

Summary
Prior to initiating DNA synthesis, E. coli oriC switches from ORC, comprising initiator DnaA bound at three high affinity sites, to pre-RC, when additional DnaA molecules interact with low affinity sites. Two types of low affinity sites exist; R boxes that bind DnaA-ATP and DnaA-ADP with equal affinity, and I-sites with a 3–4 fold preference for DnaA-ATP. To assess the regulatory role of weak DnaA interactions during pre-RC assembly in vivo, we compared the behavior of plasmid-borne wild-type oriC with mutants having an increased or decreased number of DnaA-ATP discriminatory I-sites. Increasing the number of discriminatory sites by replacing R5M with I2 inactivated extrachromosomal oriC function. Mutants with no discriminatory sites perturbed host growth and rapidly replaced wild-type chromosomal oriC, but normal function returned if one I-site was restored at either the I2, I3 or R5M position. These observations are consistent with assembly of E. coli pre-RC in vivo from mixtures of DnaA-ATP and DnaA-ADP, with I-site interactions coupling pre-RC assembly to DnaA-ATP levels.
Keywords: oriC, DnaA, pre-RC, ORC, I-sites, R boxes
Introduction
Initiation of chromosome replication is precisely regulated in all cells so that replication origins initiate at the correct time only once during each cell cycle, reviewed in (Boye et al., 2000; Diffley, 2004; Messer, 2002). Such stringent regulation is dependent, in part, on properly timed and ordered conversion of origin recognition complexes (ORC) into pre-replication complexes (pre-RC) comprising replication origin DNA and origin-binding initiator proteins (Baker and Bell, 1998; Leonard and Grimwade, 2005). For E. coli, both ORC and pre-RC assembly require oriC interactions with initiator DnaA, a AAA+ protein (Davey et al., 2002; Kaguni, 2006; Messer, 2002; Skarstad and Boye, 1994) with structural similarities to Cdc6/Orc1(Erzberger et al., 2002).
DnaA interacts with two classes of 9-mer recognition sequences within the 245 bp E. coli oriC (Figure 1A) (Bramhill and Kornberg, 1988; Fuller et al., 1984; Leonard and Grimwade, 2005; Margulies and Kaguni, 1996; McGarry et al., 2004). One class, termed R boxes (Figure 1A), carries the consensus 5′-TGTGNAA/TAA, and shows equivalent affinities for DnaA-ADP or DnaA-ATP (Fuller et al., 1984; Matsui et al., 1985; Sekimizu et al., 1987). The other class, termed I-sites (Figure 1A) with consensus 5′-TT/GGGATCAA/G, shows a 3–4 fold preference for DnaA-ATP (Kawakami et al., 2005; McGarry et al., 2004). R1, R2, and R4 are the highest affinity sites (Grimwade et al., 2000; Margulies and Kaguni, 1996), and are bound to DnaA during the majority of the cell cycle (Cassler et al., 1995; Nievera et al., 2006), forming a complex that is temporally and functionally equivalent to budding yeast ORC. Near the time of initiation, R5M and I-sites bind additional DnaA to form the pre-RC (Nievera et al., 2006; Ryan et al., 2002) with localized DNA strand separation within a region containing three A-T rich, 13-mer repeats (Bramhill and Kornberg, 1988; Gille and Messer, 1991) (Figure 1). DnaB helicase is then loaded onto the available single strands, followed by the sequential loading of DnaG primase and DNA polymerase III holoenzyme (Benkovic et al., 2001; Fang et al., 1999; Kornberg and Baker, 1992). Other DnaA-ATP interactions within the 13 mer region at a smaller recognition consensus 5′-ATGATC-3′ may enhance the stability of separated DNA strands (Speck and Messer, 2001). It is not known how DnaA-oriC interactions are able to produce localized DNA strand separation, but it was recently reported that DnaA-ATP forms right handed helical filaments with the potential to change the topology of supercoiled DNA (Erzberger et al., 2006). During pre-RC assembly, DnaA-ATP binding to sites with a preference for DnaA-ATP would be expected to play a key role in proper assembly of the helical filament.
Figure 1.

DnaA binding sites in oriC. A) Relative position of R boxes and I-sites are marked as open boxes on the line representing E. coli oriC. Position of the duplex unwinding region (DUE), as well as binding sites for architectural proteins Fis and IHF are also indicated (solid lines) below oriC. R box sequences are shown above oriC, and wt and mutated I-site sequences (oriCI2/I3ADP) are shown below oriC. The change in DMS modification pattern caused by DnaA binding is indicated by up (increased modification) or down (decreased modification) arrows. B) The preference for DnaA-ATP or DnaA-ADP at low affinity sites can be modified by site-directed mutagenesis. DMS modification patterns were measured after incubation of either wt oriC, oriCMATP, or oriCI2/I3ADP with DnaA-ATP or DnaA-ADP. Positions of binding sites R5M, I1, I2, I3 and R4 are marked, and bands representing the Gs at position 2 or 4 (when present in a site) are indicated. Relative intensities of DMS modified guanosines in all eight DnaA binding sites were quantified from scans of footprinting gels. Quantitation from a representative scan is shown.
DnaA levels remain constant throughout the E. coli cell cycle, but the proportion of active DnaA-ATP and inactive DnaA-ADP fluctuates, such that DnaA-ATP levels peak near the time of initiation (Kurokawa et al., 1999; Sekimizu et al., 1987). The increase in DnaA-ATP comes from newly synthesized DnaA, which rapidly binds ATP, and from conversion of DnaA-ADP to DnaA-ATP, mediated by acidic membrane phospholipids (Crooke, 2001). The decrease in DnaA-ATP after initiation results from replication-coupled hydrolysis (Kato, 2005; Lee and Bell, 2000). Total cellular DnaA levels are also regulated to permit E. coli cells to accommodate many copies of oriC, all of which initiate replication synchronously (Helmstetter and Leonard, 1987; Leonard and Helmstetter, 1986; Leonard et al., 1990; Skarstad et al., 1986; Skarstad et al., 2003). During rapid growth, E. coli cells contain more than one copy of oriC because new rounds of DNA replication are initiated before sufficient time has passed to complete the previous round creating multi-forked chromosomes (Helmstetter, 1996). Additionally, up to 30 extrachromosomal copies of oriC as minichromosomes or cloned into pBR322 can be tolerated. Multiple oriC copies on plasmids do not perturb host growth unless the chromosome harbors additional mutations that compromise initiation of replication or produce initiation asynchrony (dnaA, dam, seqA, ihf, fis) (Dasgupta and Lobner-Olesen, 2004; Koppes, 1987; Lobner-Olesen et al., 1994; Lobner-Olesen and von Freiesleben, 1996; Skarstad and Lobner-Olesen, 2003). In these compromised hosts, competition between chromosomal and extrachromosomal oriC copies results in plasmid integration into the genome (Jensen et al., 1990; Lobner-Olesen, 1999). In addition to the mutations in replication proteins, a wide variety of mutations in oriC have been constructed and harbored on plasmids with a pBR322 origin. Although many of these oriC mutations have resulted in loss of plasmid oriC function when placed in a wild-type host, none are reported to either integrate spontaneously into the genome or inhibit host growth when harbored at high copy number, as might be expected from mutation which causes the plasmid oriC to out-compete chromosomal oriC (Bates et al., 1995; Oka et al., 1982; Oka et al., 1984; Weigel et al., 2001).
Since weak DnaA interactions required for pre-RC assembly include both I-site sequences (preferring DnaA-ATP) and R boxes (non-discriminatory sites), we wondered if altering the number and location of the discriminatory sites would affect oriC function and host growth. We chose to examine this by using the plasmid competition system. Using site-specific mutagenesis, DnaA-ATP preference was removed from sites I2 and I3 (oriCI2/I3ADP) (McGarry et al., 2004) and an “extra I-site” mutant (oriCMATP) was also constructed by converting R5M into I2. The behavior of these mutant oriCs carried on pBR322 was examined in hosts carrying wild-type or compromised (ORC-defective oriC ΔR4) oriC on the chromosome. We found that removing DnaA-ATP preference from I2 and I3 was sufficient to produce an oriC that would rapidly out-compete wild-type chromosomal oriC, dramatically inhibit host growth, and cause replacement of the wild-type chromosomal origin with the mutant version in survivors. This phenotype was suppressed by adding back one I-site at either I2, I3, or R5M positions. An oriC with three I-sites was unable to function in the presence of wild-type chromosomal oriC, but both growth inhibition and integration into the chromosome were observed in hosts carrying compromised chromosomal oriC. Based on these behaviors we propose that both nucleotide forms of DnaA are used to assemble E. coli pre-RC in vivo, with I-sites responsible for coupling oriC function to DnaA-ATP levels during pre-RC assembly.
Results
An extrachromosomal oriC with three discriminatory I-sites is functional only when chromosomal oriC is compromised
Three low affinity DnaA binding sites in oriC, R5M, I2, and I3, simultaneously become occupied during pre-RC formation. In vitro DMS and DNase I footprinting studies have demonstrated that all three sites bind DnaA-ATP with equal affinity. However, R5M binds DnaA-ADP with the same affinity as DnaA-ATP, unlike I-sites, which discriminate between DnaA nucleotide forms (Kawakami et al., 2005; McGarry et al., 2004). DnaA binding to oriC causes distinctive changes to the modification pattern produced by DMS, comprising a hypersensitive guanine residue in the fourth position of the DnaA 9 mer sequence, and suppressed modification of the guanine in the second position (if the site contains a G in this position) (Figure 1).
To test whether or not the ability of R5M to bind DnaA-ADP played a role in pre-RC formation in vivo, we replaced R5M in wt oriC with I2, creating oriCMATP; this mutant oriC has three discriminatory I-sites. Binding of DnaA-ATP and DnaA-ADP was examined using in vitro DMS footprinting, which is likely to reflect binding preferences in vivo as well. In vitro DMS footprints verify that oriCMATP has three sites that preferentially bind DnaA-ATP: the mutated R5M position, I2, and I3 (Figure 1B, second panel). The in vitro footprints also show that occupation of R5M and I2 in the R5M position (termed I2M) is first observed at comparable DnaA concentrations (Figure 1B, see binding pattern for 40 nM DnaA-ATP in first and second panels), indicating that R5M and I2M have similar affinities for DnaA-ATP. OriCMATP was cloned into a plasmid that also contained the pBR322 origin, and oriC function was tested by transforming a polA strain with the chimeric plasmid. Since the pBR322 origin requires Pol I to replicate, transformants are obtained only if replication initiates from the plasmid oriC. Interestingly, extrachromosomal oriCMATP was not functional in the presence of wt chromosomal oriC in the polA strain (Table 1). This result could arise either because oriCMATP functions less efficiently than chromosomal wt oriC, or because the mutation reduced the ability of oriCMATP to initiate replication under any conditions. To determine if oriCMATP retained any activity, we removed the pBR322 origin from the chimeric plasmid, making a minichromosome. This minichromosome successfully transformed cells whose chromosomal oriC was made less efficient by a scrambled R4 box (oriCΔR4) (Weigel et al., 2001) indicating that oriCMATP was functional when not forced to compete with wt oriC (data not shown). Since oriCMATP differs from wild type only in the replacement of R5M with I2, it appears loss of the ability to bind DnaA-ADP at the R5M position renders oriCMATP less efficient than wt oriC, indicating that binding of DnaA-ADP during pre-RC assembly plays a role in normal oriC function.
Table 1.
Activity of oriC plasmids mutated in low affinity DnaA binding sites after transformation of cells with wt chromosomal oriC
| Mutation | Mnemonic | Discriminatory sites | polA& | Colony | Stable Integration# |
|---|---|---|---|---|---|
| Wild type | wt oriC | I2, I3 | 1.0 | normal | no |
| R5M to I2 | oriCMATP | I2, I3, R5M | <0.001 | normal | no |
| I2/I3 G3 to T | oriCI2/I3ADP | none | 0.6 | very small | yes, >85% |
| I2 G3 to T | oriCI2ADP | I3 | 0.5 | normal | no |
| I3 G3 to T | oriCI3ADP | I2 | 0.4 | normal | no |
| I2/I3 G3 to T R5M to I2 | oriCI2/I3ADP/MATP | R5M | 0.5 | normal | no |
Transformants of p3478polA/W3110 by test plasmid, normalized for transformation by wt oriC plasmid.
Measured by MAMA-PCR after overnight culture of primary transformed colonies.
An oriC mutation that allows DnaA-ADP binding to I2 and I3 (oriCI2/I3ADP) perturbs host growth
Since making R5M into a discriminatory I-site decreased oriC function, we decided to examine whether discrimination between DnaA-ATP and DnaA-ADP at I2 and I3 was also important for normal origin activity. The preference of I2 and I3 for DnaA-ATP can be eliminated by replacing the G in position 3 of I2 and I3 with a T (McGarry et al., 2004). The resulting origin, oriCI2/I3ADP, was cloned into a pBR322 plasmid. PolA cells were successfully transformed by this chimeric plasmid (Table 1), indicating that extrachromosomal oriC12/13ADP was functional, but the transformation of wild type cells resulted in heterogenous, but generally small, slow growing, transparent colonies (Table 1, Figure 2A). This perturbed phenotype is not simply due to the presence of a plasmid with two origins, since transformation of a plasmid harboring wt oriC and the pBR322 origin plasmid yielded normal-looking colonies (Figure 2A). The colonies harboring oriCI2/I3ADP also contained fewer viable cells (10–20% viable cells) than colonies harboring the wt chimeric plasmid (100% viable cells, data not shown).
Figure 2.
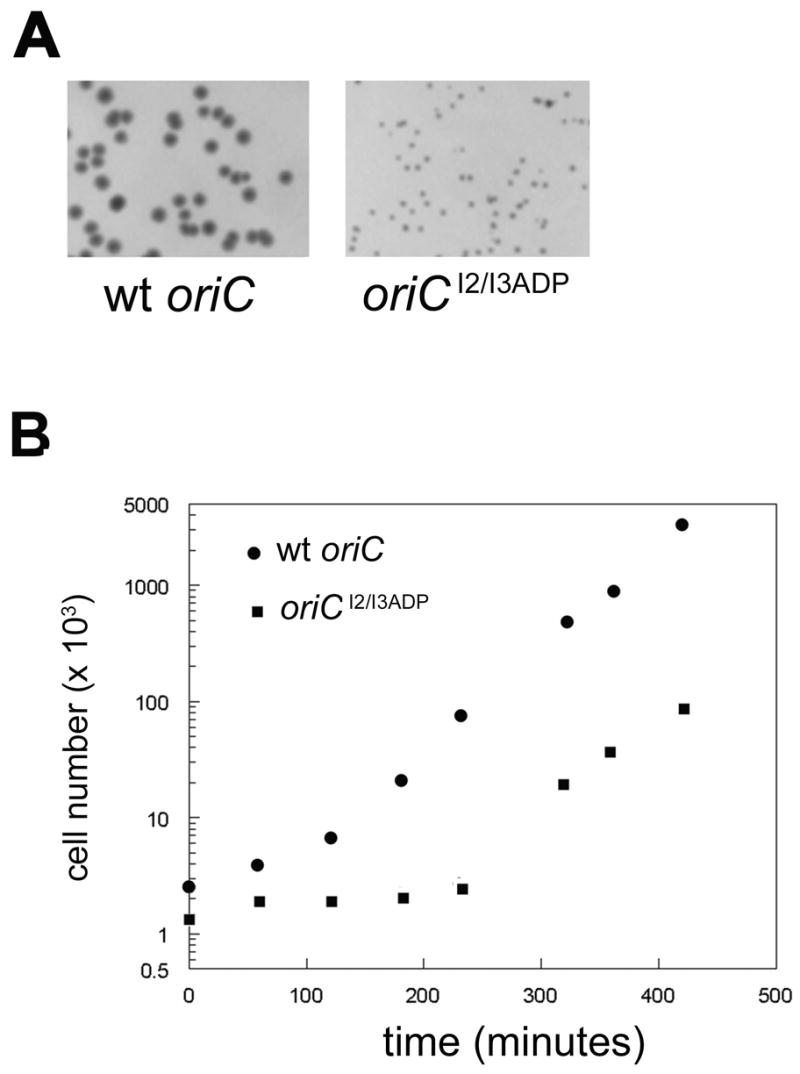
Transformation with pBR322/oriCI2/I3ADP chimeric plasmids perturbs host cell growth. A) Representative colonies after overnight incubation of W3110 cells (with wt chromosomal oriC) transformed with either wt oriC (left) or oriCI2/I3ADP (right) chimeric plasmids. B) A single colony of cells transformed with either the wt oriC or the oriCI2/I3ADP was grown in non-selective Luria-Broth. Cell number was measured at the times indicated using a model ZB Coulter Counter.
When cultured in non-selective media, cells harboring wt oriC plasmid grew normally, while cells harboring oriCI2/I3ADP plasmid had a prolonged, over 3 hour lag phase (Figure 2B). After both cultures entered exponential growth, they grew at the same rate (Figure 2B), suggesting that cells were eventually able to recover from the detrimental effects of the oriCI2/I3ADP plasmid.
Growth perturbation is due to competition with chromosomal oriC, and host survival requires integration of oriCI2/I3ADP into the genome
One possible reason for the growth perturbation caused by the oriCI2/I3ADP chimera is competition between the mutant origin and wt oriC on the host chromosome. If this is the case then the oriCI2/I3ADP chimera should not alter growth of a host strain in which chromosomal oriC is deleted. To test this, wt oriC and oriCI2/I3ADP plasmids were transformed into MM294ΔoriC::pKN1562 (clockwise), which replicates from a R1 plasmid origin (Koppes and Nordstrom, 1986). Neither plasmid altered colony morphology (not shown) or growth of this strain, although ΔoriC cells grew more slowly than cells with a normal chromosomal copy of oriC (Figure 3A). These data are consistent with the hypothesis that the growth defect caused by the oriCI2/I3ADP plasmid was due to competition with host oriC.
Figure 3.

Competition of oriCI2/I3ADP with wt host oriC causes growth perturbation and spontaneous integration into the chromosomal oriC region. A) ΔoriC cells were transformed with either wt oriC or oriCI2/I3ADP chimeric plasmids. After overnight incubation, single colonies were grown in non-selective Luria-Bertani media. Cell number was measured at the indicated times. B) Cells were transformed with chimeric plasmids containing either B/r oriC or oriCI2/I3ADP, and chromosomal oriC in cells from primary transformed colonies or after one passage in overnight culture from primary colonies was examined using MAMA-PCR. The amplification product (marked by arrow) was resolved on an agarose gel. C) Summary of data obtained by analyzing the chromosomal oriC from 100 or more colonies from either the primary transformants or the overnight cultures.
Previous studies demonstrated that wt oriC plasmids spontaneously integrate into the chromosome of cells harboring mutations that compromised initiation, due to competition between host and plasmid origins ((Skarstad et al., 2003; Skarstad and Lobner-Olesen, 2003, Lobner-Olesen, 1999). Since the oriCI2/I3ADP plasmid appears to compete with host oriC, we tested to see if this mutant oriC also integrated into the chromosome. Cells were transformed with either oriCI2/I3ADP or a plasmid harboring the E. coli B/r oriC, which differs from the published K12 oriC sequence by 2 bp changes (213A to T, and 221 T to C, (de Wind et al., 1987). The B/r oriC plasmid does not perturb host growth, and behaves identically to the K12 oriC plasmid (data not shown). Cells from at least 100 primary transformed colonies were subjected to Mismatch Amplification Mutational Analysis (MAMA-PCR), to detect single bp changes in DNA (described in Experimental Procedures). Primers were designed to detect K12 oriC, B/r oriC, or oriCI2/I3ADP in the position of chromosomal oriC. Amplification products were examined by agarose gel electrophoresis (Figure 3B, and Table 1). B/r oriC plasmids integrated infrequently, since fewer than 15% of colonies gave even a weak amplification product using primers to detect the B/r oriC (Figure 3B, second panel, and Figure 3C). These integrates were not stable, and no chromosomal B/r oriC was observed after overnight culture of the primary transformants (Figure 3B, fourth panel, and Figure 3C). In contrast, when cells were transformed with oriCI2/I3ADP plasmid, 30%–40% of colonies contained cells that had integrated the mutant origin. Further, the strong amplification signal obtained using the oriCI2/I3ADP primers indicates that most of the cells in the colony were integrates (Figure 3B, first panel, and Figure 3C). Integration of oriCI2/I3ADP was stable, since cultures from 90% of the primary transformants contained cells with the mutant origin (Figure 3B, third panel; Figure 3C). The chromosomal oriC region from a subset of the stable integrates was amplified by PCR and sequenced, verifying that the oriCI2/I3ADP had replaced wt oriC on the chromosome. We are currently investigating initiation timing of chromosomal oriCI2/I3ADP during the cell cycle (manuscript in preparation).
If integration of oriCI2/I3ADP was caused by competition with host oriC, then the growth perturbation caused by transformation of the mutant plasmid should be relieved by the integration event. This was tested in two ways. First, wt oriC and oriCI2/I3ADP plasmids were transformed into recombination defective (recA) cells. Colonies harboring the wt oriC appeared normal (Figure 4A, top panel), and grew without a notable lag phase when cultured in non-selective media (Figure 4B). In contrast, cells transformed with oriCI2/I3ADP formed extremely small, very transparent colonies (Figure 4A, bottom panel) containing cells that did not grow within 8 hours of culture in non-selective media (Figure 4B), indicating that recovery from the growth perturbation required a recombination event. We also tested to see if integration of oriCI2/I3ADP correlated with resumption of normal growth after the prolonged lag phase noted in normal cells (Figure 2). Colonies resulting from transformation with oriCI2/I3ADP were tested for integration of the mutant origin, by MAMA-PCR. A colony that harbored plasmid, but had not yet integrated the mutant oriC into the chromosome was used to inoculate a culture in non-selective media. Aliquots of the culture were taken at various time intervals, cell growth was monitored (Figure 4C), and cells were tested for mutant origin integration (Figure 4C, inset). During the prolonged lag phase, integration of the mutant origin was not observed. However, the oriCI2/I3ADP amplification product appeared coincidently with resumption of normal growth. Combined, these data indicate that RecA-mediated recombination of oriCI2/I3ADP, replacing the host oriC, was required for normal host growth and survival.
Figure 4.
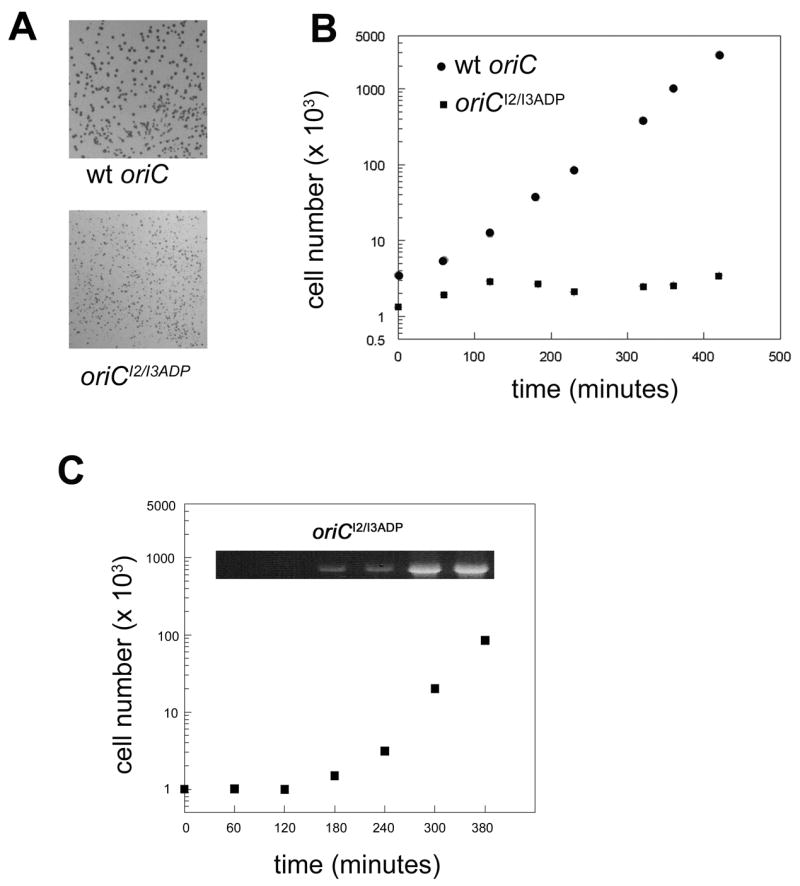
Integration relieves growth perturbation caused by oriCI2/I3ADP. A) MM294 recA was transformed with either wt oriC or oriCI2/I3ADP chimeric plasmids. Representative colonies after overnight culture are shown. B) A single colony was picked and grown in non-selective Luria-Bertani media. Cell number was measured at the indicated times. C) MM294 (recA +) were transformed with oriCI2/I3ADP/pBR322 plasmid. Colonies were tested for mutant origin integration by MAMA-PCR. A colony that tested negative for integration was picked, and grown in non-selective Luria-Bertani media. At the indicated times, cell number was measured, and an aliquot of cells from the culture were examined for integration of oriCI2/I3ADP by MAMA-PCR (inset).
Extrachromosmal oriCs containing only one discriminatory I-site are functional and do not perturb host growth
The studies described above indicate that loss of discrimination from two I-sites results in an oriC that can effectively out-compete wt chromosomal oriC. However, they do not reveal whether or not one I-site plays a more critical role in this competition than the other. To examine this, we made chimeric oriC/pBR322 plasmids in which oriC was mutated in only one I-site (oriCI2ADP, or oriCI3ADP). Both oriCI2ADP and oriCI3ADP were similar to wt oriC in their ability to transform polA cells containing wt chromosomal oriC (Table 1). Additionally, competition between the plasmid and host oriC was not detected; the plasmids did not cause altered colony morphology (Table 1, Figure 5, panels 3 and 4), and they did not form stable integrates in the host chromosome (Table 1). Thus, it appears that one discriminatory I-site is sufficient for a plasmid oriC to stably coexist with a wt chromosomal oriC.
Figure 5.
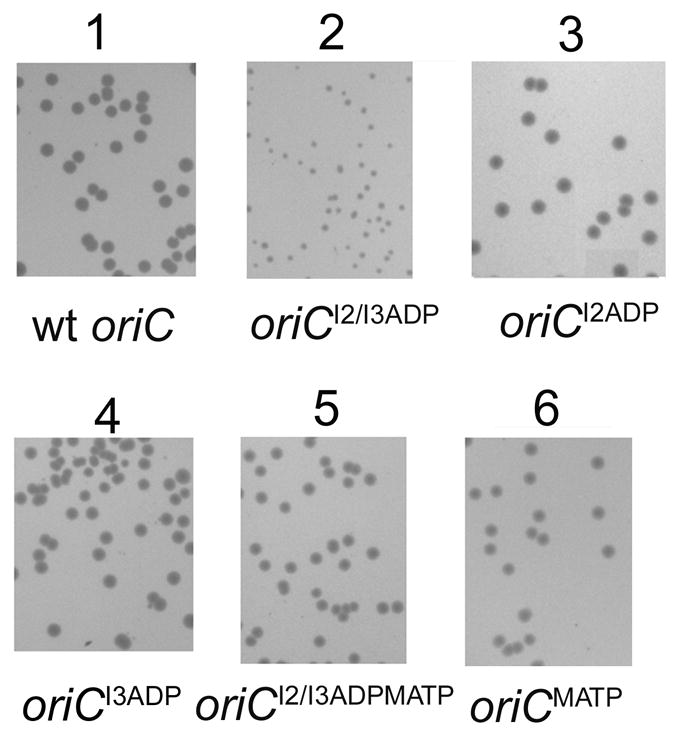
Transformation of cells with oriC chimeric plasmids with only one I-site results in normal colonies. W3110 was transformed with chimeric plamsids containing wt oriC (panel 1), oriCI2/I3ADP (panel 2), oriCI2ADP (panel 3), oriC I3ADP (panel 4), oriC I2/I3ADP/MATP (panel 5), or oriCMATP (panel 6). Colonies are shown after overnight growth.
I2 and I3 are separated by 70 bp, but the results described above indicate that the position of discriminatory I-sites is less important than their number. To test this idea further, we replaced R5M in oriCI2/I3ADP with I2 (oriCI2/I3ADP/MATP). We examined binding of DnaA-ATP and DnaA-ADP to this mutant origin using DMS footprinting (Figure 6). The footprints verify that this origin DnaA-ATP preferentially binds to the mutated R5M position, but the I2 and I3 positions bind DnaA-ATP and DnaA-ADP equally (Figure 6). The oriCI2I3ADP/MATP successfully transformed the polA strain, (Table 1), colonies formed by transformation into a wt strain appeared normal (Figure 5, panel 5), and the plasmid did not integrate into the host chromosome (Table 1). These data suggest that the position of discriminatory I-sites in oriC is not critical for function.
Figure 6.
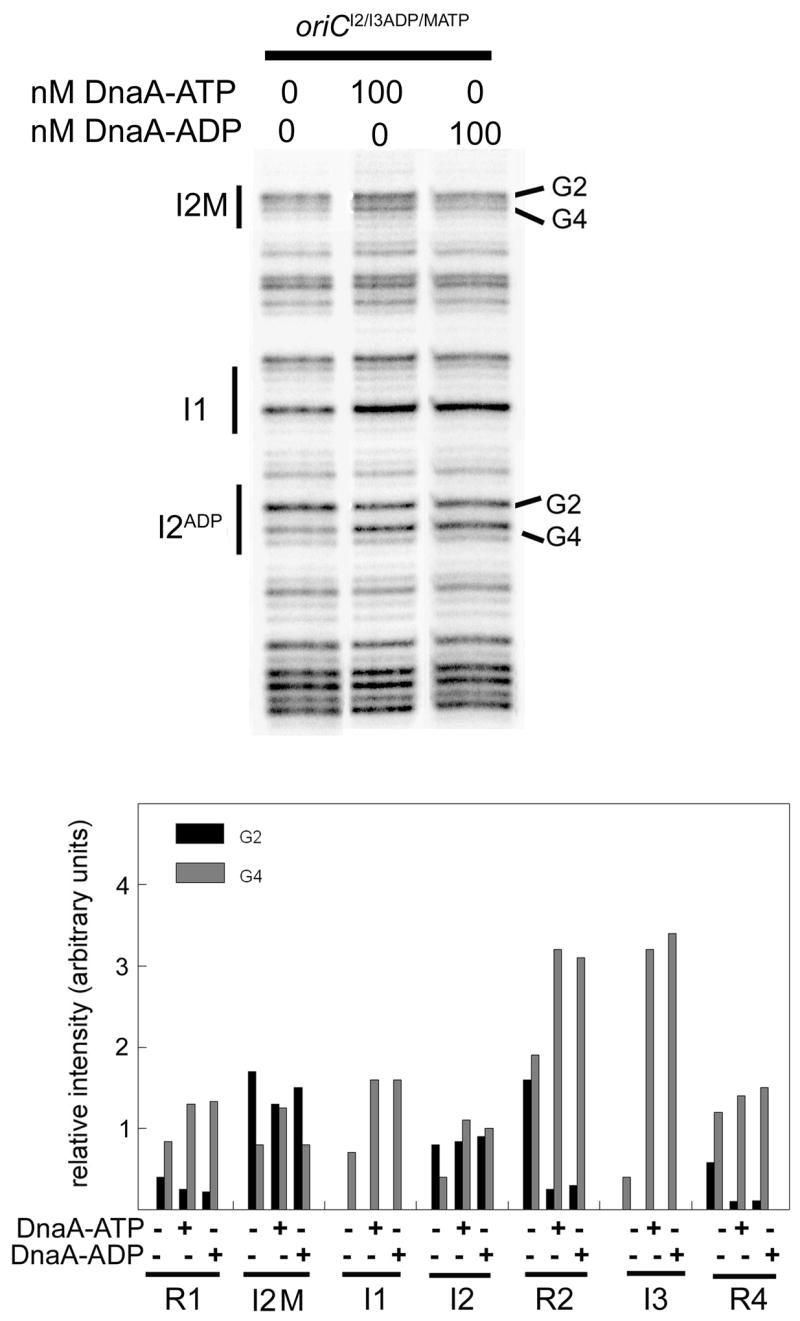
Replacement of R5M with I2 on oriCI2/I3ADP results in a DnaA-ATP site in the R5M position of oriC. R5M was replaced by I2 in oriCI2/I3ADP by site-directed mutagenesis to make oriCI2/I3ADP/MATP. DMS modification patterns were measured after incubation of oriCI2/I3ADP/MATP with DnaA-ATP or DnaA-ADP. Positions of binding sites R5M, I1, and I2 are marked, and bands representing the Gs at position 2 or 4 are indicated. Relative intensities of DMS modified guanosines in all eight DnaA binding sites were quantified from scans of footprinting gels. Quantitation from a representative scan is shown.
A new method for replacing chromosomal oriC with mutant versions
This work supports the results of previous studies in demonstrating that mutations which reduce oriC efficiency can not function as extrachromomsomal oriCs in the presence of wt chromosomal oriC (Langer et al., 1996; Lobner-Olesen, 1999; Oka et al., 1984). Many oriC mutants retain enough activity to function as the sole oriC in the cell (Bates et al., 1995; Weigel et al., 2001), but all of the processes reported to replace chromosomal oriC require multiple steps and do not give very many stable integrates (Weigel et al., 2001). The studies in this work have provided new information, demonstrating that a plasmid harboring mutations that should logically increase oriC efficiency can effectively out-compete chromosomal oriC, resulting in growth perturbation that is relieved by high frequency, stable integration of the more efficient origin. If this is generally true, then cells harboring mutations in chromosomal oriC that reduce function, such as oriCΔR4, should readily integrate normal or less perturbed mutated origins harbored on plasmids. We tested this hypothesis, by transforming oriCΔR4 cells with wt oriC/pBR322 chimeric plasmids. The transformed colonies were small and clear. MAMA-PCR analysis of cells in the colonies showed that plasmid wt oriC replaced the chromosomal oriCΔR4 with high frequency, and over 60% of the colonies contained stably integrated wt origins (data not shown). The frequency of integration was similar to that seen with the oriCI2/I3ADP mutation in wt cells. Thus, by using an origin “ranking” system, it should be possible to rapidly replace chromosomal oriC with mutant oriCs, as long as the chromosomal oriC is less efficient that the replacement copy.
Discussion
Unwinding of oriC in vitro requires DnaA-ATP (Sekimizu et al., 1987), and newly synthesized DnaA-ATP is believed to be the limiting factor for in vivo pre-RC assembly in E. coli. However, it is unclear whether every DnaA molecule comprising pre-RC is in the DnaA-ATP form, since R boxes bind DnaA-ATP and DnaA-ADP equally. In vitro, the replication activity of low levels of DnaA-ATP can be augmented by DnaA-ADP (Yung et al., 1990), suggesting that at some stage of initiation, both forms make positive contributions. We suggest that this stage is pre-RC formation, when discriminatory I-sites and non-discriminatory R5M are filled (Nievera et al., 2006; Ryan et al., 2002). The behavior of the “loss of preference” mutant oriCI2/I3ADP and the “extra I-site” mutant oriC/MATP reported here are consistent with this scenario.
Decreasing the requirement for DnaA-ATP by mutating I2 and I3 resulted in chimeric oriCI2/I3ADP plasmids that inhibited host cell growth and integrated rapidly into the chromosome. The growth perturbation caused by the high copy chimeric plasmid suggests that the plasmid titrated sufficient DnaA or other proteins to prohibit chromosomal initiation. One way this might occur is through premature initiation of the plasmid origin, triggered by filling of I-sites with DnaA-ADP. Unwinding would be followed by recruitment of DnaA-ATP to the single-stranded 13-mer region on the plasmids, since this region is reported to interact directly with DnaA-ATP (Speck and Messer, 2001; Yung and Kornberg, 1989), and DnaA-ATP is required to localize strand separation in oriCI2/I3ADP to the 13 mer region (McGarry et al., 2004). It is also possible that early unwinding of the plasmid origins titrated DnaB, or some other required factor, away from chromosomal oriC. We have observed early initiations of oriCI2/I3ADP minichromosomes (manuscript in preparation). However, since minichromosomes are harbored at a lower copy number than chimeric plasmids (Leonard et al., 1990), minichromosomes are not as effective at titrating initiation proteins; thus oriCI2/I3ADP minichromosomes do not severely inhibit host growth.
Integration of oriCI2/I3ADP relieved growth perturbation, suggesting that despite the loss of I-site preference for DnaA-ATP, sufficient negative regulation remains to prevent lethal runaway replication (Simmons et al., 2004). Blocking of pre-RC formation by the hemimethylated DNA protein, SeqA, remains functional following the onset of chromosome replication since the required GATC sequences in I2 and I3 were not altered in the “loss of preference” mutants (Boye et al., 2000; Campbell and Kleckner, 1990; Kaguni, 2006; Kato, 2005; Lu et al., 1994; Nievera et al., 2006; Skarstad et al., 2000). DNA replication-coupled hydrolysis of DnaA-ATP (Katayama, 2001) will also prevent over-initiation, since oriCI2/I3ADP requires DnaA-ATP for localized unwinding (McGarry et al., 2004).
Co-existence of wild-type and oriCI2/I3ADP required adding back just one I-site at I2, I3, or R5M. Thus, although coupling of DnaA-ATP binding to the assembly of pre-RC is an important feature for correct regulation, the location of this binding within the complex appears to be flexible. Further, the behavior of the oriCMATP mutant is consistent with the idea that DnaA-ADP normally interacts with non-discriminatory weak sites. If DnaA-ATP was the only form of the initiator protein used during normal pre-RC formation, then conversion of R5M to I2 should not alter oriCMATP function. However, oriCMATP was non-functional in the presence of wild-type oriC, indicating that the added requirement for DnaA-ATP caused a competition for DnaA. Earlier initiation of chromosomal DNA replication from wild-type oriC (containing DnaA-ADP in the pre-RC) and the resulting replication-coupled DnaA-ATP hydrolysis prevents the DnaA-ATP levels from ever being high enough to fill all three extrachromosomal I-sites in oriCMATP.
The possibility that E. coli pre-RC comprises a mixture of both inactive DnaA-ADP and active DnaA-ATP raises interesting questions about the mechanism used to unwind oriC. It was recently suggested that origin unwinding activity of DnaA is produced by assembly of a right-handed DnaA helical filament, with polymerization of the filament requiring DnaA-ATP monomers (Erzberger et al., 2006). A role for DnaA-ADP bound to oriC weak sites is difficult to understand in light of this model, but several possibilities exist. First, DnaA-ADP may normally form the end of a polymerized filament causing it to be of fixed length, or binding of DnaA-ADP may occur independently of any filament formation. Despite the lack of clarity on the mechanism to unwind oriC, further analysis of oriCI2/I3ADP and oriCMATP should provide important information on the conversion of ORC to pre-RC in E. coli and the rules for filament formation.
Although unable to compete with wt oriC, oriCMATP was functional as a sole chromosomal copy, similar to many oriC mutants with compromised function (Bates et al., 1995; Weigel et al., 2001). However, our finding that cloned oriCMATP was able to replace chromosomal copies of oriCΔR4 on the chromosome reveals a hierarchy of mutational defects. Based on our observation, the ORC-defective oriCΔR4 must require even higher levels of DnaA to initiate in vivo compared to oriCMATP. The competitive hierarchy of wild-type and mutant versions of oriC provides a convenient approach for origin swapping. As long as the chromosomal oriC is out-competed by the pBR322-cloned oriC mutant, host growth will be perturbed, and the more efficient origin will integrate.
Experimental Procedures
Bacterial strains and plasmids
pOC170 is 3,852 base pairs long and carries replication origins from both pBR322 and oriC (Weigel et al., 1997). Supercoiled plasmid used for transformations and in vitro analyses of DnaA binding was isolated by using the QIAPrep Spin plasmid preparation kit (Qiagen). Mutagenesis of oriC was performed on pOC170 using the QuikChange site-directed mutagenesis kit (Stratagene) with oligonucleotide primers (Invitrogen) of 20–28 base pairs carrying the mutation in the center. After DpnI restriction endonuclease digestion to remove parental template, mutant plasmids were transformed into XL1-Blue endA1 gyrA46 hsdR17 lac recA1 relA1 supE44 thi; F’lac: lacIqΔ(lacZ) M15 Tn10 proA+proB+. DNA sequence analysis of mutant oriC was performed by using an Applied Biosystems 373 DNA sequencer with XL upgrade and Bio-Rad sequence analysis software, version 3.4. For evaluation of plasmid oriC function in vivo, pOC170 or mutant plasmids were transformed into either P3478 polA1, thyA36 deoC2 IN(rrnD-rrnE)1, or its isogenic parent, W3110. Effect of plasmids on cell growth was determined by transforming W3110, MM294 endA, thiA, hsdR17, supE44, MM294 recA or MM294 ΔoriC. MM294ΔoriC strain was made by P1 transduction of the origin region from ΔoriC::pKN1562 (clockwise), asnA+, Km, thi-1, relA1, spoT1, λ− (Koppes and Nordstrom, 1986). Cells were made competent using CaCl2, transformed with 100 ng of plasmid DNA, and plated onto Luria-Bertani agar plates containing 100 μg/ml ampicillin. Plates were incubated overnight at 37°C before examining colonies. Cells from colonies were grown in Luria-Bertani media supplemented with 100 μg/ml thymine. Cell growth was measured by counting cells using a model ZB Coulter Counter.
Chemicals, proteins and enzymes
Reagent grade chemicals were purchased from either Midwest Scientific, Fisher Scientific, or Sigma. Media components were from Difco or Midwest Scientific. Polymerases and nucleotides were purchased from Bioline. Amino-terminal His10-tagged DnaA was purified as described (Li and Crooke, 1999).
DNA modification and primer extension
DMS modification of plasmid DNA in vitro was performed as previously described (Grimwade et al., 2000, Ryan et al., 2004). In 50 μl reactions, 0.75 μg (300 fmole) of supercoiled plasmid DNA was added to 40 mM Hepes-KOH, pH 7.6, 8 mM MgCl2, 30% (w/v) glycerol, 320 μg/ml BSA, and 5 mM ATP. DnaA was pre-incubated for 5 minutes with 1 mM ATP or ADP in reaction buffer before adding to reactions. Reactions were incubated at 38°C for at least 7 minutes before addition of DMS. DMS-treated samples were extended with radiolabelled primer as previously described (Grimwade et al., 2000). Two primers were used in extension reactions, a leftward primer hybridizing at bases 272–290 to analyze top strand modifications, and a rightward primer hybridizing at bases 124–142 to analyze bottom strand modifications. Extension products were resolved on 6% polyacrylamide sequencing gels as described (Grimwade et al., 2000), and dried gels were scanned on a Bio-Rad Molecular Imager FX. Images were analyzed using Bio-Rad’s Quantity One software. Ratios of intensities of bands in binding sites to internal standard bands were calculated to yield relative intensity of modified guanines. Deviations in band intensities among experiments were <15%. The reference bands chosen were near to the site being analyzed and when possible, of comparable intensity.
Mismatch Amplification Mutation Analysis
Integration of plasmid mutations into the chromosome was checked in primary transformed colonies and overnight cultures using Mismatch Amplification Mutation Assay (MAMA PCR) (Cha et al., 1992). Cells from the primary transformed colonies were transferred to a PCR tube containing 10 pmoles of each primer, 12.5 pl of 2x Master Mix (Bioline), and water to a final volume of 25 μl. Samples were placed in a Techne thermocycler at 96°C for 8 minutes, followed by 40 cycles of 96°C for 30 seconds, 52°C for 20 seconds, and 72°C for 3 minutes at an 80% ramp, then a final at extension of 72°C for 6 minutes. Extension products were analyzed on 1% agarose gels.
Acknowledgments
We thank Walter Messer, Elliott Crooke, Anders Lobner-Olesen, Flemming Hansen, and Kurt Nordstrom for kindly providing us with bacterial strains and plasmids. This work was supported by National Institutes of Health Grant GM54042.
References
- Baker TA, Bell SP. Polymerases and the replisome: machines within machines. Cell. 1998;92:295–305. doi: 10.1016/s0092-8674(00)80923-x. [DOI] [PubMed] [Google Scholar]
- Bates DB, Asai T, Cao Y, Chambers MW, Cadwell GW, Boye E, Kogoma T. The DnaA box R4 in the minimal oriC is dispensable for initiation of Escherichia coli chromosome replication. Nucleic Acids Res. 1995;23:3119–3125. doi: 10.1093/nar/23.16.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkovic SJ, Valentine AM, Salinas F. Replisome-mediated DNA replication. Annu Rev Biochem. 2001;70:181–208. doi: 10.1146/annurev.biochem.70.1.181. [DOI] [PubMed] [Google Scholar]
- Boye E, Lobner-Olesen A, Skarstad K. Limiting DNA replication to once and only once. EMBO Rep. 2000;1:479–483. doi: 10.1093/embo-reports/kvd116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramhill D, Kornberg A. Duplex opening by dnaA protein at novel sequences in initiation of replication at the origin of the E. coli chromosome. Cell. 1988;52:743–755. doi: 10.1016/0092-8674(88)90412-6. [DOI] [PubMed] [Google Scholar]
- Campbell JL, Kleckner N. E. coli oriC and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell. 1990;62:967–979. doi: 10.1016/0092-8674(90)90271-f. [DOI] [PubMed] [Google Scholar]
- Cassler MR, Grimwade JE, Leonard AC. Cell cycle-specific changes in nucleoprotein complexes at a chromosomal replication origin. Embo J. 1995;14:5833–5841. doi: 10.1002/j.1460-2075.1995.tb00271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha RS, Zarbl H, Keohavong P, Thilly WG. Mismatch amplification mutation assay (MAMA): application to the c-H-ras gene. PCR Methods Appl. 1992;2:14–20. doi: 10.1101/gr.2.1.14. [DOI] [PubMed] [Google Scholar]
- Crooke E. Escherichia coli DnaA protein--phospholipid interactions: in vitro and in vivo. Biochimie. 2001;83:19–23. doi: 10.1016/s0300-9084(00)01224-4. [DOI] [PubMed] [Google Scholar]
- Dasgupta S, Lobner-Olesen A. Host controlled plasmid replication: Escherichia coli minichromosomes. Plasmid. 2004;52:151–168. doi: 10.1016/j.plasmid.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Davey MJ, Jeruzalmi D, Kuriyan J, O’Donnell M. Motors and switches: AAA+ machines within the replisome. Nat Rev Mol Cell Biol. 2002;3:826–835. doi: 10.1038/nrm949. [DOI] [PubMed] [Google Scholar]
- de Wind N, Parren P, Stuitje AR, Meijer M. Evidence for the involvement of the 16kD gene promoter in initiation of chromosomal replication of Escherichia coli strains carrying a B/r-derived replication origin. Nucleic Acids Res. 1987;15:4901–4914. doi: 10.1093/nar/15.12.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffley JF. Regulation of early events in chromosome replication. Curr Biol. 2004;14:R778–786. doi: 10.1016/j.cub.2004.09.019. [DOI] [PubMed] [Google Scholar]
- Erzberger JP, Pirruccello MM, Berger JM. The structure of bacterial DnaA: implications for general mechanisms underlying DNA replication initiation. Embo J. 2002;21:4763–4773. doi: 10.1093/emboj/cdf496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erzberger JP, Mott ML, Berger JM. Structural basis for ATP-dependent DnaA assembly and replication-origin remodeling. Nat Struct Mol Biol. 2006;13:676–683. doi: 10.1038/nsmb1115. [DOI] [PubMed] [Google Scholar]
- Fang L, Davey MJ, O’Donnell M. Replisome assembly at oriC, the replication origin of E. coli, reveals an explanation for initiation sites outside an origin. Mol Cell. 1999;4:541–553. doi: 10.1016/s1097-2765(00)80205-1. [DOI] [PubMed] [Google Scholar]
- Fuller RS, Funnell BE, Kornberg A. The dnaA protein complex with the E. coli chromosomal replication origin (oriC) and other DNA sites. Cell. 1984;38:889–900. doi: 10.1016/0092-8674(84)90284-8. [DOI] [PubMed] [Google Scholar]
- Gille H, Messer W. Localized DNA melting and structural pertubations in the origin of replication, oriC, of Escherichia coli in vitro and in vivo. Embo J. 1991;10:1579–1584. doi: 10.1002/j.1460-2075.1991.tb07678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimwade JE, Ryan VT, Leonard AC. IHF redistributes bound initiator protein, DnaA, on supercoiled oriC of Escherichia coli. Mol Microbiol. 2000;35:835–844. doi: 10.1046/j.1365-2958.2000.01755.x. [DOI] [PubMed] [Google Scholar]
- Helmstetter CE, Leonard AC. Coordinate initiation of chromosome and minichromosome replication in Escherichia coli. J Bacteriol. 1987;169:3489–3494. doi: 10.1128/jb.169.8.3489-3494.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstetter CE. Timing of synthetic activities in the cell cycle. In: Neidhardt FC, Curtis R III, Ingraham J, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. Escherichia coli and Salmonella:Cellular and Molecular Biology. Washington, D.C: ASM Press; 1996. pp. 1627–1639. [Google Scholar]
- Jensen MR, Lobner-Olesen A, Rasmussen KV. Escherichia coli minichromosomes: random segregation and absence of copy number control. J Mol Biol. 1990;215:257–265. doi: 10.1016/S0022-2836(05)80344-4. [DOI] [PubMed] [Google Scholar]
- Kaguni JM. DnaA: controlling the initiation of bacterial DNA replication and more. Annu Rev Microbiol. 2006;60:351–375. doi: 10.1146/annurev.micro.60.080805.142111. [DOI] [PubMed] [Google Scholar]
- Katayama T. Feedback controls restrain the initiation of Escherichia coli chromosomal replication. Mol Microbiol. 2001;41:9–17. doi: 10.1046/j.1365-2958.2001.02483.x. [DOI] [PubMed] [Google Scholar]
- Kato J. Regulatory network of the initiation of chromosomal replication in Escherichia coli. Crit Rev Biochem Mol Biol. 2005;40:331–342. doi: 10.1080/10409230500366090. [DOI] [PubMed] [Google Scholar]
- Kawakami H, Keyamura K, Katayama T. Formation of an ATP-DnaA-specific initiation complex requires DnaA arginine-285, a conserved motif in the AAA+ protein family. J Biol Chem. 2005 doi: 10.1074/jbc.M502764200. [DOI] [PubMed] [Google Scholar]
- Koppes L, Nordstrom K. Insertion of an R1 plasmid into the origin of replication of the E. coli chromosome: random timing of replication of the hybrid chromosome. Cell. 1986;44:117–124. doi: 10.1016/0092-8674(86)90490-3. [DOI] [PubMed] [Google Scholar]
- Koppes LJ. OriC plasmids do not affect timing of chromosome replication in Escherichia coli K12. Mol Gen Genet. 1987;209:188–192. doi: 10.1007/BF00329857. [DOI] [PubMed] [Google Scholar]
- Kornberg A, Baker TA. DNA Replication. New York: W.H. Freeman and Company; 1992. [Google Scholar]
- Kurokawa K, Nishida S, Emoto A, Sekimizu K, Katayama T. Replication cycle-coordinated change of the adenine nucleotide-bound forms of DnaA protein in Escherichia coli. Embo J. 1999;18:6642–6652. doi: 10.1093/emboj/18.23.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer U, Richter S, Roth A, Weigel C, Messer W. A comprehensive set of DnaA-box mutations in the replication origin, oriC, of Escherichia coli. Mol Microbiol. 1996;21:301–311. doi: 10.1046/j.1365-2958.1996.6481362.x. [DOI] [PubMed] [Google Scholar]
- Lee DG, Bell SP. ATPase switches controlling DNA replication initiation. Curr Opin Cell Biol. 2000;12:280–285. doi: 10.1016/s0955-0674(00)00089-2. [DOI] [PubMed] [Google Scholar]
- Leonard AC, Helmstetter CE. Cell cycle-specific replication of Escherichia coli minichromosomes. Proc Natl Acad Sci U S A. 1986;83:5101–5105. doi: 10.1073/pnas.83.14.5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AC, Theisen PW, Helmstetter CE. Replication timing and copy number control of oriC plasmids. In: Dirlica K, Riley M, editors. The bacterial chromosome. Washington, D.C: American Society for Microbiology; 1990. pp. 279–286. [Google Scholar]
- Leonard AC, Grimwade JE. Building a bacterial orisome: emergence of new regulatory features for replication origin unwinding. Mol Microbiol. 2005;55:978–985. doi: 10.1111/j.1365-2958.2004.04467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Crooke E. Functional analysis of affinity-purified polyhistidine-tagged DnaA protein. Protein Expr Purif. 1999;17:41–48. doi: 10.1006/prep.1999.1094. [DOI] [PubMed] [Google Scholar]
- Lobner-Olesen A, Hansen FG, Rasmussen KV, Martin B, Kuempel PL. The initiation cascade for chromosome replication in wild-type and Dam methyltransferase deficient Escherichia coli cells. Embo J. 1994;13:1856–1862. doi: 10.1002/j.1460-2075.1994.tb06454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobner-Olesen A, von Freiesleben U. Chromosomal replication incompatibility in Dam methyltransferase deficient Escherichia coli cells. Embo J. 1996;15:5999–6008. [PMC free article] [PubMed] [Google Scholar]
- Lobner-Olesen A. Distribution of minichromosomes in individual Escherichia coli cells: implications for replication control. Embo J. 1999;18:1712–1721. doi: 10.1093/emboj/18.6.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Campbell JL, Boye E, Kleckner N. SeqA: a negative modulator of replication initiation in E. coli. Cell. 1994;77:413–426. doi: 10.1016/0092-8674(94)90156-2. [DOI] [PubMed] [Google Scholar]
- Margulies C, Kaguni JM. Ordered and sequential binding of DnaA protein to oriC, the chromosomal origin of Escherichia coli. J Biol Chem. 1996;271:17035–17040. doi: 10.1074/jbc.271.29.17035. [DOI] [PubMed] [Google Scholar]
- Matsui M, Oka A, Takanami M, Yasuda S, Hirota Y. Sites of dnaA protein-binding in the replication origin of the Escherichia coli K-12 chromosome. J Mol Biol. 1985;184:529–533. doi: 10.1016/0022-2836(85)90299-2. [DOI] [PubMed] [Google Scholar]
- McGarry KC, Ryan VT, Grimwade JE, Leonard AC. Two discriminatory binding sites in the Escherichia coli replication origin are required for DNA strand opening by initiator DnaA-ATP. Proc Natl Acad Sci U S A. 2004;101:2811–2816. doi: 10.1073/pnas.0400340101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer W. The bacterial replication initiator DnaA. DnaA and oriC, the bacterial mode to initiate DNA replication. FEMS Microbiol Rev. 2002;26:355–374. doi: 10.1111/j.1574-6976.2002.tb00620.x. [DOI] [PubMed] [Google Scholar]
- Nievera C, Torgue JJ, Grimwade JE, Leonard AC. SeqA blocking of DnaA-oriC interactions ensures staged assembly of the E. coli pre-RC. Mol Cell. 2006;24:581–592. doi: 10.1016/j.molcel.2006.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka A, Sugimoto K, Sasaki H, Takanami M. An in vitro method generating base substitutions in preselected regions of plasmid DNA: application to structural analysis of the replication origin of the Escherichia coli K-12 chromosome. Gene. 1982;19:59–69. doi: 10.1016/0378-1119(82)90189-5. [DOI] [PubMed] [Google Scholar]
- Oka A, Sasaki H, Sugimoto K, Takanami M. Sequence organization of replication origin of the Escherichia coli K-12 chromosome. J Mol Biol. 1984;176:443–458. doi: 10.1016/0022-2836(84)90171-2. [DOI] [PubMed] [Google Scholar]
- Ryan VT, Grimwade JE, Nievera CJ, Leonard AC. IHF and HU stimulate assembly of pre-replication complexes at Escherichia coli oriC by two different mechanisms. Mol Microbiol. 2002;46:113–124. doi: 10.1046/j.1365-2958.2002.03129.x. [DOI] [PubMed] [Google Scholar]
- Sekimizu K, Bramhill D, Kornberg A. ATP activates dnaA protein in initiating replication of plasmids bearing the origin of the E. coli chromosome. Cell. 1987;50:259–265. doi: 10.1016/0092-8674(87)90221-2. [DOI] [PubMed] [Google Scholar]
- Simmons LA, Breier AM, Cozzarelli NR, Kaguni JM. Hyperinitiation of DNA replication in Escherichia coli leads to replication fork collapse and inviability. Mol Microbiol. 2004;51:349–358. doi: 10.1046/j.1365-2958.2003.03842.x. [DOI] [PubMed] [Google Scholar]
- Skarstad K, Boye E, Steen HB. Timing of initiation of chromosome replication in individual Escherichia coli cells. Embo J. 1986;5:1711–1717. doi: 10.1002/j.1460-2075.1986.tb04415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K, Boye E. The initiator protein DnaA: evolution, properties and function. Biochim Biophys Acta. 1994;1217:111–130. doi: 10.1016/0167-4781(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Skarstad K, Lueder G, Lurz R, Speck C, Messer W. The Escherichia coli SeqA protein binds specifically and co-operatively to two sites in hemimethylated and fully methylated oriC. Mol Microbiol. 2000;36:1319–1326. doi: 10.1046/j.1365-2958.2000.01943.x. [DOI] [PubMed] [Google Scholar]
- Skarstad K, Boye E, Fanning E. Circles in the sand. EMBO Rep. 2003;4:661–665. doi: 10.1038/sj.embor.embor888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K, Lobner-Olesen A. Stable co-existence of separate replicons in Escherichia coli is dependent on once-per-cell-cycle initiation. Embo J. 2003;22:140–150. doi: 10.1093/emboj/cdg003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck C, Messer W. Mechanism of origin unwinding: sequential binding of DnaA to double- and single-stranded DNA. Embo J. 2001;20:1469–1476. doi: 10.1093/emboj/20.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel C, Schmidt A, Ruckert B, Lurz R, Messer W. DnaA protein binding to individual DnaA boxes in the Escherichia coli replication origin, oriC. Embo J. 1997;16:6574–6583. doi: 10.1093/emboj/16.21.6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel C, Messer W, Preiss S, Welzeck M, Morigen, Boye E. The sequence requirements for a functional Escherichia coli replication origin are different for the chromosome and a minichromosome. Mol Microbiol. 2001;40:498–507. doi: 10.1046/j.1365-2958.2001.02409.x. [DOI] [PubMed] [Google Scholar]
- Yung BY, Kornberg A. The dnaA initiator protein binds separate domains in the replication origin of Escherichia coli. J Biol Chem. 1989;264:6146–6150. [PubMed] [Google Scholar]
- Yung BY, Crooke E, Kornberg A. Fate of the DnaA initiator protein in replication at the origin of the Escherichia coli chromosome in vitro. J Biol Chem. 1990;265:1282–1285. [PubMed] [Google Scholar]