

Abstract
Nanogels are colloidal microgel carriers that have been introduced recently as a prospective drug delivery system for nucleotide therapeutics. The crosslinked protonated polymer network of nanogels binds oppositely charged drug molecules, encapsulating them into submicron particles with a core-shell structure. The nanogel network also provides a suitable template for chemical engineering, surface modification and vectorisation. This review reveals recent attempts to develop novel drug formulations of nanogels with antiviral and antiproliferative nucleoside analogs in the active form of 5′-triphosphates; discusses structural approaches to the optimisation of nanogel properties, and; discusses the development of targeted nanogel drug formulations for systemic administration. Notably, nanogels can improve the CNS penetration of nucleoside analogs that are otherwise restricted from passing across the blood–brain barrier. The latest findings reviewed here demonstrate an efficient intracellular release of nucleoside analogs, encouraging further applications of nanogel carriers for targeted drug delivery.
Keywords: accumulation enhancement, drug delivery, nucleoside 5′-triphosphates, polymeric microgels, structural modification, triggered drug release, vectorisation
1. Introduction
Nucleoside analogs and their derivatives remain the first-line chemotherapeutic agents in the fight against a broad variety of viral pathogens [1,2] and actively proliferating cells of various cancer types [3,4]. Of the 30 compounds used in the US for treatment of viral infections, 15 are nucleoside analogs. During the past decade, there has been dramatic progress in understanding intracellular uptake, drug metabolism, interaction with cellular targets and pharmacokinetics of many nucleoside analogs. These compounds are actually antimetabolites that interfere with nucleic acid synthesis either by being incorporated into viral or cellular DNA or RNA, or by modifying the metabolism of physiological nucleosides. Each nucleoside analog possesses unique drug–target interactions that help to explain the differences in their activity, in various diseases. The initial drug paradigm was that nucleoside analogs are mostly active in proliferating or virus-infected cells, and do not affect normal cells. However, in reality, severe toxic effects accompany treatments with therapeutic doses of nucleoside analogs. Given the low therapeutic index (ratio of toxic and effective drug concentrations) of most nucleoside analogs, the abundance of their molecular targets and the multitude of drug resistance mechanisms, they are among the most complex therapeutic agents. Major reasons why nucleoside analogs need to be administered in high doses are: low levels of intracellular drug accumulation; ineffective cellular phosphorylation necessary for drug activation; constitutive cellular drug efflux mechanisms; drug metabolism and deactivation; and rapid clearance from the bloodstream. Therefore, we face an unmet medical need for the improvement of the drug efficacy and bio-distribution through delivery of activated-form nucleoside analogs to the site of treatment. There are now two main directions for developing new advanced drug forms of nucleoside analogs – development of prodrugs and supramolecular drug formulations. This review focuses on recent progress in developing nanoparticulate drug carriers (nanogels) for the delivery of nucleoside analogs in their active phosphorylated form.
2. The active form of nucleoside analogs is nucleoside 5′-triphosphate
Following administration, many nucleoside analogs are rapidly subjected to enzymatic metabolism [5]; their cellular accumulation and clearance is regulated by specific nucleoside drug transporters [6–8]. The initial step in the activation of nucleoside analogs is cellular phosphorylation to nucleoside 5′-monophosphates (NMPs), performed by cellular or viral kinases [9]. The next step of intracellular enzymatic transformation of nucleoside analogs is their conversion into the active nucleoside 5′-triphosphates (NTPs), which are major substrates for DNA or RNA polymerases. Accumulation of NTP depends on the activity of corresponding cellular phosphoryl transferases and is an ineffective process for most nucleoside analogs [10]. As a result, only a small fraction of the administered drug is converted into the active form. NTPs not only terminate the chain elongation by polymerases, but also induce S-phase specific apoptosis [11]. Nucleoside analogs have also been implicated in mitochondrial toxicity. In fact, this mode of action was even suggested as a major cytotoxic mechanism of many nucleoside analogs [12]. The therapeutic effect of nucleoside analogs is determined mainly by sufficiently high intracellular concentrations of their respective NTPs and strongly depends on nucleoside structure and cell type [13]. Therefore, administration of previously phosphorylated drugs will result in bypassing the first step of intracellular activation and faster intracellular accumulation of NTP.
The prodrug (pronucleotide) concept entails administration of stable and metabolically active forms of nucleoside analogs, which are accumulated better into the targeted cells and are readily converted by in vivo biological enzymes into unstable, activated forms. Most nucleoside prodrugs include advanced oral forms or hydrophobic derivatives with superior bio-availability [14–16]. However, successful applications of this approach using NTP only include covalent phospholipid conjugates of anti-HIV drug, 3′-azidothymidine (AZT) demonstrating a higher bioavailability and lower toxicity than AZT [17]. A derivative of cytosine arabinoside, 5′-hexadecyl phosphonodiphosphate, was found to be orally active against murine lymphatic leukaemia with less pronounced side effects than the parent drug [18].
3. Drug delivery systems increase therapeutic effect of nucleoside analogs
The prodrug approach does not solve all the problems associated with intracellular delivery and activation of nucleoside analogs. Many laboratories are now focused on the development of effective drug carriers using nanotechnology [19]. Different polymeric drug delivery systems have been introduced and evaluated in recent years. Following intravenous administration, polymer-conjugated drugs was better accumulated in various tumours with leaky vasculature through an enhanced permeability and retention (EPR) effect [20], and exhibited lower systemic toxicity compared with non-conjugated drugs [21,22]. However, as the active content in such polymeric drugs may be quite low, in many cases therapeutic doses cannot be achieved in the tumour site.
Liposomes were the first delivery system recognised for drug formulations of nucleoside analogs [23]. Liposomal delivery of chemotherapeutic agents reduces normal tissue toxicity by lowering free-drug concentration in the bloodstream, and enhances drug efficacy due to better cellular and tissue accumulation. Multiple examples of liposome-encapsulated nucleoside analogs and their lipophilic derivatives were extensively discussed in recently published reviews [24–26]. Principal weaknesses of liposomal drug formulations were drug leakage, short storage time and the problems associated with oral administration.
Biodegradable nanoparticles composed of poly(D,L-lactide) or poly(D,L-lactide-co-glycolide) have been successfully applied to drug delivery and the sustained release of many drugs, including nucleoside analogs in the treatment of HIV and cerebral tumours [27–29]. As in the case of liposomes, nanoparticles display low toxicity and immunogenic effects, and are known to accumulate in phagocytic cells after intravenous administration. Therefore, they have a significant therapeutic potential for macrophage targeting [30]. Nevertheless owing to the slow kinetics of drug release from nanoparticles, it will be difficult to obtain effective local concentrations of nucleoside analogs.
Hydrophilic microgel particles composed of swollen polymer networks are now recognised as promising systemic carriers for controlled and site-specific drug delivery [31–33]. Different types of crosslinked polyacrylate microgels were earlier synthesised and used for encapsulation of nucleoside analogs. Polyhexyl-cyanoacrylate nanoparticles, alone or coated with polysorbate 80, were studied for antiviral nucleoside drug delivery to the HIV-infected monocytes [34,35]. After intravenous administration, colloidal drug delivery systems are preferentially taken up by the reticuloendothelial system (RES). Authors report an 18-fold increase of AZT concentration in organs belonging to the RES if the drug was bound to nanoparticles, compared with unbound AZT. A similar study on polyisohexylcyanoacrylate nanospheres encapsulating AZT through hydrophobic interactions demonstrated an ability of the intestinal epithelium and associated lymphoid tissues (one of the major reservoirs of the HIV) to accumulate the drug-loaded carrier and efficiently increase the tissue concentration of the drug [36]. Recently, polybutylcyanoacrylate and methylmethacrylate-sulfopropylmethacrylate nanoparticles were used for binding of anti-HIV nucleoside analogs on the external surface of these carriers [37,38]. The former carrier was found to be compatible with intravenous administration, and was also found to be 1.5 times more efficient in crossing the blood–brain barrier (BBB) than the latter carrier, using bovine brain microvessel endothelial cell monolayers as an in vitro BBB model. However, in both cases, the amount of incorporated drugs crossing the BBB was much higher than that of free drugs. Notably, the particle size of the carriers was kept nearly monodispersed and < 100 nm. Biodistribution of larger colloidal carriers could be of use in the treatment of infections involving the RES, whereas brain and tumour delivery remains at lower level [39]. Drug delivery outside the RES can be achieved by adjusting the colloidal carrier size, coating with different non-ionic surfactants or attachment of targeting vector molecules to the surface of carriers [40]. Recently, various applications of microgels for drug delivery have been reviewed [41].
4. Nanogels are a novel type of hydrophilic polymer carriers
A novel type of nanosized polymeric microgel (nanogel) (Figure 1) consisting of a polymer network of charged polyionic segments crosslinked by polyethylene glycol (PEG) segments, and having a total molecular mass of several million Daltons, was developed for encapsulation of oligonucleotides, poly-nucleotides and other charged molecules [42, 101–102]. Many drug delivery applications of Nanogel carriers have been recently reviewed [41,43]. The nanogel network offers many advanced features as a drug delivery system including: simplicity of drug formulation; high drug-loading capacity; exceptional dispersion stability; and prolonged storage of drug formulations in the freeze-dried form. Nanogels can be stored in a dried form at ambient temperature and can easily be resuspended in aqueous media, forming particulate dispersions with a hydrodynamic diameter of 120 – 350 nm. Oppositely charged compounds may be loaded into nanogels by simply mixing their solutions, with the dispersion of the carrier in aqueous media. Polyionic complexes of negatively charged oligonucleotides and protonated amines of polyethylenimine (PEI) molecules in cationic nanogels formed spontaneously in a matter of minutes. This process was accompanied by a significant compaction of the nanogel network, resulting in > 10-fold volume reduction [42]. Although the degree of initial swelling of nanogels depends on the number of crosslinks in the polymer network (or PEG to PEI molar ratio), more than a two-fold reduction of hydrodynamic diameter was observed for nanogels loaded with oppositely charged molecules. The authors have found that a minimum number of six PEG crosslinks per PEI molecule was required to obtain nanogels with desirable mechanical properties. At this PEG to PEI molar ratio, homologous macrogels demonstrated the highest (30- to 40-fold) swelling degree in water [44]. The nanogel network provides an excellent protection of labile drug molecules and is capable of carrying a high payload of drugs, up to 30% by weight. Several features of nanogels, such as high molecular mass and small particle size, can promote a passive targeting in tumours due to the previously mentioned EPR effect. In addition to passive targeting, surface modification of nanogel with tumour-specific vector molecules acting synergistically, may significantly enhance the selectivity of drug accumulation in solid tumours.
Figure 1. Schematic presentation of the cationic nanogel network.

(A) Structure of Nanogel NG(PEG) consists of branched molecules of PEI (25 kDa) cross-linked with PEG (as shown in Figure 1A) or Pluronic® molecules (as shown in Fig. 1B), activated by 1,1′-carbonyldiimidazole and forming urethane bonds with amino groups of PEI. (B) Structure of biodegradable Nanogel NG(Pluronic®)ss contains molecules of segmented PEI (30 kDa) consisting of branched PEI (2 kDa) blocks linked with disulfide bonds. Nanogel network was cross-linked the same way as in Figure 1A.
4.1 Nanogel formulations with phosphorylated nucleoside analogs
Many prospective nucleoside analogs have been discarded in earlier preclinical studies, or withdrawn later from clinical studies as their intracellular conversion into NTP was inefficient within an acceptable dosage range. The choice of available anticancer drugs could be considerably broadened given that these drugs are delivered into the cytosol in a preactivated NTP form. However, only a limited number of efforts were made to directly administer NTP encapsulated in special drug-delivery vehicles, as this drug form was found to be too unstable to be used in chemotherapy. In one example, 5′-triphosphate of 2′,3′-dideoxycytidine was found to be well-protected in circulation and delivered to mononuclear phagocyte system in PEG-stabilised liposomes [45]. Recently, the authors have demonstrated successful application of cationic nanogels for encapsulating 5′-triphosphates of natural nucleosides and nucleoside analogs. Phosphate moieties of NTP formed ionic complexes with protonated amino groups of PEI and compacted into the nanogel network core, surrounded by a layer of PEG loops (Figure 2). Based on these findings, nanogels can be used for direct delivery of immediately active NTPs (instead of nucleoside analogs) into infected or cancerous cells. Therefore, nanogels may be developed into a promising therapeutic formulation. To test this strategy, nanogel/NTP formulations of several cytotoxic nucleoside analogs, such as 2′-fluoroadenine arabinoside, AZT and cytosine arabinoside were studied. Initial nucleosides were converted into NTP, using well-developed chemical phosphorylation methods [46]. A polyionic drug complex with nanogel was formed immediately by the direct mixing of an aqueous dispersion of the carrier with the NTP solution. Complex formation could be traced by marked reduction of the hydrodynamic diameter of Nanogel particles, observed by dynamic light scattering. An alternative approach involved titration of the dispersion of nanogel-containing deprotonated amines with NTP in an acidic form, until a neutral pH of the resulting solution was reached. In both cases, lyophilised formulations were obtained that could be stored in dry form at 4°C, without any trace of NTP decomposition for at least 1 year. Drug (NTP) loading depended directly on the PEI content in nanogels, and was equal to 15 – 30% of the weight of dry formulation, or 0.5 mmol of NTP/g on average. Drug-loaded nanogel particles swelled and retained enough water in their interior to ensure a high solubility and dispersion stability in aqueous media. Experiments on in vitro drug-release demonstrated a sustained release of nucleotide drugs from nanogels in physiological medium, reaching ~ 30% of the initial drug-loading during the first 24 h of incubation at 37°C. Evidently, in vitro release of free drug from nanogels in circulation is slow enough to cause no serious problems associated with non-specific toxic effects of unbound nucleoside analogs.
Figure 2. Diagram demonstrating nanogel loading with a nucleoside 5′-triphosphate (NTP).

Aqueous dispersion of the nanogel network mixed with aqueous solution of NTP forms a polyionic complex with the drug (as shown in the insert, with AZT-TP and amino groups of PEI). The drug binding results in the immediate network compaction and formation of small particles with a core-shell structure: a dense core of the PEI-NTP complex surrounded by an envelope of polymer loops.
AZT: 3′-azidothymidine; NTP: Nucleoside 5′-triphosphate; PEI: Polyethylenimine.
Nanogels provided significant protection of encapsulated phosphorylated nucleosides from degradation by ubiquitous intra- or extra-cellular dephosphorylating enzymes. In model experiments, 30–60% of encapsulated NTP was protected and kept intact, depending on the nanogel loading capacity, compared with only 10% of the free NTP following incubation with alkaline phosphatase. In addition, ~ 60% of degraded nucleotide was found in the form of mono- and di-phosphates that are also active drug species. Drugs may be released from nanogel/NTP formulations by a multi-variant mechanism. Nanogel can slowly release a considerable amount of the encapsulated drug in the form of nucleoside 5′-phosphates, which are formed as a result of partial dephosphorylation at physiological conditions and dissociate more easily from the nanogel interior. However, a remaining fraction of the formulated NTP may be unbound only in the event of binding with competitive cellular polyanions. Previously, a putative mechanism of drug release (Figure 3) including interaction of nanogel cationic network with negatively charged phospholipids and other components of the cellular membrane has been suggested [43]. Membranotropic properties of nanogels were clearly illustrated recently by dose-dependent interactions of tritium-labelled nanogels with isolated cellular membranes, and by direct visualisation of these events using transmi sion electron and atomic force microscopy [47]. These findings could clearly be observed in a cellular system of rhodamine-labelled nanogels loaded with a fluorescein-labelled ATP (Figure 4). Initial accumulation of nanogel on the cellular membrane was evident on these confocal images taken earlier, after cell treatment. This process was accompanied with a fast release and accumulation of the green fluorescent ATP into the cytosol. At a later time point (60 min), most of the drug-loaded nanogels, as well as membrane-bound unloaded nanogels, were taken up by endocytosis (yellow and red dots) and accumulated in endosomes. However, membrane binding and the process of drug release continued inside the endosomes, and a much higher level of green fluorescent ATP was now observed in the cytosol. This process has been called membrane-triggered drug release from nanogel-ATP complexes. In this process, positively charged nanogel competitively formed more stable complexes with phospholipids of the cellular membrane, fusing into the lipid bilayer and releasing NTP directly into the cytosol on the other side of the membrane. The feature provides an evident advantage over many existing drug delivery systems, as nanogel can release the drug rapidly, by means of bursting, as soon as the carrier reaches the target site.
Figure 3. Putative mechanism of membrane-triggered drug release from nanogel.
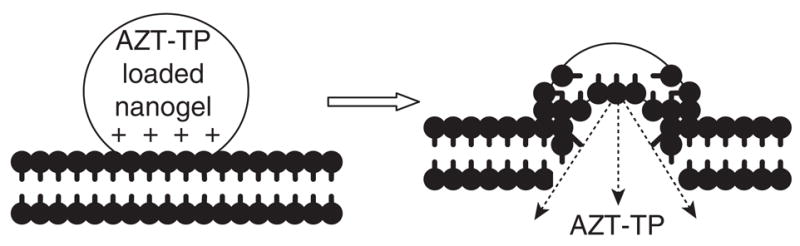
On the left side, protonated parts of drug-loaded nanogel comes in close contact and interacts with negatively charged phospholipids and components of the cellular membrane. The right side of the figure shows the nanogel network fused into the phospholipid bilayer and rapid drug release into the cytosol through competitive interactions of cationic PEI molecules with phospholipids.
PEI: Polyethylenimine.
Figure 4. Interaction of drug-loaded nanogel with cellular membrane and drug release in cancer cells.
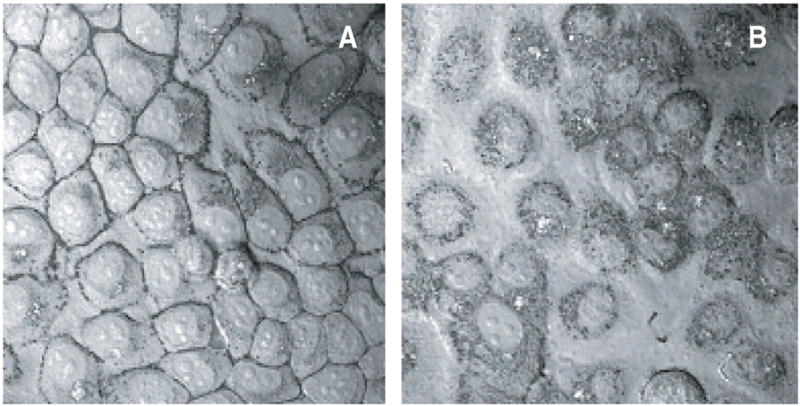
Laser confocal micrographs overlaid on contrast pictures of human breast carcinoma MCF-7 cells, following incubation with a model formulation of rhodamine-labeled nanogel with fluorescein-labeled ATP. Figure 4A shows initial accumulation of nanogel on the cellular membrane (30 min after treatment), while a part of the released drug is already visible in cytosol. Further intense drug release and accumulation of nanogel in cytosol is demonstrated in Figure 4B (60 min after treatment) where green fluorescence associated with the released ATP is profoundly distributed all over the cytosol. Separate points of yellow fluorescence show ATP-loaded nanogel locations.
Several cytotoxic nucleoside analogs in the form of NTP have been formulated with nanogel. Surprisingly, the cytotoxicity of these formulations tested in various human breast carcinoma cell lines was approximately two orders of magnitude higher compared with the parental non-phosphorylated drug (Figure 5). This additional cytotoxic effect was independent of the inherent cytotoxicity of the carrier, as similar Nanogel-ATP complexes exhibited markedly lower cytotoxicity. Based on previous findings, the authors have attributed the observed cytotoxic efficacy of Nanogel-NTP formulations to the rapid creation of an excessive inhibitory NTP concentration higher than the cytosolic pool of cellular nucleotides.
Figure 5. Comparative cytotoxicity of nucleoside analogs, NTP and Nanogel formulations.
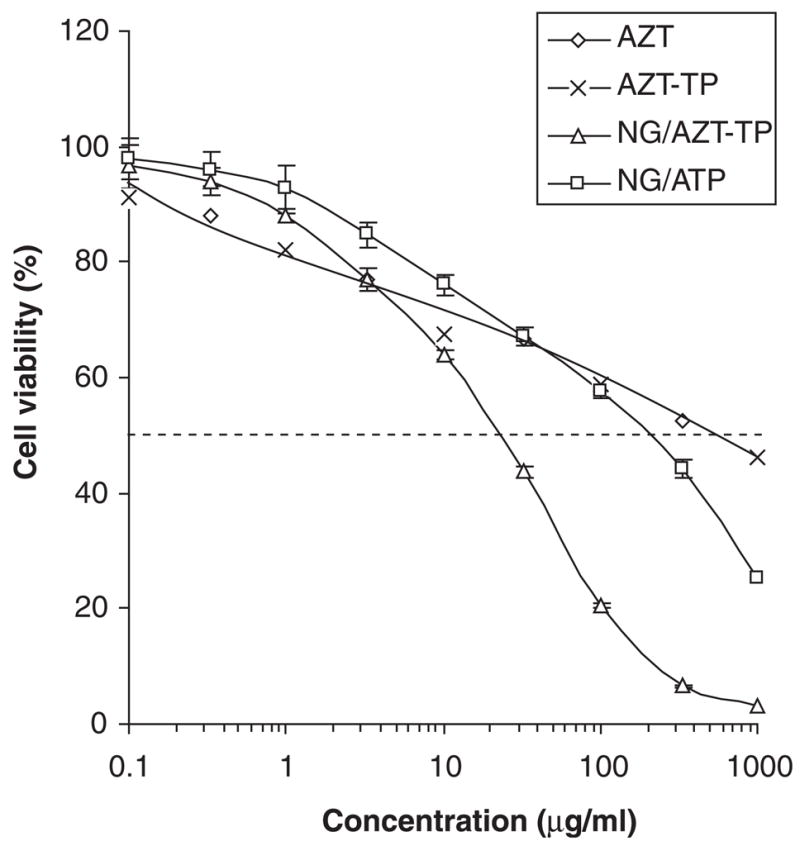
This graph shows a significant 10-fold increase of cytotoxicity for nanogel-NTP formulations at 24h-treatment of human breast carcinoma MCF-7 cells, compared to non-encapsulating drugs. Nanogel NG(PEG) loaded with 20% (w/w) of cytotoxic drug (AZT) or non-toxic nucleotide ATP, and non-encapsulated nucleoside analog AZT and its 5′-triphosphate values AZT-TP were used in the treatment. The corresponding IC50 were equal to 22, 210 and 600 μg/ml (for both compounds).
Toxicology of many novel drug carriers constitutes a serious challenge for polymer chemists and chemical engineers. By analogy to the therapeutic index of antiviral drugs (usually determined as a ratio of drug cytotoxic concentration [IC50] to the drug effective concentration [EC50]), various drug delivery systems may be also compared by their own therapeutic index, which may be determined as a ratio of cytotoxic concentration of the carrier to the cytotoxic concentration of the carrier-loaded drug. The higher the therapeutic index, the better the drug-carrier formulation. In the case of cationic nanogel-NTP formulations, the therapeutic index value was usually > 50, which is considered to be a very good ratio for many drugs.
4.2 Biodegradation reduces cytotoxicity of nanogels
One of the major concerns related to therapeutic applications of cationic carriers such as PEI, poly-L-lysine, block copolymers and others, was the cytotoxicity related to their cationic nature [48–50]. Toxic effects of polycations were attributed mainly to the damage of cellular membranes (leaking patches) [51,52] or binding with important cellular components [53]. Some immunological problems were also associated with systemic application of PEI (a major component of nanogel) for gene therapy [54]. However, modification of PEI by PEG molecules significantly reduced these responses because of the ‘shielding’ of immunogenic PEI from interaction with plasma proteins [55]. Similarly, the nanogel network (PEG-cl-PEI) demonstrated lower cytotoxicity than free PEI (by two orders of magnitude). Evidently, this was partially due to a negligibly small surface charge of nanogel compared with PEI in solution (4 versus 50 mV, respectively). Nanogel toxicity may be determined, in part, by the toxicity of its degradation products. Release of free PEI in the event of nanogel degradation would definitely enhance cytotoxicity of the carrier. Urethane bonds connecting PEI and PEG chains in nanogels are relatively stable at physiological conditions. However, when in close proximity to the highly charged amino groups of PEI, these bonds become degraded, and the whole nanogel network decomposes into biological milieu. Major initial nanogel degradation products are multiple PEG-grafted PEI copolymers (PEG-g-PEI). The authors have evaluated the rate of the nanogel degradation process and have found the half-life period to be ~ 8 days in physiological solution at 37°C. This was found to be even shorter at lower pH values of 4 or 5 (e.g., the existing pH in endosomal compartments). In this property, nanogel is similar to PLGA nanoparticles that completely degrade in the course of several weeks. Conjugates of PEG-g-PEI showed markedly lower cytotoxicity than PEI in many cell cultures [56–58]. The authors also showed that nanogels with higher crosslinking density (greater PEG to PEI ratio) are less toxic than carriers with lower ratios [44]. Low pore size associated with high cross- linking density of nanogels will not hamper encapsulation of smaller drug molecules, such as NTP, in the carrier.
Another suggestion to reduce cytotoxicity of nanogel carriers was through the application of biodegradable nanogel with a PEI-backbone consisting of short PEI (2 kDa) molecules connected via biodegradable disulfide bonds [59]. Synthesis of the segmented PEI used in preparation of the biodegradable carrier was previously described [60]. Fast degradation of disulfide bonds in a nanogel network can occur in the reductive intracellular environment, resulting in the formation of very low toxic PEG-g-PEI (2 kDa) conjugates with molecular weights of ~ 12 kDa. These reducible nanogels (NGss) demonstrated significant (up to 57-fold) decreases in cytotoxicity compared with carriers containing regular PEI (25 kDa) (Table 1). Although environmental degradation of regular nanogels with urethane bonds was slow, ~ 70% of the biodegradable nanogel network was readily converted into the above mentioned conjugates with much lower molecular weights, following a short treatment with the reducing agent, dithiothreitol (DTT). These conjugates are also expected to have good renal clearance (MWCO 40 kDa). In summary, overall cellular toxicity of biodegradable nanogels was equal to or less than the best examples of existing cationic polymer carriers [61,62].
Table 1.
Comparison of hydrodynamic diameter, cytotoxicity and cellular binding of nanogels with regular and biodegradable networks (NG and NGss, respectively)
| Nanogel (NG)* | Nanogel diameter (nm) | Diameter of NTP-loaded nanogel (nm) | Cytotoxicity, (IC50, μg/ml)‡ | Cell binding, (μg/mg protein§) |
|---|---|---|---|---|
| NG (PEG) | 152 ± 6 | 84 ± 3 | 70 | 18 ± 1.5 |
| NG (PEGss) | 100 ± 8 | n.d. | 250 | n.d. |
| NG (F127) | 132 ± 4 | 69 ± 1 | 80 | 27.3 ± 0.9 |
| NG (F127ss) | 187 ± 6 | n.d. | 190 | n.d. |
| NG (F68) | 176 ± 4 | 74 ± 0.5 | 95 | 37.5 ± 1.5 |
| NG(F68ss) | 120 ± 5 | 58 ± 2 | 250 | n.d. |
| NG(P85) | 270 ± 3 | 144 ± 4 | 25 | 34.5 ± 0.7 |
| NG (P85ss) | 99 ± 7 | n.d. | 170 | n.d. |
| NG (P123) | 102 ± 1 | 69 ± 0.5 | 30 | 48.7 ± 1.3 |
| NG (P123ss) | 75 ± 8 | n.d. | 130 | n.d. |
| PEI 25kDa | n/a | n/a | 5 | n/a |
| PEIss | n/a | n/a | 36 | n/a |
All nanogels have the same PEI content 25% (wt).
Based on MTT assay after 24h-incubation of ATP-loaded nanogels with human breast carcinoma MCF-7 cells.
Measured for ATP-loaded rhodamine-labelled nanogels following 4 h incubation with MCF-7 cells.
PEG: Polyethylene glycol; PEI: Polyethylenimine; n.d.: Not determined; NTP: Nucleoside 5′-triphosphate.
4.3 Modulation of membranotropic properties of nanogels
Evidently, interaction of the nanogel network with the cellular membrane may be a key event in triggered drug release. Once this mechanism was revealed, efforts were focused on chemical engineering of nanogels to increase binding and fusion with the lipid bilayer. The first approach consisted of introduction of lipophilic components of the cellular membrane (cholesterol moieties) into the nanogel network [63]. In this case, primary amino groups in the nanogel were modified at various rates with cholesterol chloroformate. Highly modified nanogels (> 5%) were insoluble or slightly soluble. Nanogels with a lower substitution rate (1 – 5%) demonstrated sufficient solubility and significantly increased cellular association. An average increase of cellular association for cholesterol-nanogels was three- to four-fold above the association for non-modified carriers.
In the second approach, nanogels were synthesised starting from amphiphilic polymers instead of PEG (Table 1). These water-soluble polymers contained blocks of lipophilic poly(propylene oxide) (PPO) flanked with hydrophilic poly(ethylene oxide) (PEO) blocks. It has been shown that size of the PPO block and PPO/PEO ratio are important parameters determining membranotropic properties and the ability of amphiphilic polymers to form micelles [64]. The authors compared properties of nanogels with PEG substituted for hydrophilic Pluronic® F68 or F127 (BASF AG), and the more lipophilic P85 [65]. Nanogels NG(P85), NG(F68) and NG(F127) consisted of lipophilic PPO segments of similar sizes, and the hydrophilic components and total molecular weights increase in the order listed. All the amphiphilic nanogels demonstrated a 40 – 170% higher degree of cellular binding compared with PEG-based nanogel NG(PEG) (Table 1). At the same time, no decrease in water-solubility was noted for all Pluronic-based nanogels. Evidently, these amphiphilic nanogels may represent a valuable alternative to NG(PEG) as drug delivery vehicles. NG(F68) was one of the best binding nanogels, probably because of an optimal balance between the sizes of PPO and PEO segments. Besides, Pluronic F68 has an excellent toxicity profile, and under the name of Poloxamer 188, is approved for human applications.
4.4 Surface modification by targeting moieties (vectorisation)
Targeted nanosized drug carriers could be thought of as microscopic pills delivered directly to disease-affected sites (organs, tumours or cancer metastases). The most effective method is to inject/infuse dispersion of these targeted drug-loaded particles into the bloodstream. Drug-loaded nanogel carriers, sized ~ 100 nm, are able to reach the smallest capillaries, and consequently accumulate in the sites with a high density of targeted receptors. An optimal drug delivery system will show a reduced level of interaction with serum proteins, extended circulation time and optimised renal clearance. All these requirements have been met by PEG-covered (stealth) liposomes with sizes 100–200 nm [66]. When comparing nanogels with stealth liposomes, many common properties can be observed. Preliminary data on the in vivo biodistribution of nanogels have shown that these carriers have a shorter plasma half-life than stealth liposomes, although they have a longer half-life than the previously described nanoparticles. Nanogels can also be optimised for prolonged blood circulation in terms of particle size and PEG coverage; factors extensively studied previously for other cationic micellar systems [67–69].
Previously, a preparation of biotinylated nanogel that could be modified via streptavidin by biotinylated targeting ligands (e.g., transferrin or insulin) was described [70]. Using these ligands, a significant enhancement of the BBB permeability for nucleotide drug-loaded carriers was recently observed in bovine brain microvessel endothelial cell monolayers, a cellular model of the BBB. Transferrin and insulin were nearly equal in potency, demonstrating a two-fold increase in the transcellular transport of protein-conjugated nanogels versus non-vectorised nanogel, or even a 12-fold increase compared to the free oligonucleotide. Nanogel by itself markedly increased systemic drug transport and brain accumulation of anionic oligonucleotides, usually crosses the BBB poorly, in a mouse model. A small residual positive charge of the carrier resulted in anapproximately 10-fold increase of brain/plasma ratio for an intravenously injected drug-loaded nanogel, compared with free oligonucleotide. Although this system has demonstrated the targeting efficacy of nanogels, complications associated with slow aggregation of the nanogel avidin constructs made the authors overlook alternative targeting approaches. Recently, various chemical approaches for covalent attachment of targeting ligands to the nanogel surface have been developed. These include:
Modification of the nanogel surface with folate moieties; polymetacrylate microgels modified with folate moieties demonstrated an increased and selective cellular uptake in cancer cell lines overexpressing folate receptors [71]. As a proof of principle, folate-vectorised nanogels were obtained and loaded with a fludarabine 5′-triphosphate. These vectorised carriers demonstrated a significant increase in transport of the NTP across Caco-2 cell monolayers, a cellular model of the gastro intestinal tract, compared with non-vectorised nanogels [44]. However, not all folate moieties could be equally available as part of them would remain hidden in the interior of nanogel network. To reduce problems associated with folate accessibility for cellular folate receptors, an insertion of a polymer linker between the folate moiety and drug carrier was deemed as the necessary way to expose the vector [72,73]. A simple modification of 5 – 10% of primary amino groups in nanogel can be performed using bis-amino-PEG and folic acid, activated by water-soluble carbodiimide. Our preliminary data demonstrated a approximate 10-fold increase of binding for the folate-modified Nanogel compared to the initial carrier (Figure 6). These folate-PEG-nanogels are now under evaluation for drug delivery in various cellular models.
Nanogel modification with targeting proteins; a limited amount of amino acid groups in the protein were initially converted by reaction with 2-iminothiolane (Trout’s reagent) into thiol groups. Then, the nanogel was modified by a PEG linker flanked with N-hydroxy-succinimide and maleimide moieties. Finally, conjugation between thiolated protein and maleimide-PEG-nanogels yielded vectorised nanogels carrying 4 – 12 protein molecules per particle. Due to protein size, most vector molecules do not penetrate the nanogel network and are located on the surface, allowing for easy access to cellular receptors [74]. The authors have demonstrated that transferrin-modified nanogels are extremely more efficient in binding with cancer cells than non-modified nanogels (Figure 6). This fact, together with an approximate hundred-fold increase of transfection efficacy for transferrin-modified nanogel versus non-modified nanogel, observed in cultured cancer cells, confirmed the significant potential of targeted nanogels for drug delivery and improvement of chemotherapy (manuscript in preparation).
Vectorisation of the nanogel surface with multiple peptide ligands; vectorisation of nanogel with homing or other receptor-specific small peptides may provide an efficient targeting of the carrier and drug release in metastases or tumours, at the same time maintaining a low level of free drug in circulation [63]. Initially, a bifunctional PEG linker was used to modify a fraction of amino groups in the nanogel. The second moiety on the linker was thiol-specific, and could react with synthetic peptide C-amides carrying terminal cysteine residues. Tumour-specific, epithelial growth factor receptor-binding peptides [75], several brain-specific homing peptides [76], and other peptides were conjugated to nanogels in amounts of 40 – 90 molecules per particle using this synthetic approach [59]. The author’s preliminary in vivo data demonstrated statistically significant increases of accumulation of nanogels targeted with selected peptides in murine breast tumour or brain, correspondingly (Figure 7). Other targeting peptides in linear and cyclic form, and postsynthetic modification procedures intended to ameliorate the bio-distribution profiles of drug-loaded, targeted nanogels are now under investigation.
Figure 6. Binding of vectorized nanogels with cancer cells.
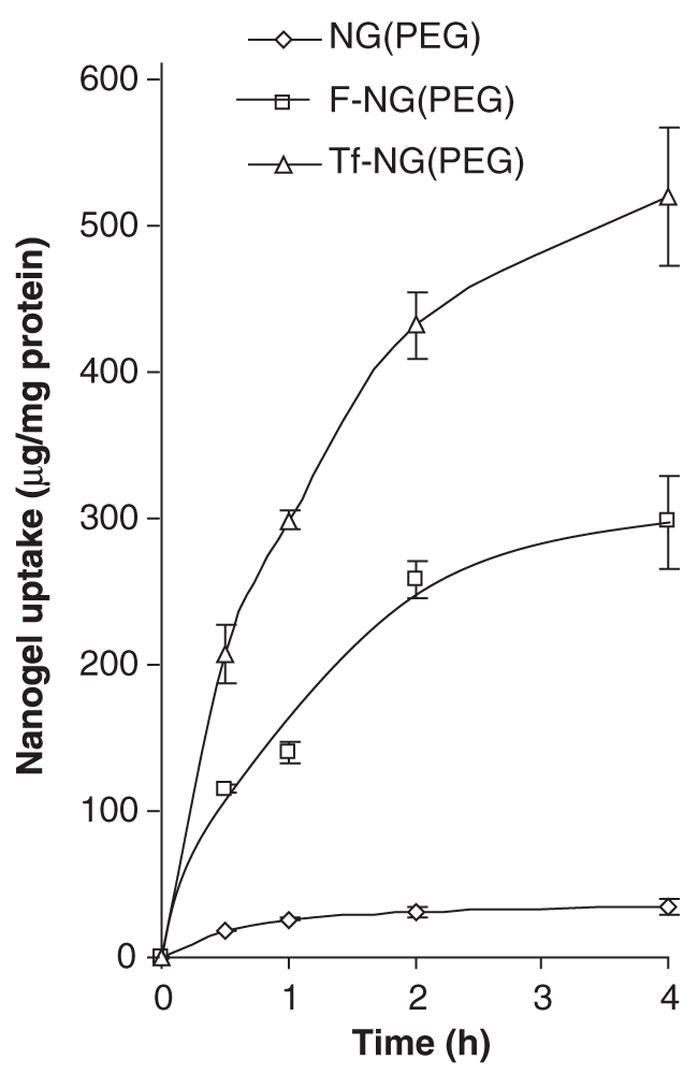
Human breast carcinoma MCF-7 cells were treated with rhodamine-labeled Nanogel, NG(PEG), and its folate-vectorized, F-NG(PEG), or human transferrin-vectorized, Tf-NG(PEG) derivatives, loaded with model drug ATP. Nanogel uptake was calculated based on the fluorescence associated with the cells and calibration curves for separate rhodamine-labeled carriers.
Figure 7. Selected biodistribution of non-modified and multiple peptides-modified nanogels.
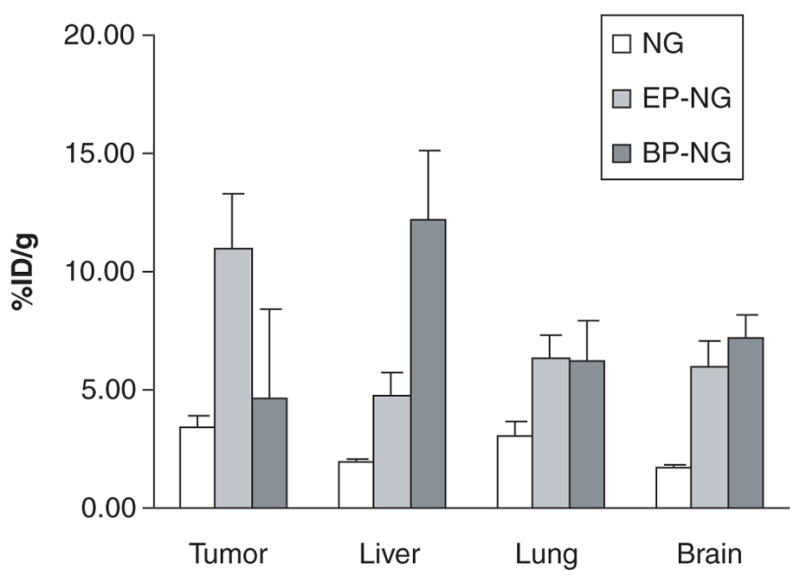
Tritium-labeled nanogel, NG(PEG), and its derivatives modified with tumour-specific peptide, (EP-NG), or brain-specific peptide, (BP-NG), were loaded with model NTP drug, and intravenously injectedin mice with L66-murine breast carcinoma. Organ/tumour accumulation was measured 4 h post-injection and expressed as a percentage of the injected dose (%ID).
5. Conclusion
In conclusion, nanogel carriers offer considerable potential for development of stable pharmaceutical formulations and organ/tissue specific delivery of activated nucleoside analogs in their phosphorylated form. Formulations of nanogels with nucleotide 5′-triphosphates described here represent a therapeutic formulation with unique properties, including high drug loading, prolonged storage stability, lyophilisable and well-soluble drug form, injectability, circulation safety and controllable biodistribution pattern. However, several obscure areas exist, which have not been satisfactorily studied to draw definitive conclusions about the pros and cons of nanogel. Perhaps serious modification of the cationic element of nanogels is required before these carriers could be considered safe for therapeutic application. Nevertheless, this review has clearly shown that additional structural optimisation of nanogel and introduction of receptor-specific targeting moieties on the surface of the carrier is a promising way to obtain drug delivery systems with advanced properties and may overcome many side effects of regular drugs in human chemotherapy.
6. Expert opinion
Evidently, the progress in chemical design and development of nucleoside analogs and their prodrugs will continue. However, the major shortcoming of these drugs (i.e., lack of targeting capabilities) cannot be avoided. Introduction of miniature drug delivery vehicles that are capable of easily navigating inside the body is just a matter of time away, and should be considered a a mainstream way of continuing nano-medicine progress. The nanogel drug carrier, with its unique ability to upload NTP, convenience of freeze-dried formulations, opportunity for targeting tissues of interest and capability for triggered drug release, is clearly one of few delivery systems that can be considered as a first-generation nanomedicine device. Future developments of the nanogel drug delivery system are required in order to include features that could optimise biodistribution, degradation pattern, component toxicity and manufacturing variance of nanogels in combination with nucleoside analogs. In all these developmental goals, the nanogel delivery system reflects the same limitations, and faces the same challenges that were encountered by a great number of other polymeric drug carriers, and that are common for nanoparticles in general. To address these issues, several key problems should first be solved.
The first important obstacle is associated with systemic administration and fast disappearance of nanoparticles from the bloodstream due to RES/macrophage uptake. Surface binding of plasma proteins (opsonisation) is the first step to macrophage recognition of exogenous nanoparticles. Following NTP drug loading, the nanogel network forms a neutral polymer envelope of laterally stabilised PEG loops around the core containing the complexed drug. A simple increase of PEG to PEI ratio in nanogels can suppress the entrapment of the carrier in RES-related organs. Systemic in vivo studies and evaluation of circulation parameters of nanogels with chemically engineered surfaces (PEG, carbohydrates and short peptides) are now in progress.
The second problem is associated with biodegradation of nanogels and concerns two major issues: systemic toxicity and efficient drug release. Evidently, products of degradation of drug delivery systems can add to the toxicity of a drug formulation on the whole, and so should be accounted for. Major products of nanogel degradation, PEG/Pluronic-g-PEI conjugates were shown to be hundreds of times less toxic than corresponding PEI molecules. Also these molecules are < 40 kDa in size, which is considered an upper limit for fast renal exclusion of different compounds. Nanogels with reducible bonds will also provide a faster intracellular drug release over a short time period, and will offer an enhanced therapeutic effect compared with conventional biodegradable carriers, which have drug release kinetics measuring of several weeks.
The third problem is directly associated with a search for an alternative to PEI. Recently, several new polymers that are less toxic and have similar properties as PEI have been suggested [61,62]. Several new candidates for nanogel synthesis will be assayed shortly by in vitro toxicological tests.
The fourth issue of manufacturing variance is the most complicated one. Surely, one cannot avoid polydispersity of polymers, and real characteristics of nanogels are only possible within a certain range. However, state-of-the-art chemical procedures allow us to obtain reproducible polymer to polymer ratios, mass yields, particle-size ranges and drug loading. Polydispersity of nanogels could be reduced by using either narrow fractionation or advanced polymerisation methods. Regulatory issues will necessarily be changed with further introduction of novel polymer therapeutics, so that proper characterisation methods and parameters of polymer drug delivery systems will be developed.
It is clear that the field of nanomedical applications and nanoparticulate drug delivery approaches will expand tremendously in the near future [19]. Nanogel-drug formulations will be further evaluated in application to other drug delivery routes, such as oral, aerosolic or transdermal administration. A promising application for nanogel-encapsulated NTP is aerosolic pulmonary drug delivery. In this mode of administration, the fastest and most direct drug delivery can be achieved, especially for the treatment of lung cancer and pulmonary infections such as influenza, which are increasing worldwide. Aerosolic drug-loaded nanogel particles can be easily administered deep into the lungs at first signs of infection. This timely treatment will save the airway epithelium from rapid degradation and development of severe secondary infections.
Specific targeted nanogels will be developed as soon as novel receptors and ligands are discovered in the disease/virus-affected tissues and organs. On the other hand, strong metal-chelating properties of nanogels can provide additional modalities in post-synthetic modifications of the carriers for diagnostic and therapeutic purposes (e.g., PET/SPECT scans or radionuclide treatments) [44]. Subsequent research will reveal the actual potential of nanogels and nanogel formulations with nucleoside analogs in achieving more complex aims associated with methodical preclinical studies.
Acknowledgments
This research was supported by grants from National Institutes of Health (R01 CA102791 and R01 NS050660). Michael Jackobsen’s assistance in preparation of the manuscript is greatly appreciated.
Footnotes
Patent
101. KABANOV AV, VINOGRADOV SV, ALAKHOV YV: US6333051 (2001).
102. KABANOVAV, VINOGRADOV SV: US6696089 (2004).
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.VIVET-BOUDOU V, DIDIERJEAN J, ISEL C, MARQUET R. Nucleoside and nucleotide inhibitors of HIV-1 replication. Cell Mol Life Sci. 2006;63(2):163–186. doi: 10.1007/s00018-005-5367-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DE CLERCQ E, NEYTS J. Therapeutic potential of nucleoside/nucleotide analogs against poxvirus infections. Rev Med Virol. 2004;14(5):289–300. doi: 10.1002/rmv.439. [DOI] [PubMed] [Google Scholar]
- 3••.GALMARINI CM, JORDHEIM L, DUMONTET C. Pyrimidine nucleoside analogs in cancer treatment. Expert Rev Anti-Cancer Ther. 2003;3(5):717–728. doi: 10.1586/14737140.3.5.717. Detailed review on nucleoside analogs currently used in chemotherapy. [DOI] [PubMed] [Google Scholar]
- 4••.PARKER WB, SECRIST JA, 3RD, WAUD WR. Purine nucleoside antimetabolites in development for the treatment of cancer. Curr Opin Investig Drugs. 2004;5(6):592–596. Detailed review on nucleoside analogs currently used in chemotherapy. [PubMed] [Google Scholar]
- 5.HUNSUCKER SA, MITCHELL BS, SPYCHALA J. The 5′-nucleotidases as regulators of nucleotide and drug metabolism. Pharmacol Ther. 2005;107(1):1–30. doi: 10.1016/j.pharmthera.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 6.CLARKE ML, MACKEY JR, BALDWIN SA, YOUNG JD, CASS CE. The role of membrane transporters in cellular resistance to anticancer nucleoside drugs. Cancer Treat Res. 2002;112:27–47. doi: 10.1007/978-1-4615-1173-1_2. [DOI] [PubMed] [Google Scholar]
- 7•.BORST P, BALZARINI J, ONO N, et al. The potential impact of drug transporters on nucleoside-analog-based antiviral chemotherapy. Antiviral Res. 2004;62(1):1–7. doi: 10.1016/j.antiviral.2003.11.002. Overview on mechanisms of nucleoside drug efflux. [DOI] [PubMed] [Google Scholar]
- 8.PASTOR-ANGLADA M, CANO-SOLDADO P, MOLINA-ARCAS M, et al. Cell entry and export of nucleoside analogs. Virus Res. 2005;107(2):151–164. doi: 10.1016/j.virusres.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 9.GALMARINI CM, MACKEY JR, DUMONTET C. Nucleoside analogs and nucleobases in cancer treatment. Lancet Oncol. 2002;3(7):415–424. doi: 10.1016/s1470-2045(02)00788-x. [DOI] [PubMed] [Google Scholar]
- 10•.VAN ROMPAY AR, JOHANSSON M, KARLSSON A. Substrate specificity and phosphorylation of antiviral and anticancer nucleoside analogs by human deoxyribonucleoside kinases and ribonucleoside kinases. Pharmacol Ther. 2003;100(2):119–139. doi: 10.1016/j.pharmthera.2003.07.001. Summary of processes leading to the cellular activation of nucleoside analogs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.GRANT S. Ara-C: cellular and molecular pharmacology. Adv Cancer Res. 1998;72:197–233. doi: 10.1016/s0065-230x(08)60703-4. [DOI] [PubMed] [Google Scholar]
- 12•.BRINKMAN K, KAKUDA TN. Mitochondrial toxicity of nucleoside analog reverse transcriptase inhibitors: a looming obstacle for long-term antiretroviral therapy. Curr Opin Infect Dis. 2000;13(1):5–11. doi: 10.1097/00001432-200002000-00002. Review on side effects associated with treatment by nucleoside analogs. [DOI] [PubMed] [Google Scholar]
- 13.MCGUIGAN C, KINCHINGTON D, WANG MF, et al. Nucleoside analogs previously found to be inactive against HIV may be activated by simple chemical phosphorylation. FEBS Lett. 1993;322(3):249–252. doi: 10.1016/0014-5793(93)81580-s. [DOI] [PubMed] [Google Scholar]
- 14••.WAGNER CR, IYER VV, MCINTEE EJ. Pronucleotides: toward the in vivo delivery of antiviral and anticancer nucleotides. Med Res Rev. 2000;20(6):417–451. doi: 10.1002/1098-1128(200011)20:6<417::aid-med1>3.0.co;2-z. Summary of the prodrug approach in application to phosphorylated nucleoside analogs. [DOI] [PubMed] [Google Scholar]
- 15.MEIER C, AUBERTIN AM, DE MONTE M, et al. Synthesis and antiviral evaluation of SATE-foscarnet prodrugs and new foscarnet-AZT conjugates. Antivir Chem Chemother. 1998;9(1):41–52. doi: 10.1177/095632029800900105. [DOI] [PubMed] [Google Scholar]
- 16.KUKHANOVA M, KRAYEVSKY A, PRUSOFF W, CHENG YC. Design of anti-HIV compounds: from nucleoside to nucleoside 5′-triphosphate analogs. Problems and perspectives. Curr Pharm Des. 2000;6(5):585–598. doi: 10.2174/1381612003400687. [DOI] [PubMed] [Google Scholar]
- 17.MORRIS-NATSCHKE SL, ISHAQ KS, KUCERA LS. Phospholipid analogs against HIV-1 infection and disease. Curr Pharm Des. 2003;9(18):1441–1451. doi: 10.2174/1381612033454702. [DOI] [PubMed] [Google Scholar]
- 18.BRACHWITZ H, BERGMANN J, FICHTNER, et al. 1-beta-D-Arabinofuranosylcytosine-5′-alkyl phosphonophosphates and diphosphates: new orally active derivatives of ara-C. J Lipid Res. 1998;39(1):162–172. [PubMed] [Google Scholar]
- 19••.COUVREUR P, VAUTHIER C. Nanotechnology: intelligent design to treat complex disease. Pharm Res. 2006;23(7):1417–1450. doi: 10.1007/s11095-006-0284-8. Recent advances of biomedical nanotechnology and development of drug delivery systems. [DOI] [PubMed] [Google Scholar]
- 20.MAEDA H, SAWA T, KONNO T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Control Release. 2001;74(1–3):47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- 21.KOPECEK J, KOPECKOVA P, MINKO T, LU ZR, PETERSON CM. Water soluble polymers in tumor targeted delivery. J Control Release. 2001;74(1–3):147–158. doi: 10.1016/s0168-3659(01)00330-3. [DOI] [PubMed] [Google Scholar]
- 22•.GREISH K, FANG J, INUTSUKA T, NAGAMITSU A, MAEDA H. Macromolecular therapeutics: advantages and prospects with special emphasis on solid tumour targeting. Clin Pharmacokinet. 2003;42(13):1089–1105. doi: 10.2165/00003088-200342130-00002. Passive targeting of polymeric drug delivery systems. [DOI] [PubMed] [Google Scholar]
- 23.HOFHEINZ RD, GNAD-VOGT SU, BEYER U, HOCHHAUS A. Liposomal encapsulated anti-cancer drugs. Anti-Cancer Drugs. 2005;16(7):691–707. doi: 10.1097/01.cad.0000167902.53039.5a. [DOI] [PubMed] [Google Scholar]
- 24••.DUZGUNES N, SIMOES S, SLEPUSHKIN V, et al. Delivery of antiviral agents in liposomes. Methods Enzymol. 2005;391:351–373. doi: 10.1016/S0076-6879(05)91020-3. Review of liposomal drug delivery. [DOI] [PubMed] [Google Scholar]
- 25.RUEDA DOMINGUEZ A, OLMOS HIDALGO D, VICIANA GARRIDO R, TORRES SANCHEZ E. Liposomal cytarabine (DepoCyte) for the treatment of neoplastic meningitis. Clin Transl Oncol. 2005;7(6):232–238. doi: 10.1007/BF02710168. [DOI] [PubMed] [Google Scholar]
- 26•.VIROVIC L, WU CH, KONISHI M, WU GY. Novel delivery methods for treatment of viral hepatitis: an update. Expert Opin Drug Deliv. 2005;2(4):707–717. doi: 10.1517/17425247.2.4.707. Review on application of nanoparticulate carriers. [DOI] [PubMed] [Google Scholar]
- 27.BENDER A, SCHFER V, STEFFAN AM, et al. Inhibition of HIV in vitro by antiviral drug-targeting using nanoparticles. Res Virol. 1994;145(3–4):215–220. doi: 10.1016/s0923-2516(07)80025-2. [DOI] [PubMed] [Google Scholar]
- 28.KRYCZKA T, BERO M, KASPERCZYK J, et al. In vitro release of cytotoxic nucleoside analogs from lactide-caprolactone and lactide-glycolide copolymers. Acta Biochim Pol. 2002;49(1):205–210. [PubMed] [Google Scholar]
- 29.BALA I, HARIHARAN S, KUMAR MN. PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carrier Syst. 2004;21(5):387–422. doi: 10.1615/critrevtherdrugcarriersyst.v21.i5.20. [DOI] [PubMed] [Google Scholar]
- 30.CHELLAT F, MERHI Y, MOREAU A, YAHIA L. Therapeutic potential of nanoparticulate systems for macrophage targeting. Biomaterials. 2005;26(35):7260–7275. doi: 10.1016/j.biomaterials.2005.05.044. [DOI] [PubMed] [Google Scholar]
- 31.LOPEZ VC, HADGRAFT J, SNOWDEN MJ. The use of colloidal microgels as a (trans)dermal drug delivery system. Int J Pharm. 2005;292(1–2):137–147. doi: 10.1016/j.ijpharm.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 32•.BROMBERG L. Intelligent hydrogels for the oral delivery of chemotherapeutics. Expert Opin Drug Deliv. 2005;2(6):1003–1013. doi: 10.1517/17425247.2.6.1003. Description of polymer hydrogels for drug delivery. [DOI] [PubMed] [Google Scholar]
- 33.ZHANG H, MARDYANI S, CHAN WC, KUMACHEVA E. Design of biocompatible chitosan microgels for targeted pH-mediated intracellular release of cancer therapeutics. Biomacromolecules. 2006;7(5):1568–1572. doi: 10.1021/bm050912z. [DOI] [PubMed] [Google Scholar]
- 34.BENDER AR, VON BRIESEN H, KREUTER J, DUNCAN IB, RUBSAMEN-WAIGMANN H. Efficiency of nanoparticles as a carrier system for antiviral agents in human immunodeficiency virus-infected human monocytes/macrophages in vitro. Antimicrob Agents Chemother. 1996;40(6):1467–1471. doi: 10.1128/aac.40.6.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.LOBENBERG R, ARAUJO L, VON BRIESEN H, RODGERS E, KREUTER J. Body distribution of azidothymidine bound to hexyl-cyanoacrylate nanoparticles after i.v injection to rats. J Control Release. 1998;50(1–3):21–30. doi: 10.1016/s0168-3659(97)00105-3. [DOI] [PubMed] [Google Scholar]
- 36.DEMBRI A, MONTISCI MJ, GANTIER JC, CHACUN H, PONCHEL G. Targeting of 3′-azido 3′-deoxythymidine (AZT)-loaded poly(isohexylcyanoacrylate) nanospheres to the gastrointestinal mucosa and associated lymphoid tissues. Pharm Res. 2001;18(4):467–473. doi: 10.1023/a:1011050209986. [DOI] [PubMed] [Google Scholar]
- 37.KUO YC. Loading efficiency of stavudine on polybutylcyanoacrylate and methylmethacrylate-sulfopropylmethacrylat e copolymer nanoparticles. Int J Pharm. 2005;290(1–2):161–172. doi: 10.1016/j.ijpharm.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 38.KUO YC, CHUNG CY. Transport of zidovudine- and lamivudine-loaded polybutylcyanoacrylate and methylmethacrylate-sulfopropylmethacryl ae nanoparticles across the in vitro blood-brain barrier: characteristics of the drug-delivery system. Journal of the Chinese Institute of Chemical Engineers. 2005;36(6):627–638. [Google Scholar]
- 39.SIMEONOVA M, IVANOVA T, RAIKOV Z, KONSTANTINOV H. Tissue distribution of polybutylcyanoacrylate nanoparticles loaded with spin-labelled nitrosourea in Lewis lung carcinoma-bearing mice. Acta Physiol Pharmacol Bulg. 1994;20(3–4):77–82. [PubMed] [Google Scholar]
- 40.RONEY C, KULKARNI P, ARORA V, et al. Targeted nanoparticles for drug delivery through the blood-brain barrier for Alzheimer’s disease. J Control Release. 2005;108(2–3):193–214. doi: 10.1016/j.jconrel.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 41.VINOGRADOV SV. Colloidal microgels in drug delivery applications. Curr Pharm Des. 2006;12:36. doi: 10.2174/138161206779026254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42•.VINOGRADOV S, BATRAKOVA E, KABANOV A. Poly(ethylene glycol)-polyethylenimine NanoGel particles: novel drug delivery systems for antisense oligonucleotides. Coll Surf B: Biointerfaces. 1999;16(1–4):291–304. The first paper describing cationic polymeric nanogels. [Google Scholar]
- 43•.VINOGRADOV SV, BRONICH TK, KABANOV AV. Nanosized cationic hydrogels for drug delivery: preparation, properties and interactions with cells. Adv Drug Deliv Rev. 2002;54(1):135–147. doi: 10.1016/s0169-409x(01)00245-9. Review on application of Nanogels for drug delivery. [DOI] [PubMed] [Google Scholar]
- 44.VINOGRADOV SV, ZEMAN AD, BATRAKOVA EV, KABANOV AV. Polyplex Nanogel formulations for drug delivery of cytotoxic nucleoside analogs. J Control Release. 2005;107:143–157. doi: 10.1016/j.jconrel.2005.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.OUSSOREN C, MAGNANI M, FRATERNALE A, et al. Liposomes as carriers of the antiretroviral agent dideoxycytidine-5′-triphosphate. Int J Pharm. 1999;180(2):261–270. doi: 10.1016/s0378-5173(99)00016-2. [DOI] [PubMed] [Google Scholar]
- 46.VAGHEFI MM. Chemical synthesis of nucleoside 5′-triphosphate. In: Vaghefi M, editor. Nucleoside Triphosphates and their Analogs. Taylor and Francis, Boca-Raton; London, New York, Singapore: 2005. pp. 1–22. [Google Scholar]
- 47•.VINOGRADOV SV, KOHLI E, ZEMAN AD. Cross-linked polymeric nanogel formulations of 5′-triphosphates of nucleoside analogs: role of the cellular membrane in drug release. Mol Pharm. 2005;2(6):449–461. doi: 10.1021/mp0500364. Demonstration of high affinity nanogels to the cellular membrane. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48••.NEU M, FISCHER D, KISSEL T. Recent advances in rational gene transfer vector design based on poly(ethylene imine) and its derivatives. J Gene Med. 2005;7(8):992–1009. doi: 10.1002/jgm.773. Description of PEI-based drug delivery systems. [DOI] [PubMed] [Google Scholar]
- 49.LUNGWITZ U, BREUNIG M, BLUNK T, GOPFERICH A. Polyethylenimine-based non-viral gene delivery systems. Eur J Pharm Biopharm. 2005;60(2):247–266. doi: 10.1016/j.ejpb.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 50.TIERA MJ, WINNIK FO, FERNANDES JC. Synthetic and natural polycations for gene therapy: state of the art and new perspectives. Curr Gene Ther. 2006;6(1):59–71. doi: 10.2174/156652306775515510. [DOI] [PubMed] [Google Scholar]
- 51.BASHFORD CL, ALDER GM, MENESTRINA G, et al. Membrane damage by hemolytic viruses, toxins, complement, and other cytotoxic agents. A common mechanism blocked by divalent cations. J Biol Chem. 1986;261(20):9300–9308. [PubMed] [Google Scholar]
- 52.ZHANG ZY, SMITH BD. High-generation polycationic dendrimers are unusually effective at disrupting anionic vesicles: membrane bending model. Bioconjug Chem. 2000;11(6):805–814. doi: 10.1021/bc000018z. [DOI] [PubMed] [Google Scholar]
- 53.MISLICK KA, BALDESCHWIELER JD. Evidence for the role of proteoglycans in cation-mediated gene transfer. Proc Natl Acad Sci USA. 1996;93(22):12349–12354. doi: 10.1073/pnas.93.22.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.CHOLLET P, FAVROT MC, HURBIN A, COLL JL. Side-effects of a systemic injection of linear polyethylenimine-DNA complexes. J Gene Med. 2002;4(1):84–91. doi: 10.1002/jgm.237. [DOI] [PubMed] [Google Scholar]
- 55.KIRCHEIS R, BLESSING T, BRUNNER S, WIGHTMAN L, WAGNER E. Tumor targeting with surface-shielded ligand-polycation DNA complexes. J Control Release. 2001;72(1–3):165–170. doi: 10.1016/s0168-3659(01)00272-3. [DOI] [PubMed] [Google Scholar]
- 56.VINOGRADOV S, BATRAKOVA E, LI S, KABANOV A. Polyion complex micelles with protein-modified corona for receptor-mediated delivery of oligonucleotides into cells. Bioconjug Chem. 1999;10(5):851–860. doi: 10.1021/bc990037c. [DOI] [PubMed] [Google Scholar]
- 57.LEMIEUX P, VINOGRADOV SV, GEBHART CL, et al. Block and graft copolymers and NanoGel copolymer networks for DNA delivery into cell. J Drug Target. 2000;8(2):91–105. doi: 10.3109/10611860008996855. [DOI] [PubMed] [Google Scholar]
- 58.NGUYEN HK, LEMIEUX P, VINOGRADOV SV, et al. Evaluation of polyether-polyethyleneimine graft copolymers as gene transfer agents. Gene Ther. 2000;7(2):126–138. doi: 10.1038/sj.gt.3301052. [DOI] [PubMed] [Google Scholar]
- 59.VINOGRADOV SV, KOHLI E, ZEMAN A, KABANOV AV. Chemical engineering of nanogel drug carriers: increased bioavailability and decreased cytotoxicity. Polymer Preprints (American Chemical Society, Division of Polymer Chemistry) 2006;47(2):27–28. [PMC free article] [PubMed] [Google Scholar]
- 60.GOSSELIN MA, GUO W, LEE RJ. Efficient gene transfer using reversibly cross-linked low molecular weight polyethylenimine. Bioconjug Chem. 2001;12(6):989–994. doi: 10.1021/bc0100455. [DOI] [PubMed] [Google Scholar]
- 61.KANAYAMA N, FUKUSHIMA S, NISHIYAMA N, et al. A PEG-based biocompatible block catiomer with high buffering capacity for the Construction of polyplex micelles showing efficient gene transfer toward primary cells. Chem Med Chem. 2006;1(4):439–444. doi: 10.1002/cmdc.200600008. [DOI] [PubMed] [Google Scholar]
- 62.CHRISTENSEN LV, CHANG CW, KIM WJ, KIM SW. Reducible poly(amido ethylenimine)s designed for triggered intracellular gene delivery. Bioconjug Chem. 2006;17(5):1233–1240. doi: 10.1021/bc0602026. [DOI] [PubMed] [Google Scholar]
- 63.VINOGRADOV SV, BERNARD K, KOHLI E, KABANOV AV. Nanogel drug carriers: Surface and chemical engineering of increased bioavailability. Annual Meeting of the Controlled Release Society; Vienna, Austria. ( 22 - 26 July 2006); p. A61. [Google Scholar]
- 64••.KABANOV AV, LEMIEUX P, VINOGRADOV S, ALAKHOV V. Pluronic block copolymers: novel functional molecules for gene therapy. Adv Drug Deliv Rev. 2002;54(2):223–233. doi: 10.1016/s0169-409x(02)00018-2. Interesting review on application of Pluronic for drug delivery. [DOI] [PubMed] [Google Scholar]
- 65.VINOGRADOV SV, KOHLI E, ZEMAN AD. Comparison of nanogel drug carriers and their formulations with nucleoside 5′-triphosphates. Pharm Res. 2006;23(5):920–930. doi: 10.1007/s11095-006-9788-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66•.MOGHIMI SM, SZEBENI J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42(6):463–478. doi: 10.1016/s0163-7827(03)00033-x. Review on drug delivery potential of PEGylated stealth liposomes. [DOI] [PubMed] [Google Scholar]
- 67.OUPICKY D, OGRIS M, HOWARD KA, et al. Importance of lateral and steric stabilization of polyelectrolyte gene delivery vectors for extended systemic circulation. Mol Ther. 2002;5(4):463–472. doi: 10.1006/mthe.2002.0568. [DOI] [PubMed] [Google Scholar]
- 68.OGRIS M, WAGNER E. Tumor-targeted gene transfer with DNA polyplexes. Somat Cell Mol Genet. 2002;27(1–6):85–95. doi: 10.1023/a:1022988008131. [DOI] [PubMed] [Google Scholar]
- 69.TORCHILIN VP, LUKYANOV AN, GAO Z, PAPAHADJOPOULOS-STERNBERG B. Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc Natl Acad Sci USA. 2003;100(10):6039–6044. doi: 10.1073/pnas.0931428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70••.VINOGRADOV SV, BATRAKOVA EV, KABANOV AV. Nanogels for oligonucleotide delivery to the brain. Bioconjug Chem. 2004;15(1):50–60. doi: 10.1021/bc034164r. Demonstration of prospective nanogel applications for drug delivery into the brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.NAYAK S, LEE H, CHMIELEWSKI J, LYON LA. Folate-mediated cell targeting and cytotoxicity using thermoresponsive microgels. J Am Chem Soc. 2004;126(33):10258–10259. doi: 10.1021/ja0474143. [DOI] [PubMed] [Google Scholar]
- 72.LEAMON CP, COOPER SR, HARDEE GE. Folate-liposome-mediated antisense oligodeoxynucleotide targeting to cancer cells: evaluation in vitro and in vivo. Bioconjug Chem. 2003;14(4):738–747. doi: 10.1021/bc020089t. [DOI] [PubMed] [Google Scholar]
- 73.SHIOKAWA T, HATTORI Y, KAWANO K, et al. Effect of polyethylene glycol linker chain length of folate-linked microemulsions loading aclacinomycin A on targeting ability and antitumor effect in vitro and in vivo. Clin Cancer Res. 2005;11(5):2018–2025. doi: 10.1158/1078-0432.CCR-04-1129. [DOI] [PubMed] [Google Scholar]
- 74.VINOGRADOV SV, KABANOV AV. Synthesis of nanogel carriers for delivery of active phosphorylated nucleoside analogs. Polymer Preprints (American Chemical Society, Division of Polymer Chemistry) 2004;45(2):378–379. [PMC free article] [PubMed] [Google Scholar]
- 75.LUTSENKO SV, FELDMAN NB, SEVERIN SE. Cytotoxic and antitumor activities of doxorubicin conjugates with the epidermal growth factor and its receptor-binding fragment. J Drug Target. 2002;10(7):567–571. doi: 10.1080/1061186021000038058. [DOI] [PubMed] [Google Scholar]
- 76.VINOGRADOV SV, KOHLI E, BATRAKOVA EV, KABANOV AV, et al. Synthetic vehicles for drug delivery to brain capillary endothelial cells. Gene Ther. 2006 in press. [Google Scholar]