

Abstract
The mechanism of proton transfer from the bulk into the membrane protein interior was studied. The light-induced reduction of a bound ubiquinone molecule QB by the photosynthetic reaction center is accompanied by proton trapping. We used kinetic spectroscopy to measure (i) the electron transfer to QB (at 450 nm), (ii) the electrogenic proton delivery from the surface to the QB site (by electrochromic carotenoid response at 524 nm), and (iii) the disappearance of protons from the bulk solution (by pH indicators). The electron transfer to QB− and the proton-related electrogenesis proceeded with the same time constant of ≈100 μs (at pH 6.2), whereas the alkalinization in the bulk was distinctly delayed (τ ≈ 400 μs). We investigated the latter reaction as a function of the pH indicator concentration, the added pH buffers, and the temperature. The results led us to the following conclusions: (i) proton transfer from the surface-located acidic groups into the QB site followed the reduction of QB without measurable delay; (ii) the reprotonation of these surface groups by pH indicators and hydronium ions was impeded, supposedly, because of their slow diffusion in the surface water layer; and (iii) as a result, the protons were slowly donated by neutral water to refill the proton vacancies at the surface. It is conceivable that the same mechanism accounts for the delayed relaxation of the surface pH changes into the bulk observed previously with bacteriorhodopsin membranes and thylakoids. Concerning the coupling between proton pumps in bioenergetic membranes, our results imply a tendency for the transient confinement of protons at the membrane surface.
Living organisms use various energy sources to generate the transmembrane electrochemical potential difference of the proton, Δμ̃H+, which is then used for productive work (1). These reactions are catalyzed by specialized membrane enzymes that serve as proton pumps and/or users of Δμ̃H+. There is a continuing debate in the literature as to whether protons are able to migrate between membrane enzymes along the membrane surface faster than they equilibrate with the bulk (see refs. 2–4). Recently, several research groups reported studies on bacteriorhodopsin (BR) membrane sheets and showed that the response of the pH indicator in the bulk to a pulse of proton pumping (τ ≈ 1 ms) lagged behind the response of a pH indicator that was attached either to the same side of the purple membrane where protons were ejected or to the other side, ≈0.1 μm away (τ ≈ 100 μs in both cases; refs. 2, 5, and 6).
We used the photosynthetic reaction center (RC) of Rhodobacter sphaeroides to investigate in detail both the intraprotein proton transfer from the membrane surface and the pH changes in the bulk during a single turnover. RCs are situated in chromatophores, vesicular invaginations of the inner cell membrane of phototrophic bacteria. Crystal structures of the R. sphaeroides RC are available (see ref. 7 for a recent review). Subunits L and M form a heterodimer in the membrane, which is capped by the H subunit from the cytoplasmic side. Illumination by a light flash causes a transmembrane electron transfer from a bacteriochlorophyll dimer P (the primary electron donor that is located at the interface of the L and M subunits close to the periplasmic side of the chromatophore membrane) to a pair of quinone acceptors (see refs. 8 and 9 for reviews). They are located in the protein, below the interface between the LM heterodimer and the H subunit and ≈15 Å away from the RC/water boundary. The primary quinone acceptor (QA), a ubiquinone molecule bound by the M subunit, is reduced in ≈200 ps. The electron moves farther, in parallel to the membrane, to another ubiquinone, the secondary acceptor QB, which is bound to the L subunit. QB is reduced to the semiquinone anion QB−:
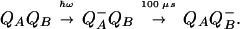 |
1 |
Although QB− itself stays deprotonated, its negative charge causes a pK shift of amino acid residues in its vicinity, which triggers proton binding from the bulk (10–13). If QB is absent or the electron transfer from QA− to QB is blocked, proton binding still occurs, in response to the negative charge on QA− (11, 14).
The delivery of the second electron to QB− is kinetically coupled to its protonation, which yields the anion QBH− (15). The subsequent binding of the second proton produces the neutral ubiquinol QBH2.
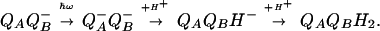 |
2 |
Protons reach QB via a diffuse water cluster, a kind of water lens, that is positioned at the interface between the LM heterodimer and the H subunit. The water lens is connected with the surface by several water channels that could serve as proton inlets (16–19). The intrinsic proton transfer from the surface into the QB site can be traced in chromatophore preparations from R. sphaeroides by monitoring the changes of the transmembrane voltage, Δψ, both spectrophotometrically (20) and electrometrically (21–24). Complementary to the cited studies of proton release by BR, the proton binding by the RC may be considered as a model of a “proton inlet.”
In this work, R. sphaeroides chromatophores were excited with one or two short flashes of light with the aim to resolve the partial steps of proton transfer from the bulk into the QB site. Three observables were monitored, namely the electron transfer, the Δψ generation, and the pH changes in the bulk. The electron transfer to QB− and electrogenesis revealed the same rise time (τ ≈ 100 μs), whereas the response of pH indicator in the bulk lagged distinctly behind (τ ≈ 400 μs). From the dependence of the latter reaction on the concentration of pH indicators, added pH buffers, and the temperature, we concluded that the retardation was due to conveyance of protons by slowly reacting neutral water.
Materials and Methods
Cells of R. sphaeroides R2 and Rhodobacter capsulatus B10 (purple wild-type strains) were grown photoheterotrophically at +30°C; chromatophores were isolated by French press cell disruption as described in ref. 24 and stored frozen in 250 mM sucrose at −80°C. Before the measurements, chromatophores were washed in 100 mM KCl in the presence of 10% (vol/vol) sucrose, pelleted, and resuspended in the same medium. The RC concentration in chromatophores was estimated from the extent of absorption changes of P at 603 nm (25).
Binary oscillations of the semiquinone anion formation at the site QB in response to a train of flashes were maintained by using 20 μM methylene blue (midpoint redox potential at pH 7.0, Em,7 = 11 mV) to oxidize the residual QB− in the course of dark adaptation (26). With 20 μM methylene blue, the life time of QB− was less than 20 s. Hence, with 3–5 min of dark adaptation between flash series, the concentration of QB− before the first flash was expected to be negligible.
Flash-spectrophotometry was performed with a homemade single-beam differential spectrophotometer, which has been described elsewhere (27). A Xenon flash lamp [full width at half maximum ≈ 4 μs; Schott RG 780-nm filter (Mainz, Germany); saturation under given conditions ≈ 95%] was used for excitation. The dwell time of the setup was 1 μs. The optical path length was 2 cm. Routinely, 12–16 transients were digitized and averaged on a Nicolet Pro10 recorder. The redox changes of QB− were monitored at 450 nm versus 480 nm as in ref. 26. The voltage transients across the chromatophore membrane were monitored by the electrochromic bandshifts of carotenoid pigments at 524 nm. Flash-induced pH changes were measured with following pH indicators monitored at their appropriate wavelengths: bromcresol purple (BCP; pH 5.3–7.3; 578 nm), neutral red (NR; pH 5.6–7.6; 545 nm), and cresol red (CR; pH 7.2–9.2; 578 nm). The pH transients were obtained by subtraction of the transients obtained in the presence of pH indicator and pH buffer from those obtained in the presence of the pH indicator alone.
The kinetic traces were analyzed by using the gim software package developed by A. L. Drachev (Dr. Achev Development, Tempe, AZ) and the microcal origin 4.1 software package (Microcal Software, Northampton, MA).
Results
Fig. 1A shows the binary oscillations of QB− in R. sphaeroides chromatophores measured at 450–480 nm in response to a series of flashes. After odd-numbered flashes, the semiquinone anion QB− was formed; after even-numbered ones, it was reduced to ubiquinol, which does not absorb at 450 nm. The Insert shows the kinetics of QB− reduction to ubiquinol after the second flash (τ ≈ 100 μs) at higher time resolution.
Figure 1.

Absorption changes (ΔA) of R. sphaeroides chromatophores after excitation with a series of saturating flashes (1 s apart) in a pH buffer-free chromatophore suspension. (A) QB− oscillations as traced at 450–480 nm. Arrows indicate light flashes. (A Insert) Expansion of the absorption changes after the second light flash on the time scale of microseconds. (B) Transients of the transmembrane voltage after the first and second flashes in the absence and in the presence of 100 nM stigmatellin (measured by electrochromic absorption transients at 524 nm). (C) pH indicating absorption changes at 578 nm of BCP with and without pH buffer. (D) Difference voltage traces (+/− stigmatellin). (E) Difference pH transients (+/− pH buffer). The suspending medium contained 300 mM KCl, 2 mM potassium ferrocyanide/ferricyanide, 2 mM KCN, 50 μM dimethylferrocene, 20 μM methylene blue, 3 μM myxothiazol, 5 μM antimycin A, and 1 μM oligomycin (pH 6.2; Eh ≈ 300 mV; 20°C).
Fig. 1B shows the voltage changes across the membrane of R. sphaeroides chromatophores measured under the conditions of binary QB− oscillations. A typical response consisted of a fast, unresolved phase A (time constant τ < 1 μs in the setup used), attributable to the formation of the P+QA− dipole, and slower components. To discriminate the kinetic components attributable to electrogenic reactions at the QB site, we applied a QB antagonist, stigmatellin (see ref. 7 for the discussion of its binding by the RC). Stigmatellin suppressed the slower components of the voltage transients after both the first and the second flash (see Fig. 1B). Fig. 1C shows the absorption transients at 578 nm of the pH indicator BCP (pK = 6.3). Both the first and the second flash produced a slow rise of the BCP absorption (τ ≈ 400 μs; upper traces in Fig. 1C) that was almost fully attributable to an alkalinization, as evident from its abolition by the added pH buffer (lower trace in Fig. 1C; the residual fast step was due to the formation of P+).
The respective difference traces are presented in Fig. 1 D and E. The lower trace in Fig. 1D is the difference between the voltage traces obtained after the first flash in the absence and in the presence of stigmatellin (hereafter, phase B1). A similar rise time of the electrogenic reaction, namely ≈100 μs, has been detected previously by electrometry (22, 23). The difference between the voltage traces obtained in the absence of stigmatellin after the second flash and those obtained in the presence of inhibitor after the first one reflects the electrogenic reaction coupled to QBH2 formation (hereafter, phase B2; Fig. 1D, upper trace). Again, the rise time was the same as previously recorded electrometrically (22, 24). The time constant of B2 corresponded to that of the electron transfer after the second flash (we plotted the kinetic trace from Fig. 1A Insert as a dashed line in Fig. 1 D and E). In agreement with the previous observations (21), the extent of the electrogenic reaction caused by the QB− formation after the first flash was smaller than that after the second one, which yielded QBH2.
The extent of proton trapping from the bulk (Fig. 1E) was also smaller after the first flash than it was after the second one, indicating that the electrogenic events B1 and B2 depicted in Fig. 1D originated from proton transfer to the QB site. The kinetics of pH changes in the bulk, however, lagged distinctly behind the electrogenesis and electron transfer, after both the first and the second flash (τ > 400 μs).
To test whether the delay was caused by a low rate of proton exchange between the intrinsic proton-buffering groups on the surface of the chromatophore membrane, we added a mobile pH buffer to accelerate the protonic equilibration (28, 29). The addition of Mes [2-(N-morpholino)ethanesulfonic acid] at a concentration of 10–60 μM did not affect the rate of pH transients (Fig. 2A) but decreased their extent, as expected (by 50% at 40 μM Mes; not shown).
Figure 2.

Dependence of the rate of pH transients on the concentration of pH buffer, pH indicator, and temperature. (A) Dependence of the rate constants of pH transients on the concentration of pH buffer: □, B1; ■, B2. (B) Dependence of the rate constants of pH transients on the concentration of the pH indicator: ▵, B1; ▴, B2. In A and B and in Fig. 3, the fitting error was less than the symbol size. (C and D) pH transients of BCP after the first and second flashes at +22°C (C) and +10°C (D), respectively, in the presence of 500 μM of pH indicator. Conditions were the same as those described for Fig. 1.
To test whether the rate of pH transients was limited by collisional interaction of the indicator dye with the buffers at the surface, we increased the concentration of the pH indicator. We found no effect up to a concentration of 60 μM of BCP (Fig. 2B), in agreement with the earlier work by Petty and Dutton (30), who applied several other pH indicators in the concentration range of 5–100 μM with similar chromatophore preparations from R. sphaeroides.
The rate of pH transients was accelerated only at very high concentrations of BCP. On the addition of 500 μM BCP¶, the time constant decreased from τ ≈ 400 μs to τ ≈ 280 μs (compare Fig. 2C with Fig. 1E). It was still slower than the electron transfer. When the temperature was decreased from 22°C to 10°C, the BCP response slowed down from τ ≈ 280 μs to τ ≈ 420 μs (Fig. 2 C and D). Such a slowing corresponds to an activation energy (Ea) of ≈30 kJ/mol.
In Fig. 3, we plotted the rates of pH transients after the second flash measured with various pH dyes as function of the pH for both R. sphaeroides and R. capsulatus chromatophores, together with the data on the rates of electron transfer and electrogenesis (see the figure legend for details). The kinetic discrepancy between the electron transfer and electrogenesis, on the one hand, and the proton binding measured by pH dyes in the bulk, on the other, was common to both R. sphaeroides and R. capsulatus. It was independent of the chemical nature of the pH indicator.
Figure 3.

pH dependence of the rate constants of (i) the electron transfer to QB− after the second flash (as measured at 450–480 nm) in the presence (□) and in the absence (⋄) of pH buffer; (ii) the difference voltage transients (as measured at 524 nm) in the presence (▿) and in the absence (▵,▴) of pH buffer; and (iii) pH transients (○,●) with no pH buffer. Open symbols, R. sphaeroides; closed symbols, R. capsulatus. Hepes (2 mM) was added as pH buffer at pH 7.2 with NR as pH indicator. Tris (3 mM) was used as pH buffer with CR at pH 8.1. Other conditions were the same as those described for Fig. 1, except that only 50 mM KCl was added in the presence of pH buffer.
Discussion
Retarded Proton Transfer from the Bulk to the Surface of the R. sphaeroides RC.
In bacterial RCs, the reduction of QB causes proton uptake from the bulk. Proton uptake, as seen by hydrophilic indicators, is delayed against the electron transfer; this kinetic discrepancy has been observed by several authors working with photosynthetic bacteria (see ref. 31 for a review). The origin of the delay has not been elucidated. By monitoring the kinetics of Δψ generation in addition to electron transfer and proton uptake, we attempted to discriminate between external and intraprotein proton transfer steps around the QB site.
The data in Figs. 1–3 indicated that proton equilibration between the QB site and the bulk includes two distinct stages.
First stage.
In response to the appearance of an electron on QB, protons are attracted into the QB site at first from the nearest inner surface groups. The proton vacancies are then distributed among surface-located ionizable groups to dwell finally on the “terminal” groups with pK values close to the ambient pH. This reaction is electrogenic. Its time constant is ≈100 μs at < 0.1 mM pH buffer (pH 6.0–6.5) but decreases up to ≈60 μs when the buffer concentration is increased from 0.1 mM up to 5 mM (32). This acceleration by pH buffers indicates that the proton vacancies indeed reach the surface groups that are accessible to a bimolecular reaction with the added pH buffer (at high concentration of the latter).
Second stage.
The pH-indicating dyes in the bulk respond rather slowly, with τ ≈ 400 μs at pH 6.2. This observation is fully compatible with the previous paragraph, because the usually applied concentration of a pH indicator, pH buffer per se, is less than 50 μM and thus below the one where the bimolecular reaction at the surface became competitive. The observation of the same delay after the first and second flashes might be taken as an evidence of the involvement of proton exchange between the surface groups and the pH dyes and not of some impediment that was particular to the QB site.
After delivering its proton to QB, the formed terminal base can be protonated by three different pathways (see refs. 14 and 33 for related considerations). It can receive a proton in a bimolecular collision either with a molecule of the pH indicator (reaction A) or with a hydronium ion (reaction B). In homogeneous solutions, the bimolecular rate constant is, for reaction A, ≤109 M−1⋅s−1 (34) and, for reaction B, ≤4 × 1010 M−1⋅s−1 (35). Alternatively, a neutral water molecule could serve as a proton donor (reaction C). The pseudo first-order rate constant of such a protolysis is determined by the pK of the terminal base, pKB, as ≤4 × 1010+pKB−14 s−1. To estimate roughly the expected rates of reactions A–C in our system, we assumed (i) that the pK values of the respective dominating terminal group(s) are close to the medium pH and (ii) that the effective concentration of terminal bases created at the surface on QB reduction, [B−], is equal to 5 × 10−8 M, the RC concentration in our samples. At 20 μM pH indicator, reaction A is expected to proceed with the rate constant of 2 × 104 s−1. This rate is expected to depend on the concentration of the pH dye. Such a dependence has not been observed at pH indicator concentrations below 100 μM either by our group (Fig. 2B) or by others in the earlier work (30). Only when BCP was added at 500 μM (Fig. 2C) did the time constant decrease slightly from ≈400 μs to ≈280 μs. If BCP started to compete with the dominating proton transfer pathway only at such a high concentration, the apparent bimolecular rate constant of this interaction must be ≈107 M−1⋅s−1, two orders of magnitude lower than expected in homogenous solution. Because the mobility of pH indicator was lower than in the bulk, either hydronium ions or water molecules acted as the main proton donors to the surface bases. At pH ≈6.5, the expected rates of the interaction of a terminal group B− with hydronium ions and with neutral water are expected to differ by one order of magnitude (≈104 s−1 and ≈103 s−1, respectively). The actual reaction rate, however, is determined by the unknown local pH in the vicinity of the particular terminal group. Besides, the protolysis is sensitive to the polarization of water: charged species can increase its rate by orders of magnitude (see ref. 33 and references therein). Thus, the two mechanisms can hardly be discriminated based on the rates alone. The Ea values, however, are expected to differ greatly. The reaction with hydronium is diffusion controlled and characterized by much lower Ea (<10 kJ⋅mol−1; ref. 36) than is proton donation by neutral water. The Ea of the latter reaction is pKB controlled:
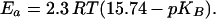 |
3 |
Ea amounts to ≈50 kJ/mol at pH 6.5. This value is close to the value of ≈45 kJ/mol that was reported by Petty and Dutton (30) for the deprotonation of BCP and CR added at 50 μM to chromatophores of R. sphaeroides. The high Ea identifies the deprotonation of neutral water as the rate-limiting step in the transfer of a proton vacancy from the membrane surface into the bulk. At 500 μM BCP, we observed Ea of ≈30 kJ/mol. It was conceivable that Ea was decreased compared with the earlier reported value, because the direct collision with BCP came into play (see above). The weak pH dependence of the rate of pH transients (see Fig. 3) also points to water as major proton donor to the surface-located terminal bases. Water has been previously recognized as a proton donor in the case of the membrane ionophore S-13 (37) and of the SnO2 and ZnO films at the semiconductor/water interface at 5 < pH < 10 (38). An ineffective competition both of the pH indicator and of the hydronium ion with water may be due to their slower diffusion in the thin layer of surface water compared with the bulk. A decrease in the diffusion coefficients at a surface by a factor of 50 compared with those in the bulk has been reported for the negatively charged eosin (39) and the positively charged tetraethyl- and tetramethyl-ammonium ions (40, 41) passing through the water/dichloroethane and water/nitrobenzene interfaces. Such a decrease may reflect the lower affinity of the structured surface water for the electrically charged species and/or the lower mobility of the water layer at the interface (the diffusion coefficient of water along the interface of BR membranes is reportedly five times smaller than in the bulk; ref. 42).
The three pH indicators used (see Fig. 3 and Materials and Methods) have different affinities to chromatophores. Although CR does not bind to chromatophores of R. sphaeroides (30), some binding of BCP to chromatophores has been reported (30). Even stronger binding of the amphiphilic NR could be expected, based on studies of its interaction with the chloroplast thylakoids (43) and the chromatophores of R. sphaeroides (44). Still, neither we nor others (30) found any dependence of the rate pH transients on the chemical nature of the pH dye. If some molecules of CR and NR were bound to chromatophores, they seemed to be “silent,” because they did not accelerate the pH transients. In the absence of any information on where and how strong these particular pH indicator molecules were bound, we can only guess as to the possible reasons for this silence. It is conceivable that the bound pH indicator molecules were out of a fast equilibrium with the surface ionizable groups of the RC because of their binding to the lipid patches far away from the latter. The insulation of the RC could be due to its encircling by the pigment–protein light-harvesting complexes in the native membrane (45). Otherwise, the bound fraction of a pH dye might accumulate, not at the surface, but inside the chromatophore (see ref. 44).
A high Ea of ≈50 kJ/mol at pH 8.0 was also reported by Maroti and Wraight (14) for the proton binding by isolated RC of R. sphaeroides in response to QA− formation. In this case, the delivery of an electron to QA has occurred in ≈200 ps and could not limit the binding of protons. Still, they were trapped from the bulk much more slowly, i.e., within ≈100–200 μs at neutral pH values. As suggested by the crystal structure, the proton redistribution in response to QA− formation is mediated by the same internal water lens as the protonation in response to QB reduction (16, 18). Therefore, we consider the elaborate study of Maroti and Wraight (14) to be directly related to our work.‖ Maroti and Wraight (14) have shown (i) that the Ea of pH transients decreases at alkaline pH; (ii) that the kinetic isotope effect of H/D substitution is small at neutral pH and increases up to a value of ≈3 at pH 10; and (iii) that the dependence of the rate on the viscosity is low. The authors (14) have taken their data as evidence of a conformational gating of the reaction between pH dye and the proton-accepting group(s). Two sets of our data qualify this notion: namely, (i) the appearance of the proton vacancy at the surface long before the onset of pH indicator response (Fig. 1) and (ii) the acceleration of the pH transients at 500 μM BCP (Fig. 2). Thus, we prefer another interpretation: namely, the involvement of water as proton donor. This interpretation conforms both with the decrease of Ea on alkalinization (which actually follows Eq. 3) and with the weak viscosity dependence (14).
The rate of pH transients in the bulk is somewhat slowed with alkalinization, in spite of the decreasing Ea (see Fig. 3 and refs. 14 and 30). This slowing may reflect the gradual involvement of buried residues as terminal groups at pH > 7.0, because there are few, if any, surface-exposed amino acid residues with pK values between 7.0 and 10.0. The proton transfer between water molecules and buried groups is expected to necessitate a quantum tunneling across a potential barrier, the height of which is modulated by the protein dynamics as shown in Fig. 4. Tunneling would slow the pH response in the bulk and, simultaneously, increase the H/D isotope effect, in agreement with the experimental data of Maroti and Wraight (14).
Figure 4.

Hypothetical energy profiles along the reaction coordinate of proton transfer from neutral water (left) to a terminal base (right) at different pH values. The difference between energy levels at two pH values is roughly determined by Eq. 3. (A) At neutral pH, the surface terminal groups (supposedly histidines) are in a direct contact with the surface water; thus, a low isotope effect is expected for the proton transfer from water. (B) At alkaline pH values, the buried amino acid residues overtake the function of terminal groups. The dashed arrow shows a proton tunneling through a potential barrier.
It is noteworthy that the pH dependence of the rate of proton uptake in response to QA− formation was reportedly the same in the absence of pH buffers when measured by pH indicators and in their presence at 10 mM when measured by electrical conductance (14). It is also noteworthy that the studies on whole cells of halobacteria have revealed the absence of intrinsic pH buffers with a mobility comparable to that of imidazole, at least in the periplasmic space of the cells (2). Hence, it seems plausible that neutral water may be the prime proton donor to the surface both in the presence of the added pH buffers and in vivo.
Outlook: Proton Release by BR and Implications for Other Systems.
A retarded proton exchange between the surface and the bulk seems to be a general phenomenon for membrane generators of ΔμH+, because such a slow exchange has been observed with membrane preparations from various phototrophic bacteria (30, 31, 46), thylakoid membranes (47, 48), and BR membranes (2, 5, 6, 49, 50). In the latter case, the excitation with a short laser flash caused the ejection of protons. The acidification was detected by covalently bound pH indicator fluorescein in ≈100 μs. In contrast, the pH dye in solution, pyranine (8-hydroxypyrene-1,3,6-trisulfonate), detected a pH change in only ≈1.0 ms (2, 5, 6, 49, 50). Originally, these observations have been explained by the existence of a nonspecified diffusion barrier for protons between the surface and the bulk (5, 49). Later, however, it has been hypothesized that the experimentally observed delay in the protonation of pyranine by the BR-ejected protons was due to the kinetic “retention” of protons by the carboxyls at the surface. The molecules of pH indicator in the bulk were assumed to compete with these surface groups for protons (4). The retention hypothesis, however, cannot account for the retarded proton transfer from the bulk to the surface of the RC: the surface carboxyls are expected rather to attract protons from the bulk and accelerate their binding but not to slow it down. The rate of the pyranine protonation by the BR-ejected protons has been shown to be independent both of the pyranine concentration (up to 150 μM, at least) and of the pH (49). These data are hardly compatible with the retention hypothesis. Similarly, the retention model can hardly explain the observed high Ea of pyranine protonation in these experiments (54 kJ/mol at pH 7.5; see ref. 2). However, the latter value is in a good agreement with the expected Ea for the protonation of neutral water. In the case of BR, a water molecule would be protonated by a terminal acidic group A with pKA close to the given pH (7.5) with Ea of
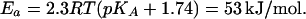 |
4 |
Thus, the delayed proton transfer from the surface of BR to pyranine in the bulk could be due to the slow, rate-limiting protonation of neutral water. The involvement of the latter seems to be caused by the slow diffusion in the surface water layer, as in the case of the RC. The independence of the pyranine protonation rate of its concentration (49) is compatible with an impeded diffusion of the pH indicator to the surface of BR. The independence of this rate of the pH (49), on the other hand, suggests that hydroxyls are not involved in the rate-limiting step. It is conceivable that their diffusion at the surface is impeded as well.
Fig. 5 shows the resemblance between the steps of proton release by BR membranes (2, 5, 6, 49) and of proton binding by the RC in chromatophores (this work). Protons/proton vacancies seem to propagate to the surface and to be distributed along it with τ < 100 μs. On their way into the bulk, they interact with neutral water, because pH buffers and charged water species seem to diffuse too slowly at the surface to compete kinetically with neutral water. The slowness of proton exchange between the surface and neutral water retards the time constant of the proton/proton vacancy transfer into the bulk up to ≈1 ms at neutral pH.
Figure 5.

Comparative presentation of the partial steps of proton release into the bulk by BR and of proton binding from the bulk by the RC of R. sphaeroides.
The protonic equilibration between the interior of membrane enzymes and their surface may be fairly fast. The time constant of the electrogenic proton transfer to QB− (B2, see above) is ≈10 μs in chromatophores of R. sphaeroides at pH 5.0 (24). It is even smaller, (≈3 μs) at pH 8.1 in the membrane vesicles from Chloroflexus aurantiacus (51). By contrast, the retardation of the water-mediated transversal proton exchange between the surface and the bulk, as revealed in this work, implies the slowing of the water-mediated component of the lateral proton transfer (in all its forms—namely by Grotthus mechanism, hydronium self-diffusion, and water diffusion) between proton “generators” and “consumers.” For a general proton generator/consumer couple, however, the lateral proton exchange has to be at least an order of magnitude faster than the rate-limiting step of the transmembrane proton transfer (typically, 1 ms). Hence, its time constant should be <100 μs. Otherwise, a resistance to the lateral proton transfer along the surface may lead to “Ohmic” losses between proton generators and consumers. This question has been addressed previously in stacked thylakoids in which the photosystem II and the ATP synthase are at least 300 nm laterally separated from each other. There was evidence for only very small Ohmic losses by lateral proton transfer (29, 52, 53). This evidence points to the kinetic efficiency of protonic communication even between rather remote membrane proteins. For halobacteria, both the proton transfer along and around the surface of purple membrane particles with τ < 100 μs has been established experimentally (2, 5), and a direct proton delivery from BR to the ATP-synthase in vivo has been claimed (54). It is conceivable that the function of water in the lateral proton transfer is taken over by a continuous web of charged amino acid residues and, perhaps, phospholipid groups at the surface. The particular mechanism and the quantitative picture of the surface-channeled proton flow have to be established yet.
Acknowledgments
We thank Drs. O. Y. Dmitriev, M. Gutman, J. Heberle, A. D. Kaulen, A. A. Konstantinov, A. B. Kotlyar, P. Maroti, D. Oesterhelt, M. I. Verkhovsky, and C. A. Wraight for valuable discussions. This work was supported by Deutsche Forschungsgemeinschaft Grants SFB-171-A2, SFB-431-D8, RUS-436, and Mu-1285/1 and by European Commission Grant INTAS 93-2852-Ex.
Abbreviations
- BCP
pH indicator bromcresol purple
- BR
bacteriorhodopsin
- CR
pH indicator cresol red
- Ea
activation energy
- pyranine
8-hydroxypyrene-1,3,6-trisulfonate
- QA
primary quinone acceptor
- NR
pH indicator neutral red
- QB
secondary quinone acceptor
- RC
photosynthetic reaction center of purple bacteria
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Higher concentrations of BCP were impractical because of too high absorption of the indicator.
The studies of proton binding in response to QB reduction in isolated RC preparations (see refs. 9 and 13 for reviews) provide little help in clarifying the delay of the pH transients in studies on chromatophores. The rate of the second electron transfer from QA to QB in isolated RC (which predominantly causes the proton binding) is much slower than in chromatophores; at pH of ≈7.0, the respective time constant is ≈500 μs. Not surprisingly, the rates of electron transfer, electrogenesis, and proton binding are then very similar.
References
- 1.Mitchell P. Physiol Rev. 1966;41:445–502. doi: 10.1111/j.1469-185x.1966.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 2.Heberle J, Riesle J, Thiedemann G, Oesterhelt D, Dencher N A. Nature (London) 1994;370:379–382. doi: 10.1038/370379a0. [DOI] [PubMed] [Google Scholar]
- 3.Gutman M, Nachliel E. Biochim Biophys Acta. 1995;1231:123–138. [Google Scholar]
- 4.Nachliel E, Gutman M. FEBS Lett. 1996;393:221–225. doi: 10.1016/0014-5793(96)00870-8. [DOI] [PubMed] [Google Scholar]
- 5.Heberle J, Dencher N A. Proc Natl Acad Sci USA. 1992;89:5996–6000. doi: 10.1073/pnas.89.13.5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alexiev U, Mollaaghababa R, Scherrer P, Khorana H G, Heyn M P. Proc Natl Acad Sci USA. 1995;92:372–376. doi: 10.1073/pnas.92.2.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lancaster C R D, Michel H. Photosynth Res. 1996;48:65–74. doi: 10.1007/BF00040997. [DOI] [PubMed] [Google Scholar]
- 8.Shinkarev V P, Wraight C A. In: The Photosynthetic Reaction Center. Deisenhofer J, Norris J R, editors. Vol. 1. San Diego: Academic; 1993. pp. 193–255. [Google Scholar]
- 9.Okamura M Y, Feher G. In: Anoxygenic Photosynthetic Bacteria. Blankenship R E, Madigan M T, Bauer C E, editors. Dordrecht: Kluwer; 1995. pp. 577–594. [Google Scholar]
- 10.Wraight C A. Biochim Biophys Acta. 1979;548:309–327. doi: 10.1016/0005-2728(79)90138-5. [DOI] [PubMed] [Google Scholar]
- 11.Maroti P, Wraight C A. Biochim Biophys Acta. 1988;934:329–347. [Google Scholar]
- 12.McPherson P H, Okamura M Y, Feher G. Biochim Biophys Acta. 1988;934:348–368. doi: 10.1016/0005-2728(93)90116-w. [DOI] [PubMed] [Google Scholar]
- 13.Maroti P. Photosynth Res. 1993;31:1–17. doi: 10.1007/BF02185435. [DOI] [PubMed] [Google Scholar]
- 14.Maroti P, Wraight C A. Biophys J. 1997;73:367–381. doi: 10.1016/S0006-3495(97)78077-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graige M S, Paddock M L, Bruce J M, Feher G, Okamura M Y. J Am Chem Soc. 1996;118:9005–9016. [Google Scholar]
- 16.Ermler U, Fritzsch G, Buchanan S K, Michel H. Structure (London) 1994;2:925–936. doi: 10.1016/s0969-2126(94)00094-8. [DOI] [PubMed] [Google Scholar]
- 17.Stowell M H, McPhillips T M, Rees D C, Soltis S M, Abresch E, Feher G. Science. 1997;276:812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- 18.Fritzsch G, Kampmann L, Kapaun G, Michel H. Photosynth Res. 1998;55:127–132. [Google Scholar]
- 19.Abresch E C, Paddock M L, Stowell M B, McPhillips T M, Axelrod H L, Soltis S M, Rees D C, Okamura M Y, Feher G. Photosynth Res. 1998;55:119–125. [Google Scholar]
- 20.Drachev L A, Mamedov M D, Mulkidjanian A Y, Semenov A Y, Shinkarev V P, Verkhovsky M I. FEBS Lett. 1988;233:315–318. [Google Scholar]
- 21.Drachev L A, Semenov A Y, Skulachev V P, Smirnova I A, Chamorovsky S K, Kononenko A A, Rubin A B, Uspenskaya N Y. Eur J Biochem. 1981;117:483–489. doi: 10.1111/j.1432-1033.1981.tb06363.x. [DOI] [PubMed] [Google Scholar]
- 22.Drachev L A, Mamedov M D, Mulkidjanian A Y, Semenov A Y, Shinkarev V P, Verkhovsky M I. FEBS Lett. 1990;259:324–326. [Google Scholar]
- 23.Gopta O A, Bloch D A, Cherepanov D A, Mulkidjanian A Y. FEBS Lett. 1997;412:490–494. doi: 10.1016/s0014-5793(97)00842-9. [DOI] [PubMed] [Google Scholar]
- 24.Gopta O A, Cherepanov D A, Semenov A Y, Mulkidjanian A Y, Bloch D A. Photosynth Res. 1998;55:309–316. [Google Scholar]
- 25.Mulkidjanian A Y, Mamedov M D, Drachev L A. FEBS Lett. 1991;284:227–231. doi: 10.1016/0014-5793(91)80691-u. [DOI] [PubMed] [Google Scholar]
- 26.Mulkidjanyan A Y, Shinkarev V P, Verkhovsky M I, Kaurov B S. Biochim Biophys Acta. 1986;849:150–161. [Google Scholar]
- 27.Junge W. In: Chemistry and Biochemistry of Plant Pigments. Goodwin T W, editor. Vol. 2. London: Academic; 1976. pp. 233–333. [Google Scholar]
- 28.Drachev L A, Kaulen A D, Skulachev V P. FEBS Lett. 1984;178:331–335. [Google Scholar]
- 29.Polle A, Junge W. Biophys J. 1989;56:27–31. doi: 10.1016/S0006-3495(89)82649-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petty K M, Dutton P L. Arch Biochem Biophys. 1976;172:335–345. doi: 10.1016/0003-9861(76)90085-0. [DOI] [PubMed] [Google Scholar]
- 31.Wraight C A, Cogdell R J, Chance B. In: The Photosynthetic Bacteria. Clayton R K, Sistrom W R, editors. New York: Plenum; 1978. pp. 471–511. [Google Scholar]
- 32.Gopta O A, Semenov A Y, Bloch D A. In: Photosynthesis: Mechanism and Effects. Garab G, editor. Dordrecht: Kluwer; 1999. pp. 873–876. [Google Scholar]
- 33.Gutman M, Nachliel E. Biochim Biophys Acta. 1990;1015:391–414. [Google Scholar]
- 34.Bell R P. The Proton in Chemistry. London: Chapman & Hall; 1973. [Google Scholar]
- 35.Eigen M. Angew Chem. 1963;75:489–588. [Google Scholar]
- 36.Pomes R, Roux B. Biophys J. 1998;75:33–40. doi: 10.1016/S0006-3495(98)77492-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kasianowicz J, Benz R, McLaughlin S. J Membr Biol. 1987;95:73–89. doi: 10.1007/BF01869632. [DOI] [PubMed] [Google Scholar]
- 38.Lemon B I, Hupp J T. J Phys Chem. 1997;101:2426–2429. [Google Scholar]
- 39.Kakiuchi T, Takasu Y. J Phys Chem. 1997;101:5963–5968. [Google Scholar]
- 40.Beattie P D, Delay A, Girault H H. Electrochim Acta. 1995;40:2961–2969. [Google Scholar]
- 41.Marecek V, Lhotsky A, Racinsky S. Electrochim Acta. 1995;40:2905–2908. [Google Scholar]
- 42.Lechner R E, Dencher N A, Fitter J, Büldt G, Belushkin A V. Biophys Chem. 1994;49:91–99. [Google Scholar]
- 43.Hong Y Q, Junge W. Biochim Biophys Acta. 1983;722:197–208. [Google Scholar]
- 44.Mulkidjanian A Y, Junge W. FEBS Lett. 1994;353:189–193. doi: 10.1016/0014-5793(94)01031-5. [DOI] [PubMed] [Google Scholar]
- 45.Jungas C, Ranck J L, Rigaud J L, Joliot P, Vermeglio A. EMBO J. 1999;18:534–542. doi: 10.1093/emboj/18.3.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halsey Y D, Parson W W. Biochim Biophys Acta. 1974;347:404–416. doi: 10.1016/0005-2728(74)90079-6. [DOI] [PubMed] [Google Scholar]
- 47.Ausländer W, Junge W. Biochim Biophys Acta. 1974;357:285–298. doi: 10.1016/0005-2728(74)90067-x. [DOI] [PubMed] [Google Scholar]
- 48.Haumann M, Junge W. FEBS Lett. 1994;347:45–50. doi: 10.1016/0014-5793(94)00495-1. [DOI] [PubMed] [Google Scholar]
- 49.Heberle J. Zeitauflösende Untersuchung der Protonentranslokationsschritte von Bakteriorhodopsin mittels chemisch-gekoppelter pH Indikatoren. Berlin: Freien Universität; 1991. [Google Scholar]
- 50.Dioumaev A K, Richter H T, Brown L S, Tanio M, Tuzi S, Saito H, Kimura Y, Needleman R, Lanyi J K. Biochemistry. 1998;37:2496–2506. doi: 10.1021/bi971842m. [DOI] [PubMed] [Google Scholar]
- 51.Mulkidjanian A Y, Hochkoeppler A, Zannoni D, Drachev L A, Melandri B A, Venturoli G. Photosynth Res. 1998;56:75–82. doi: 10.1007/BF02184153. [DOI] [PubMed] [Google Scholar]
- 52.Haraux F, de Kouchkovsky Y. Biochim Biophys Acta. 1982;679:235–247. [Google Scholar]
- 53.Junge W, Polle A. Biochim Biophys Acta. 1986;848:265–273. [Google Scholar]
- 54.Michel H, Oesterhelt D. Biochemistry. 1980;19:4615–4619. doi: 10.1021/bi00561a012. [DOI] [PubMed] [Google Scholar]