for Prx-SO2/3- (Woo et al., 2003). With this method the decrease in the sulfinic acid form of hPrxI was monitored over time following treatment with the recombinant forms of rat, mouse, and human Srx. All Srx variants exhibited the same specific activity in the presence or absence of an N-terminal GST affinity tag (Chang et al., 2004). Moreover, Srx purified from A549 human lung epithelial cells had the same specific activity. A screen of a panel of electron donors (DTT, glutathione, E. coli Trx1, Trp14 and SpTrx) indicated that glutathione (GSH) and Trx are most likely the in vivo reductant. The apparent Km values for GSH and Trx1 were 1.8 mM and 1.2 μM, respectively, when 5 μM human PrxI-SO2- was used as the substrate in the assay. The kcat value was estimated to be between 0.1-0.18 min-1, thus Srx is an inefficient enzyme. The pKa of Cys99 of hSrx was also determined to be ∼7.3 which is consistent with its proposed function as a nucleophile under physiological conditions.
The nucleotide dependence of the rat Srx reaction was investigated further by testing ATP, GTP, AMP-PNP, ADP, CTP, UTP, dATP, dGTP, dCTP, and dUTP. Reduction was supported with ATP and GTP in both nucleotide forms, but not ADP, AMP-PNP as expected. Activity with CTP, UTP, and the corresponding deoxyribonucleotide forms were significantly lower. Using saturating concentrations of Srx and PrxI-SO2-, the apparent Km for ATP was determined to be 30 μM. In a subsequent study the Kd for ATP was determined to be ∼6 μM by monitoring the change in the intrinsic fluorescence emission spectra for wild-type and the C99S variant of human Srx (Jeong et al., 2006).
6.2 SUBSTRATE SPECIFICITY AND MUTATIONAL ANALYSIS
Several lines of evidence support the selective binding of typical 2-Cys Prxs by Srx (Woo et al., 2005): (i) the GST-Srx fusion protein can bind the reduced and oxidized forms of PrxI-IV from HeLa cells with and without H2O2 treatment, but not the atypical 2-Cys PrxV or the 1-Cys PrxVI; (ii) reduced hPrxI can compete for hyperoxidized hPrxI in the reductase antibody assay; (iii) a yeast two-hybrid screen indicated interactions only between Srx and PrxI-IV; and (iv) overexpression of Srx in A549 cells reduces the levels of hyperoxidzed PrxI-IV upon H2O2 treatment, but not the other Prxs or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Activity analyses using recombinant PrxI-6 have further confirmed that only the typical 2-Cys PrxI-IV are reduced. Moreover, Srx is not able to repair the sulfinic acid form of GAPDH. It is important to note here that while Srx can repair PrxI-IV in vitro, Srx most likely only repairs PrxI and PrxII since both molecules are localized to the cytosol (Chang et al., 2004). In contrast, PrxIII and PrxIV are located in the mitochondria and the extracellular space, respectively. Interestingly, PrxVI and GAPDH are located in the cytosol and are not reduced by Srx. Based on this analysis it is possible that the sestrins are responsible for the repair of the other Prx isozymes and other proteins. Another possibility is that the hyperoxidation of some proteins is irreversible (Woo et al., 2005).
The structure of human Srx and sequence alignments have enabled the design of site-directed mutagenesis experiments to evaluate the role of conserved residues in Prx binding and Srx catalysis. Six conserved, charged amino acids were assessed by making the following mutations in rat Srx (rSrx): Arg51Met, Asp58Asn, Lys61Arg, Asp80Asn, His100Asn, Arg101Met (Jeong et al., 2006). For ease of comparison, the residue numbers correspond to hSrx; numbers in rSrx are lower by one. The Asp67Ala and Ser75Ala variants were also generated, but these residues are only conserved in mammalian Srxs. The GST-fusion form of each rSrx variant was tested for its ability to bind to either recombinant PrxI or PrxI from HeLa cell extracts using the GSH-sepharose pulldown method. The Arg51Met and Asp80Asn mutants were unable to bind PrxI. The Asp58Asn variant exhibited reduced binding while the remaining mutants were similar to wild-type rSrx. The decreased binding ability of the Asp58 and Asp80 variants is consistent with their presence on the surface of hSrx near the ATP binding site (Figure 3C). The Arg51 variant is more difficult to interpret since it is not on the surface of hSrx. One possibility for this finding is that the hydrogen bond between Arg51 and the carbonyl oxygen of Gly98 stabilizes the Gly-Gly component of the FGGCHR active site motif.
The panel of rSrx mutants was also assessed for their ability to stimulate ATP hydrolysis and Cys sulfinic acid reduction. A marked loss in both activities was observed for all residues that contact the ATP molecule; Lys61, His100, Arg101 (Figure 3B). The mutation of Cys99, Arg51 and Asp80 also lead to a reduction in both activities. The loss in activity for Cys99 is consistent with the inactivity of the Cys84Ser mutant of yeast Tsa1 (Biteau et al., 2003). The decrease in activity for the Arg51 mutation is the result of either the loss of active site stabilization described above or the decreased activation of Cys99 to act as the nucleophile in the reaction (Jönsson et al., 2005). The diminished activity for the Asp80 variant suggests that this residue may play a key role in the Prx:Srx interaction in contrast to the other surface residues tested (i.e. Asp58, Asp67, and Ser75) which showed wild-type levels of activity (Jeong et al., 2006).
6.3 EXPLORING THE REACTION MECHANISM
The original reaction mechanism proposed for yeast Srx1 (Figure 1 and Figure 7, Path 1) was based on the requirements for ATP/Mg2+ and the formation of DTT-reducible complex between Srx1 and Tsa1. Sulfinic phosphoryl ester and thiosulfinate intermediates were proposed to be formed. In this scheme Srx would function as a phosphotransferase (Path 1, Step 1) and a thioltransferase (Path 1, Step 2) (Biteau et al., 2003). Support for this reaction scheme came from the structure of hSrx in complex with ADP and an ATP model (Figure 3). In these models the γ-phosphate of ATP is near Cys99, but does not directly interact. Based on these observations it has been proposed that the sulfinic acid group of Prx directly attacks the γ-phosphate yielding the sulfinic phosphoryl ester intermediate (Jönsson et al., 2005). In this proposal it is not clear why the thiolate of Cys99, most likely generated by its interaction with Arg51, is essential for the first step of the reaction.
Figure 7.
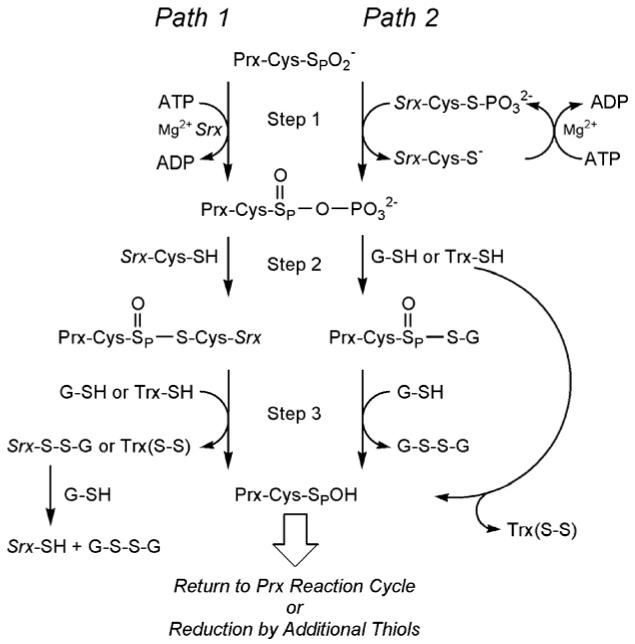
Comparison of the proposed reaction mechanisms for Srx. Path 1 represents the original mechanism as proposed for yeast Srx1. Path 2 incorporates potential modifications to the reaction pathway based on biochemical experiments with mammalian Srxs in both the wild-type and mutant forms. Step 1 involves the formation of the sulfinic phosphoryl ester intermediate. In step 2 of the reaction, the addition of a thiol group leads to the formation of alternative thiosulfinate intermediates. Breakdown of the latter intermediate occurs in steps 3 and following via reduction by additional thiol-disulfide exchange reactions.
Efforts to detect the proposed sulfinic phosphoryl ester intermediate from reactions containing hPrxI-SO2-, [γ-32P]ATP, hSrx, and reducing agents by immunoprecipitation, SDS-PAGE, and autoradiography have not been successful (Jeong et al., 2006). One possibility for this outcome is that the formation of the thiosulfinate intermediate in the original mechanistic proposal (Path 1, Step 2) with Srx is too fast. Therefore, Jeong et al. rationalized that the phosphorylated intermediate may be stabilized by mutating Cys99 to Ser, a mutation as described earlier that results in the inactive form of hSrx. Even though the Cys99Ser mutant and wild-type hSrx exhibit similar affinities for ATP (∼6 μM), the phosphorylated Prx intermediate was not observed. In contrast, phosphorylation of Ser99 of hSrx was found in <1% of the sample after four hours incubation. Interestingly, the reduced form of PrxI was not able to induce the phosphorylation of Ser99. These findings suggest that the sulfinic acid group on Prx is necessary for phosphorylation to occur. Moreover, Cys99 of hSrx may be phosphorylated by ATP first followed by transfer of the phosphate group to the sulfinic acid moiety of Prx (Path 2, Step 1) yielding the same phosphoryl ester intermediate formed in Path 1.
ATP hydrolysis was analyzed further by monitoring the time-dependent release of Pi as a function of the presence and absence of a reductant, and the hSrx and hPrxI-SO2- concentrations (Jeong et al., 2006). In the presence of GSH, Trx and DTT, the amount of Pi released is proportional to the amount of Prx-SO2- in the reaction. If a reductant is not added, the amount of Pi released is several fold greater than the amount of hyperoxidized Prx in the reaction. Based on these findings, the authors of this study suggested that the extra Pi released comes from a futile cycle between the sulfinic phosphoryl ester intermediate and its collapse back to the sulfinic acid group. The extra Pi released is thought not to come from the hydrolysis of the Cys99-S-PO32- intermediate (Path 2, Step 1) since this route would be unable to account for the observed GSH dependence. Since there was no apparent difference in the rates of sulfinic acid reduction by either GSH, DTT or Trx in vitro, it was also proposed that the sulfinic phosphoryl intermediate is fully accessible and reacts readily to form a thiosulfinate intermediate with GSH or Trx in vivo (Path 2, Step 2). The resulting thiosulfinate intermediates for Paths 1 and 2 would then be further reduced (Step 3 and following) by thiol equivalents from the second Cys residue present in Trx (e.g. Cys35 in eTrx) or additional GSH molecules. It should be noted, however, that an excess of Pi release would also be predicted for Step 1 of Path 1 if the most inefficient step of the reaction is the attack of Cys99 onto the sulfinic phosphoryl ester intermediate.
One other aspect of the Srx:Prx interaction has been investigated: the apparent ability of the proteins to form disulfide-bonded intermediates in vitro and in vivo. Yeast Srx1 and Tsa1 isolated from cells formed oligomers that were sensitive to DTT treatment (Biteau et al., 2003). This observation suggested as described previously that the Srx reaction progressed through a thiosulfinate intermediate. One other possibility is that these complexes actually contain a disulfide bond between the two molecules. Evidence for this proposal has been obtained from the analysis of mixtures containing the recombinant proteins. In these analyses a disulfide bond was established by mass spectrometry (Jeong et al., 2006; Jönsson and Lowther, unpublished results). In the presence of the hyperoxidized PrxI, a disulfide was found between PrxI-Cys52, the peroxidatic Cys, and Cys99 of hSrx. When reduced PrxI was used in the Rhee laboratory, a Srx:Prx complex was also found to be mediated between Cys99 and Cys173, the resolving Cys residue. It is unclear at this time how these intermediates fit, if at all, into the sulfinic acid reductase mechanism of Srx. It is not surprising, however, that disulfide intermediates can be trapped since once the sulfininc acid group of the Prx molecule has been repaired to the sulfenic acid moiety, this latter group can readily react with adjacent thiols. Cys99 of hSrx would be a prime candidate for such a reaction.
7. IMPORTANCE OF SRX IN CELL SIGNALLING AND DEFENSE
Intracellular levels of peroxide are tightly regulated and kept at nM levels (Seaver et al., 2001). In contrast to bacteria where peroxide generally is viewed as toxic and rapidly eliminated, higher organisms utilize intermediate levels of peroxide in cell signaling (Rhee, 2006). In an effort to designate a cellular role for the hyperoxidation of typical 2-Cys peroxiredoxin in cell signaling, the “floodgate” hypothesis was proposed (Wood et al., 2003). In this proposal the Prx molecule and its enzymatic activity constitute a wall that prevents peroxide to reach peroxide-sensitive targets. Elevated levels of peroxide, however, would promote Prx hyperoxidation and allow peroxide to breach the wall enabling the activation or inactivation of key targets. The interplay between heme/thiol/selenium-dependent peroxidases, peroxide levels, and the hyperoxidation of typical Prxs is currently an area of investigation. In contrast to the floodgate model, evidence is also accumulating which suggests that typical 2-Cys Prxs work as a peroxide dosimeter. In this scenario the formation of hyperoxidized Prx is itself an oxidative stress signal that can be turned off by the reduction or repair of the sulfinic acid group. We are currently in the early stages of understanding the role of the Prx-sulfinic acid switch in oxidative stress-mediated signaling.
7.1 PRX-DEPENDENT PEROXIDE SENSING IN Schizosaccharomyces pombe
In the yeast S. pombe at least two independent pathways have evolved to respond to different levels of H2O2: the Pap1 and Sty1 pathways. Pap1 is an AP-1-like transcription factor that under normal conditions associates with the nuclear export factor Crm1 in the cytoplasm (Toone et al., 1998). Sty1 is a mitogen-activated protein kinase (MAPK) that is known to phosphorylate the transcription factor Atf1 (Shiozaki et al., 1995; Degols et al., 1996; Shiozaki et al., 1996; Wilkinson et al., 1996). Under stress conditions with low levels of H2O2, Pap1 is activated by the formation of an intramolecular disulfide bond that prevents association with Crm1 and results in the nuclear accumulation (Toone et al., 1998; Kudo et al., 1999; Castillo et al., 2002; Quinn et al., 2002). In contrast, elevated levels of peroxide prevent nuclear accumulation and the expression of Pap1-dependent genes (Quinn et al., 2002; Vivancos et al., 2004). Sty1 and Aft1 are also activated at elevated levels of peroxide (Quinn et al., 2002).
It was until recently unclear how these disparate pathways are activated in S. pombe. The reversibility of the peroxide-sensitive inactivation of typical 2-Cys Prx and the subsequent discovery of Srx in yeast suggested a potential role for Prx in peroxide sensing in S. pombe. Two different groups, the Morgan and Hidalgo laboratories, simultaneously showed that at low levels of H2O2 (0.2 mM) Pap1 became oxidized, i.e. the disulfide-bonded form, within 5 min and accumulated in the nucleus. With high levels of H2O2 (1 mM) Pap1 did not become oxidized until 30 minutes after treatment (Bozonet et al., 2005; Vivancos et al., 2005). When the TPx1 gene was knocked out, however, Pap1 remained reduced in the cytosol independent of the peroxide levels. This observation suggests that TPx1 could transfer a redox signal to Pap1. In contrast, two other thiol peroxidases, Gpx1 and Pmp20, did not influence the oxidation status of Pap1. Rapid hyperoxidation of TPx1 was seen at the high level of peroxide treatment which after one hour returned to the reduced form. This time-dependent repair of TPx1 correlated with the delayed oxidation of Pap1. Consistent with these observations, TPx1 in cells lacking Srx stayed in the Cys-SO2- form after the peroxide insult and more importantly Pap1 remained reduced. These studies suggested that reduced TPx1, but not hyperoxidized TPx1, was involved in introducing a disulfide bond in Pap1. Studies on cysteine mutants in TPx1 and Pap1 suggested that a disulfide bond between TPx1 and Pap1 could be formed when the resolving cysteine, Cys169, was mutated in TPx1. However, this complex was never able to promote a disulfide bond in Pap1, suggesting that further factors are important in mediating oxidation on Pap1.
The observation that Sty1 and Aft1 are activated at elevated levels of peroxide led investigators to look at the role of this pathway in the Pap1/TPx1 relay (Bozonet et al., 2005; Vivancos et al., 2005). In cells lacking Sty1, TPx1 accumulated in the hyperoxidized form after the addition of high levels of peroxide, suggesting that the Sty1 pathway activated Srx expression. Indeed, Srx expression was shown to be dependent on both Pap1 and Aft1, but Sty1 is essential at high peroxide levels. In a previous study Veal et al. showed that TPx1 at high peroxide levels can form a stable disulfide bond with Sty1 via the peroxidatic cysteine to promote Sty1 activation (Veal et al., 2004). However, it is unclear to what extent this disulfide bond can be formed at high peroxide levels when TPx is rapidly undergoing hyperoxidation of the peroxidatic cysteine.
7.2 SRX AND CHLROPLAST PROTECTION
2-Cys Prxs also detoxify peroxides within the chloroplast (Dietz et al., 2002; Konig et al., 2002). The chloroplast is the main source of ROS due to its role in photosynthesis and the synthesis of amino acids, fatty acids and nucleotides (Apel et al., 2004). Given the importance of Prxs in plants, the molecular and functional characterization of Arabidopsis and rice Srx was investigated (Liu et al., 2006). As shown in Figure 2, Arabidopsis Srx (AtSrx) contains all the conserved residues necessary for the binding of ATP and catalysis. The same characteristics are also found in a variety of other dicot and monocot plant species. Plant Srxs contain a putative chloroplast targeting peptide. AtSrx has been shown to co-localize with chlorophyll using C-terminal GFP fusion constructs in vivo. Moreover, AtSrx and rice Srx were able to complement the yeast Srx1 knockout thus enabling growth on media containing H2O2. All these observations suggest that plant Srxs may play a key role in the retroreduction of plant 2-Cys Prxs.
In an effort to characterize the function of Srx in plants, the transcriptional patterns of gene expression were determined during resting and stress conditions using semiquantitative PCR (Liu et al., 2006). Under normal conditions the highest levels of Srx mRNA were observed in leaves and stems of Arabidopsis. The Srx transcript levels were dramatically increased upon treatment with H2O2 (4 mM), polyethylene glycol 8000 (10 %) to induce dehydration, and cold (4 °C). Plants containing a T-DNA insert to inactivate the endogenous AtSrx were more susceptible to oxidative stress induced by 10 μM paraquat, as indicated by a chlorosis or bleaching phenotype. AtSrx reintroduced via an Agrobacterium-mediated transformation rescued the hypersensitive phenotype. Taken together the data suggest that plant Srxs provide a protective function under stress conditions by repairing hyperoxidized, 2-Cys peroxiredoxins. Further experiments are required to confirm that AtSrx can repair hyperoxidized Prxs in vivo and in vitro via either the Prx-SO2/3- antibody or AMS assays.
7.3 SRX AND THE MODULATION OF PRX CHAPERONE FUNCTION
The peroxidase activity of typical 2-Cys Prxs have been studied extensively in vitro, however, the in vivo significance of this activity is unknown. Studies in yeast have indicated that cells lacking cPrxI (TPxI) and cPrxII (TPxII) were more susceptible to heat shock. Surprisingly, the ΔcPrxI/II cells expressing the non-cysteine containing mutant Cys47/170Ser of cPrxI were more resistant to heat shock stress than the double knockout cell line. These observations suggest that cPrxI has a protective, non-peroxidatic activity (Jang et al., 2004). In vitro studies with purified proteins showed that cPrxI was able to protect citrate synthase from thermal aggregation and insulin from DTT-dependent denaturation, leading the authors to propose that cPrxI has molecular chaperone activity. Similar to many other molecular chaperones cPrxI was shown to exist in complexes ranging from 40 to 1000 kDa. Analysis of the different molecular weight species showed that: (i) the high molecular weight (HMW) complexes had chaperone activity, but no peroxidase activity; (ii) the low molecular weight complexes (LMW) had peroxidase activity, but no chaperone activity; and (iii) increasing the temperature promotes formation of HMW species. In addition, hyperoxidized cPrxI predominantly formed HMW complexes. Therefore, Prx-SO2- formation may be important in promoting molecular chaperone activity in the cell.
In order to demonstrate the peroxidase-to-chaperone functional switch in vivo, yeast cells were exposed to various stresses (Jang et al., 2004). Wild-type cells showed greater survival rates than ΔcPrxI/II knock out cells under heat stress. Wild-type cells were also shown to promote the formation of HMW complexes concomitant with the increased intracellular levels of reactive oxygen species as judged by DCF staining. The addition of 0.5 mM peroxide shifted cPrxI from LMW to HMW complexes which correlated with the formation of hyperoxidized cPrxI. However, after 20 minutes of recovery, cPrxI-SO2- was repaired and the HMW species reverted to LMW species. Yeast lacking Srx were unable to repair cPrxI-SO2- after H2O2 exposure and only HMW species were observed. Therefore, Srx appears to play a critical role in cell survival by regulating the beneficial functions of peroxidase activity versus chaperone activity of Prxs. In addition to the traditional toroid shaped decamer, EM studies of the HMW complexes also revealed large spherically-shaped particles which appeared to lack symmetry. The ability of Srx to access different oligomeric sizes of cPrxI and to promote their disruption and reduction is unclear.
Further studies by the Lee laboratory on recombinant, human PrxII showed that it can also function as a molecular chaperone by protecting citrate synthase, insulin and α-synuclein from stress-induced aggregation (Moon et al., 2005). As observed in yeast cPrxI, hPrxII was shown to exist in multiple forms of LMW species that upon peroxide treatment resulted in overoxidation and complete conversion into HMW complexes. Removal of the C-terminal helix containing the essential hyperoxidation YF-motif (Figure 5) preserved the peroxide activity and prevented HMW species formation and chaperone function. In HeLa cells, exposure to 0.2 mM H2O2 converted all of hPrxII to HMW complexes. After a 40 min recovery period the HMW complexes returned to LMW species. This data suggests a role for Srx or sestrin in terminating the Prx-dependent chaperone activity in HeLa cells as seen in the yeast system.
Human PrxI had previously been shown to be phosphorylated at Thr90 by Cdc2, a cyclin dependent kinase, during the mitotic phase of the cell cycle (Chang et al., 2002). Studies on the recombinant analogue hPrxI-T90D which mimics phosphothreonine showed decreased peroxidase activity. It was hypothesized that the insertion of a charged moiety in close proximity to the hydrophobic dimer-dimer interfaces would promote disassociation of the decamer and thus decrease the peroxidase activity (Wood et al., 2003). However, Jang et al. showed that the recombinant hPrxI-T90D mutant almost exclusively purified as HMW complexes (Jang et al., 2006). In contrast to wild type hPrxI, the hPrxI-T90D exhibited reduced peroxidase activity and six-fold higher chaperone activity. The role of decreased peroxidase activity, increased chaperone activity, and the possible involvement of Srx repair in cell cycle regulation is at present unclear.
Given the connection between the formation of Prx-SO2- and chaperone activity, the report by Chuang et al. which proposed that H. pylori AhpC can switch from a peroxide reductase to a molecular chaperone function under oxidative stress was surprising (Chuang et al., 2006). AhpC has not been shown to be substrate inactivated nor does it contain the essential YF-motif Moreover, H. pylori lacks both Srx and sestrin genes. Harsh pre-treatment of recombinant AhpC with 10 mM H2O2 for 12 hours may have resulted in the apparent loss of peroxidase activity and the gain of chaperone activity. H. pylori cells exposed to aerobic stress (20% O2 rather than the normal 5%) forced AhpC to oligomerize into HMW species as seen in the human and yeast system. Unfortunately, the authors did not address whether Cys sulfinic acid was formed under these conditions. Thiol quantitation found no free thiols, however, suggesting that either a disulfide bond had been formed or both the resolving and peroxidatic cysteines had been hyperoxidized.
7.4 SRX AND (DE)GLUTATHIONYLATION PHENOMENA
Another curious role of human Srx is to modulate the post-translational modification of proteins by glutathione. HEK293 cells stably transfected with the Srx gene show a significant reduction in the levels of protein glutathionylation after treatment with PABA/NO, a potent inducer of oxidative stress via the release of nitric oxide (Findlay et al., 2006). In this study the level of glutathionylation was determined by Western blot analysis using an antibody specific for the protein-S-S-G linkage. In order to investigate this observation further, actin and PTP1B, known glutathionylation targets in cells, were treated in vitro with GSH and PABA/NO in the absence and presence of native and heat-inactivated, wild-type hSrx and the C99S mutant. In the absence of hSrx, both actin and PTP1B showed a time-dependent increase in glutathionylation. The presence of native and C99S Srx, however, led to a significant decrease in the protein modification. The addition of unfolded Srx resulted in the accumulation of glutathione adducts. Moreover, Srx was able to prevent the loss of the phosphatase activity of PTP1B upon PABA/NO treatment. These observations suggest that Srx can either prevent or reverse glutathionylation of a variety of proteins in a non-specific manner.
Two additional pieces of evidence suggest that hSrx does play a role in influencing glutathionylation levels in vivo. First, PABA/NO-treated, HEK293 cell extracts show a decreased level of adduct formation when treated with wild-type Srx in vitro, but not the C99S variant. Second, hSrx decreases the G/F-actin ratio leading to cell morphology changes. It is clear that more studies are needed in this area. For example, in order to confirm that Srx removes the glutathione moiety instead of protecting or blocking the site of glutathionylation, GSH adducts need to be formed in the absence of Srx, purified away from the PABA/NO and excess GSH, verified by mass spectrometry, and then treated with Srx. At the end of the incubation period with Srx, the glutathione protein target and Srx need to be reanalyzed by mass spectrometry to verify GSH removal or addition. It will also be important to assess whether this activity has the same cofactor requirements (ATP and Mg2+) as the sulfinic acid reductase activity. These initial studies suggest that Srx has therapeutic potential for a variety of diseases with aberrant protein glutathionylation (Findlay et al., 2005; Findlay et al., 2006).
8. CONCLUSIONS
The peroxide-dependent hyperoxidation of the typical 2-Cys Prxs and subsequent repair by Srx and sestrins is emerging as a unique response to severe oxidative stress. The signaling event attributed to this process is prolonged by the slow repair. It is entirely possible that the large conformational changes that must take place for Srx to access the Prx active site contribute to the inefficiency of the retroreduction process. In addition, the formation of the large hyperoxidized Prx chaperone complexes further impedes access of Srx to Prx. The peroxide-to-chaperone switch is one example of how the oligomeric state of Prx can influence cellular events. We suspect that redox-dependent conformational changes, and especially the formation of the sulfinic acid moiety, in Prx are important in discriminating their interactions with biological partners. It remains to be seen if Srx or sestrin has additional activities in addition to the sulfinic acid repair.
ACKNOWLEDGEMENTS
The authors acknowledge the technical help of Lynnette C. Johnson and Michael S. Murray for his work in the determination of the crystal structure of human Srx. This work was supported by funds from the National Institutes of Health, U.S.A. (R01-GM072866 to W.T.L.)
REFERENCES
- Apel K, Hirt H. Reactive oxygen species: metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant Biol. 2004;55:373–399. doi: 10.1146/annurev.arplant.55.031903.141701. [DOI] [PubMed] [Google Scholar]
- Baker LM, Raudonikiene A, Hoffman PS, Poole LB. Essential thioredoxin-dependent peroxiredoxin system from Helicobacter pylori: genetic and kinetic characterization. J. Bacteriol. 2001;183:1961–1973. doi: 10.1128/JB.183.6.1961-1973.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu MK, Koonin EV. Evolution of eukaryotic cysteine sulfinic acid reductase, sulfiredoxin (Srx), from bacterial chromosome partitioning protein ParB. Cell Cycle. 2005;4:947–952. doi: 10.4161/cc.4.7.1786. [DOI] [PubMed] [Google Scholar]
- Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- Bozonet SM, Findlay VJ, Day AM, Cameron J, Veal EA, Morgan BA. Oxidation of a eukaryotic 2-Cys peroxiredoxin is a molecular switch controlling the transcriptional response to increasing levels of hydrogen peroxide. J. Biol. Chem. 2005;280:23319–23327. doi: 10.1074/jbc.M502757200. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. doi: 10.1126/science.1095569. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, Skaliter R, Gudkov AV, Chumakov PM, Feinstein E. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017–6031. doi: 10.1038/sj.onc.1205877. [DOI] [PubMed] [Google Scholar]
- Budde H, Flohe L, Hecht HJ, Hofmann B, Stehr M, Wissing J, Lunsdorf H. Kinetics and redox-sensitive oligomerisation reveal negative subunit cooperativity in tryparedoxin peroxidase of Trypanosoma brucei. Biol. Chem. 2003;384:619–633. doi: 10.1515/BC.2003.069. [DOI] [PubMed] [Google Scholar]
- Castillo EA, Ayte J, Chiva C, Moldon A, Carrascal M, Abian J, Jones N, Hidalgo E. Diethylmaleate activates the transcription factor Pap1 by covalent modification of critical cysteine residues. Mol. Microbiol. 2002;45:243–254. doi: 10.1046/j.1365-2958.2002.03020.x. [DOI] [PubMed] [Google Scholar]
- Castro H, Budde H, Flohe L, Hofmann B, Lunsdorf H, Wissing J, Tomas AM. Specificity and kinetics of a mitochondrial peroxiredoxin of Leishmania infantum. Free Radic. Biol. Med. 2002;33:1563–1574. doi: 10.1016/s0891-5849(02)01088-2. [DOI] [PubMed] [Google Scholar]
- Cesaratto L, Vascotto C, D’Ambrosio C, Scaloni A, Baccarani U, Paron I, Damante G, Calligaris S, Quadrifoglio F, Tiribelli C, Tell G. Overoxidation of peroxiredoxins as an immediate and sensitive marker of oxidative stress in HepG2 cells and its application to the redox effects induced by ischemia/reperfusion in human liver. Free Radic. Res. 2005;39:255–268. doi: 10.1080/10715760400029603. [DOI] [PubMed] [Google Scholar]
- Chae HJ, Kim K, Kim IH. Redox Regulation of cell signaling and its clinical application. Marcel Dekker, Inc.; New York: 1999. pp. 85–92. [Google Scholar]
- Chae HZ, Chung SJ, Rhee SG. Thioredoxin-dependent peroxide reductase from yeast. J. Biol. Chem. 1994;269:27670–27678. [PubMed] [Google Scholar]
- Chae HZ, Kang SW, Rhee SG. Isoforms of mammalian peroxiredoxin that reduce peroxides in presence of thioredoxin. Methods Enzymol. 1999;300:219–226. doi: 10.1016/s0076-6879(99)00128-7. [DOI] [PubMed] [Google Scholar]
- Chae HZ, Kim HJ, Kang SW, Rhee SG. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pract. 1999;45:101–112. doi: 10.1016/s0168-8227(99)00037-6. [DOI] [PubMed] [Google Scholar]
- Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TS, Jeong W, Choi SY, Yu S, Kang SW, Rhee SG. Regulation of peroxiredoxin I activity by Cdc2-mediated phosphorylation. J. Biol. Chem. 2002;277:25370–25376. doi: 10.1074/jbc.M110432200. [DOI] [PubMed] [Google Scholar]
- Chang TS, Jeong W, Woo HA, Lee SM, Park S, Rhee SG. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J. Biol. Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- Chevallet M, Wagner E, Luche S, van Dorsselaer A, Leize-Wagner E, Rabilloud T. Regeneration of peroxiredoxins during recovery after oxidative stress: only some overoxidized peroxiredoxins can be reduced during recovery after oxidative stress. J. Biol. Chem. 2003;278:37146–37153. doi: 10.1074/jbc.M305161200. [DOI] [PubMed] [Google Scholar]
- Christman MF, Morgan RW, Jacobson FS, Ames BN. Positive control of a regulon for defenses against oxidative stress and some heat-shock proteins in Salmonella typhimurium. Cell. 1985;41:753–762. doi: 10.1016/s0092-8674(85)80056-8. [DOI] [PubMed] [Google Scholar]
- Chuang MH, Wu MS, Lo WL, Lin JT, Wong CH, Chiou SH. The antioxidant protein alkylhydroperoxide reductase of Helicobacter pylori switches from a peroxide reductase to a molecular chaperone function. Proc. Natl. Acad. Sci. U.S.A. 2006;103:2552–2557. doi: 10.1073/pnas.0510770103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, 3rd, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- Degols G, Shiozaki K, Russell P. Activation and regulation of the Spc1 stress-activated protein kinase in Schizosaccharomyces pombe. Mol. Cell Biol. 1996;16:2870–2877. doi: 10.1128/mcb.16.6.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz KJ, Horling F, Konig J, Baier M. The function of the chloroplast 2-cysteine peroxiredoxin in peroxide detoxification and its regulation. J. Exp. Bot. 2002;53:1321–1329. [PubMed] [Google Scholar]
- Dietz KJ, Jacob S, Oelze ML, Laxa M, Tognetti V, de Miranda SM, Baier M, Finkemeier I. The function of peroxiredoxins in plant organelle redox metabolism. J. Exp. Bot. 2006;57:1697–1709. doi: 10.1093/jxb/erj160. [DOI] [PubMed] [Google Scholar]
- Eichhorn E, van der Ploeg JR, Leisinger T. Characterization of a two-component alkanesulfonate monooxygenase from Escherichia coli. J. Biol. Chem. 1999;274:26639–26646. doi: 10.1074/jbc.274.38.26639. [DOI] [PubMed] [Google Scholar]
- Findlay VJ, Tapiero H, Townsend DM. Sulfiredoxin: a potential therapeutic agent? Biomed. Pharmacother. 2005;59:374–379. doi: 10.1016/j.biopha.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66:6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlayson AJ, MacKenzie SL, Finley FW. Reaction of alanine-3-sulfinic acid with 2-mercaptoethanol. Can. J. Chem. 1979;57:2073–2077. [Google Scholar]
- Flohe L, Budde H, Bruns K, Castro H, Clos J, Hofmann B, Kansal-Kalavar S, Krumme D, Menge U, Plank-Schumacher K, Sztajer H, Wissing J, Wylegalla C, Hecht HJ. Tryparedoxin peroxidase of Leishmania donovani: molecular cloning, heterologous expression, specificity, and catalytic mechanism. Arch. Biochem. Biophys. 2002;397:324–335. doi: 10.1006/abbi.2001.2688. [DOI] [PubMed] [Google Scholar]
- Hamann M, Zhang T, Hendrich S, Thomas JA. Quantitation of protein sulfinic and sulfonic acid, irreversibly oxidized protein cysteine sites in cellular proteins. Methods Enzymol. 2002;348:146–156. doi: 10.1016/s0076-6879(02)48634-x. [DOI] [PubMed] [Google Scholar]
- Hofmann B, Hecht H-J, Flohé L. Peroxiredoxins. Biol. Chem. 2002;383:347–364. doi: 10.1515/BC.2002.040. [DOI] [PubMed] [Google Scholar]
- Isermann K, Liebau E, Roeder T, Bruchhaus I. A peroxiredoxin specifically expressed in two types of pharyngeal neurons is required for normal growth and egg production in Caenorhabditis elegans. J. Mol. Biol. 2004;338:745–755. doi: 10.1016/j.jmb.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Jacob C, Holme AL, Fry FH. The sulfinic acid switch in proteins. Org. Biomol. Chem. 2004;2:1953–1956. doi: 10.1039/b406180b. [DOI] [PubMed] [Google Scholar]
- Jang HH, Kim SY, Park SK, Jeon HS, Lee YM, Jung JH, Lee SY, Chae HB, Jung YJ, Lee KO, Lim CO, Chung WS, Bahk JD, Yun DJ, Cho MJ. Phosphorylation and concomitant structural changes in human 2-Cys peroxiredoxin isotype I differentially regulate its peroxidase and molecular chaperone functions. FEBS Lett. 2006;580:351–355. doi: 10.1016/j.febslet.2005.12.030. [DOI] [PubMed] [Google Scholar]
- Jang HH, Lee KO, Chi YH, Jung BG, Park SK, Park JH, Lee JR, Lee SS, Moon JC, Yun JW, Choi YO, Kim WY, Kang JS, Cheong GW, Yun DJ, Rhee SG, Cho MJ, Lee SY. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Jeong W, Park SJ, Chang TS, Lee DY, Rhee SG. Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J. Biol. Chem. 2006;281:14400–14407. doi: 10.1074/jbc.M511082200. [DOI] [PubMed] [Google Scholar]
- Jönsson TJ, Murray MS, Johnson LC, Poole LB, Lowther WT. Structural basis for the retroreduction of inactivated peroxiredoxins by human sulfiredoxin. Biochemistry. 2005;44:8634–8642. doi: 10.1021/bi050131i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Rhee SG, Stadtman ER. Nonenzymatic cleavage of proteins by reactive oxygen species generated by dithiothreitol and iron. J. Biol. Chem. 1985;260:15394–15397. [PubMed] [Google Scholar]
- Konig J, Baier M, Horling F, Kahmann U, Harris G, Schurmann P, Dietz KJ. The plant-specific function of 2-Cys peroxiredoxin-mediated detoxification of peroxides in the redox-hierarchy of photosynthetic electron flux. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5738–5743. doi: 10.1073/pnas.072644999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo KH, Lee S, Jeong SY, Kim ET, Kim HJ, Kim K, Song K, Chae HZ. Regulation of thioredoxin peroxidase activity by C-terminal truncation. Arch. Biochem. Biophys. 2002;397:312–318. doi: 10.1006/abbi.2001.2700. [DOI] [PubMed] [Google Scholar]
- Kudo N, Taoka H, Yoshida M, Horinouchi S. Identification of a novel nuclear export signal sensitive to oxidative stress in yeast AP-1-like transcription factor. Ann. N. Y. Acad. Sci. 1999;886:204–207. doi: 10.1111/j.1749-6632.1999.tb09417.x. [DOI] [PubMed] [Google Scholar]
- Lee DY, Rhee SG, Ferretti J, Gruschus JM. 1H, 15N, and 13C chemical shift assignments of the human Sulfiredoxin (hSrx) J. Biomol. NMR. 2005;32:339. doi: 10.1007/s10858-005-0472-6. [DOI] [PubMed] [Google Scholar]
- Liu XP, Liu XY, Zhang J, Xia ZL, Liu X, Qin HJ, Wang DW. Molecular and functional characterization of sulfiredoxin homologs from higher plants. Cell Res. 2006;16:287–296. doi: 10.1038/sj.cr.7310036. [DOI] [PubMed] [Google Scholar]
- Martin IV, MacNeill SA. ATP-dependent DNA ligases. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-4-reviews3005. REVIEWS3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsumoto A, Nakagawa Y, Takeuchi A, Okawa K, Iwamatsu A, Takanezawa Y. Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat. Free Radic. Res. 2001;35:301–310. doi: 10.1080/10715760100300831. [DOI] [PubMed] [Google Scholar]
- Mitsumoto A, Takanezawa Y, Okawa K, Iwamatsu A, Nakagawa Y. Variants of peroxiredoxins expression in response to hydroperoxide stress. Free Radic. Biol. Med. 2001;30:625–635. doi: 10.1016/s0891-5849(00)00503-7. [DOI] [PubMed] [Google Scholar]
- Moon JC, Hah YS, Kim WY, Jung BG, Jang HH, Lee JR, Kim SY, Lee YM, Jeon MG, Kim CW, Cho MJ, Lee SY. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J. Biol. Chem. 2005;280:28775–28784. doi: 10.1074/jbc.M505362200. [DOI] [PubMed] [Google Scholar]
- Moore RB, Mankad MV, Shriver SK, Mankad VN, Plishker GA. Reconstitution of Ca(2+)-dependent K+ transport in erythrocyte membrane vesicles requires a cytoplasmic protein. J. Biol. Chem. 1991;266:18964–18968. [PubMed] [Google Scholar]
- Morgan RW, Christman MF, Jacobson FS, Storz G, Ames BN. Hydrogen peroxide-inducible proteins in Salmonella typhimurium overlap with heat shock and other stress proteins. Proc. Natl. Acad. Sci. U.S.A. 1986;83:8059–8063. doi: 10.1073/pnas.83.21.8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y, Poole LB, Massey V. Amphibacillus xylanus NADH oxidase and Salmonella typhimurium alkyl-hydroperoxide reductase flavoprotein components show extremely high scavenging activity for both alkyl hydroperoxide and hydrogen peroxide in the presence of S. typhimurium alkyl-hydroperoxide reductase 22-kDa protein component. J. Biol. Chem. 1995;270:25645–25650. doi: 10.1074/jbc.270.43.25645. [DOI] [PubMed] [Google Scholar]
- Nogoceke E, Gommel DU, Kiess M, Kalisz HM, Flohe L. A unique cascade of oxidoreductases catalyses trypanothione-mediated peroxide metabolism in Crithidia fasciculata. Biol. Chem. 1997;378:827–836. doi: 10.1515/bchm.1997.378.8.827. [DOI] [PubMed] [Google Scholar]
- Pannifer AD, Flint AJ, Tonks NK, Barford D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by x-ray crystallography. J. Biol. Chem. 1998;273:10454–10462. doi: 10.1074/jbc.273.17.10454. [DOI] [PubMed] [Google Scholar]
- Poole LB. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 2. Cystine disulfides involved in catalysis of peroxide reduction. Biochemistry. 1996;35:65–75. doi: 10.1021/bi951888k. [DOI] [PubMed] [Google Scholar]
- Quinn J, Findlay VJ, Dawson K, Millar JB, Jones N, Morgan BA, Toone WM. Distinct regulatory proteins control the graded transcriptional response to increasing H(2)O(2) levels in fission yeast Schizosaccharomyces pombe. Mol. Biol. Cell. 2002;13:805–816. doi: 10.1091/mbc.01-06-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- Schröder E, Littlechild JA, Lebedev AA, Errington N, Vagin AA, Isupov MN. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 Å resolution. Structure Fold Des. 2000;8:605–615. doi: 10.1016/s0969-2126(00)00147-7. [DOI] [PubMed] [Google Scholar]
- Seaver LC, Imlay JA. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J. Bacteriol. 2001;183:7182–7189. doi: 10.1128/JB.183.24.7182-7189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki K, Russell P. Cell-cycle control linked to extracellular environment by MAP kinase pathway in fission yeast. Nature. 1995;378:739–743. doi: 10.1038/378739a0. [DOI] [PubMed] [Google Scholar]
- Shiozaki K, Russell P. Conjugation, meiosis, and the osmotic stress response are regulated by Spc1 kinase through Atf1 transcription factor in fission yeast. Genes Dev. 1996;10:2276–2288. doi: 10.1101/gad.10.18.2276. [DOI] [PubMed] [Google Scholar]
- Timson DJ, Singleton MR, Wigley DB. DNA ligases in the repair and replication of DNA. Mutat. Res. 2000;460:301–318. doi: 10.1016/s0921-8777(00)00033-1. [DOI] [PubMed] [Google Scholar]
- Toone WM, Kuge S, Samuels M, Morgan BA, Toda T, Jones N. Regulation of the fission yeast transcription factor Pap1 by oxidative stress: requirement for the nuclear export factor Crm1 (Exportin) and the stress-activated MAP kinase Sty1/Spc1. Genes Dev. 1998;12:1453–1463. doi: 10.1101/gad.12.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traut TW. The functions and consensus motifs of nine types of peptide segments that form different types of nucleotide-binding sites. Eur. J. Biochem. 1994;222:9–19. doi: 10.1111/j.1432-1033.1994.tb18835.x. [DOI] [PubMed] [Google Scholar]
- Veal EA, Findlay VJ, Day AM, Bozonet SM, Evans JM, Quinn J, Morgan BA. A 2-Cys peroxiredoxin regulates peroxide-induced oxidation and activation of a stress-activated MAP kinase. Mol. Cell. 2004;15:129–139. doi: 10.1016/j.molcel.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Velasco-Miguel S, Buckbinder L, Jean P, Gelbert L, Talbott R, Laidlaw J, Seizinger B, Kley N. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene. 1999;18:127–137. doi: 10.1038/sj.onc.1202274. [DOI] [PubMed] [Google Scholar]
- Vivancos AP, Castillo EA, Biteau B, Nicot C, Ayte J, Toledano MB, Hidalgo E. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc. Natl. Acad. Sci U.S.A. 2005;102:8875–8880. doi: 10.1073/pnas.0503251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivancos AP, Castillo EA, Jones N, Ayte J, Hidalgo E. Activation of the redox sensor Pap1 by hydrogen peroxide requires modulation of the intracellular oxidant concentration. Mol. Microbiol. 2004;52:1427–1435. doi: 10.1111/j.1365-2958.2004.04065.x. [DOI] [PubMed] [Google Scholar]
- Wilkinson MG, Samuels M, Takeda T, Toone WM, Shieh JC, Toda T, Millar JB, Jones N. The Atf1 transcription factor is a target for the Sty1 stress-activated MAP kinase pathway in fission yeast. Genes Dev. 1996;10:2289–2301. doi: 10.1101/gad.10.18.2289. [DOI] [PubMed] [Google Scholar]
- Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K, Rhee SG. Reversing the inactivation of peroxiredoxins caused by cysteine sulfinic acid formation. Science. 2003;300:653–656. doi: 10.1126/science.1080273. [DOI] [PubMed] [Google Scholar]
- Woo HA, Jeong W, Chang TS, Park KJ, Park SJ, Yang JS, Rhee SG. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-Cys peroxiredoxins. J. Biol. Chem. 2005;280:3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- Woo HA, Kang SW, Kim HK, Yang KS, Chae HZ, Rhee SG. Reversible oxidation of the active site cysteine of peroxiredoxins to cysteine sulfinic acid. Immunoblot detection with antibodies specific for the hyperoxidized cysteine-containing sequence. J. Biol. Chem. 2003;278:47361–47364. doi: 10.1074/jbc.C300428200. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Schröder E, Harris RJ, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- Yang KS, Kang SW, Woo HA, Hwang SC, Chae HZ, Kim K, Rhee SG. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J. Biol. Chem. 2002;277:38029–38036. doi: 10.1074/jbc.M206626200. [DOI] [PubMed] [Google Scholar]