

Abstract
Tyrosine phosphorylation is an important means of regulating ion channel function. Our previous gene expression studies using the Xenopus laevis oocyte system suggested that tyrosine phosphorylation of G-protein-gated inwardly rectifying potassium channels (Kir3 or GIRK) suppressed basal channel conductance and accelerated channel deactivation. To assess whether similar mechanisms regulate Kir3 function in mammalian cells, we developed and characterized a phosphoselective antibody recognizing Kir3.1 phosphorylated at tyrosine 12 in the N-terminal domain and then probed for evidence of Kir3.1 phosphorylation in cultured mammalian cells and spinal cord. The antibody was found to discriminate between the phospho-Tyr12 of Kir3.1 and the native state in transfected cell lines and in primary cultures of mouse atria. Following either mouse hindpaw formalin injection or sciatic nerve ligation, pY12-Kir3.1 immunoreactivity was enhanced unilaterally in the superficial layers of the spinal cord dorsal horn, regions previously described as expressing Kir3.1 channels. Mice lacking Kir3.1 following targeted gene disruption did not show specific pY12-Kir3.1 immunoreactivity after sciatic nerve ligation. Further, mice exposed to repeatedly forced swim stress showed bilateral enhancement in pY12-Kir3.1 in the dorsal horn. This study provides evidence that Kir3 tyrosine phosphorylation occurred during acute and chronic inflammatory pain and under behavioral stress. The reduction in Kir3 channel activity is predicted to enhance neuronal excitability under physiologically relevant conditions and may mediate a component of the adaptive physiological response.
G-protein-gated inwardly rectifying potassium channels (Kir3)4 modulate excitability by hyperpolarizing the plasma membrane (1, 2), thereby reducing heart rate (3, 4) and nociception (5, 6). The molecular mechanisms regulating these activation processes, however, remain unclear. Using Xenopus laevis oocytes, our previous studies suggested that phosphorylation of N-terminal Kir3 tyrosine residues accelerated channel deactivation kinetics and inhibited basal potassium current amplitude (7, 8), but whether Kir3 N-terminal tail tyrosine phosphorylation occurs in mammalian systems remained to be elucidated. Because Kir3 channels play an important role in regulating cardiac and neuronal signaling (1–4), modulation of channel function mediated by tyrosine phosphorylation could influence cardiac and CNS excitability. Similar tyrosine kinase mechanisms regulate other inwardly rectifying potassium channels (9–10).
Of the four Kir3 subtypes identified in mammals (Kir3.1, 3.2, 3.3, and 3.4), Kir3.1 is expressed in the greatest range of tissues, forming heterotetramers with other Kir3 subunits in heart, brain, and endocrine cells (1). Recent studies in mice with genetically ablated Kir3.1 have shown that Kir3 plays a role in attenuating opioid-mediated antinociception by activating heterotetramers of Kir3.1 and Kir3.2 in the dorsal horn of the spinal cord (4, 5). Because tyrosine kinases are up-regulated and activated in animal models of spinally mediated acute and chronic pain (11), it is reasonable to hypothesize that Kir3 may be phosphorylated at N-terminal tyrosine residues in response to these stimuli.
To identify physiological stimuli promoting Kir3 tyrosine phosphorylation in the spinal cord, in this study we developed an antibody selective for Kir3.1 when phosphorylated at tyrosine 12 (hereafter pY12-Kir3.1), a residue located in the cytoplasmic N-terminal domain. After characterizing pY12-Kir3.1 specificity and phosphoselectivity in primary cardiac myocyte cultures and transfected cell lines, we evaluated phosphorylation of Tyr12-Kir3.1 in spinal cord slices from mice subjected to hindpaw formalin injection or sciatic nerve ligation, models of inflammatory and neuropathic pain, respectively. We further investigated pY12-Kir3.1 in a mouse model of chronic stress to determine whether Kir3.1 Tyr12 phosphorylation occurred in the dorsal horn in response to stressful stimuli independently of nociception. This study provides evidence that Kir3.1 tyrosine phosphorylation occurs in response to nociceptive stimuli and physiological stress.
EXPERIMENTAL PROCEDURES
DNA Clones
Plasmid vectors containing coding regions for Kir3.1 (GenBank™ U01071) were obtained from Dr. Henry Lester (California Institute of Technology). Kir3.1 was point-mutated by PCR-based site-directed mutagenesis to create Kir3.1[F137S] according to the manufacturer’s specifications (Stratagene, La Jolla, CA). The F137S form of Kir3.1 was used because it expresses functional homotetramers in the absence of other Kir3 subunits, whereas Kir3.1 expressed alone is non-functional and gets trapped in Golgi (7). PCR-based site-directed mutagenesis was also used to mutate Tyr12 to Phe. Fluorescently tagged fusion proteins were created by cloning the construct into a pEYFP-C1 vector (Clontech Laboratories, Palo Alto, CA), which fused YFP to the Kir3.1 N terminus.
Cell Lines
SH-SY5Y cells were a gift from Dr. Zhengui Xia (University of Washington). NIH-3t3 fibroblasts stably transfected with full-length trkB were a gift from Dr. Mark Bothwell (University of Washington). Chinese hamster ovary cells and AtT20 mouse pituitary cells were from American Type Culture Collection (Manassas, VA) and maintained according to recommended protocols.
Pharmacological Agents and Antibodies
BDNF was a gift from AMGEN Corporation. K252A was from Calbiochem. Concentrated stocks were made by dilution in Me2SO. Working aliquots were diluted such that Me2SO concentration did not exceed 0.1% of the final solution in cell culture experiments. Formalin was from Fisher Scientific (Fair Lawn, NJ). Actin antibody was from Ab-Cam (Cambridge, MA). Unmodified Kir3.1 antibody was from Chemicon Corporation (Temecula, CA). Phospho-ERK antibody was from Cell Signaling (Beverly, MA). Phalloidin-688 toxin was from Molecular Probes (Eugene, OR). Secondary antibodies were from Jackson Immunoresearch (West Grove, PA). Hydrogen peroxide concentration was determined by Amplex Red assays (Molecular Probes).
Polyclonal Antibody Generation
A polypeptide-containing residues 1–17 (MSALRRKFGDDpYQVVTT) of rodent Kir3.1 phosphorylated at tyrosine residue 12 was generated by PeptidoGenic Research & Co, Inc. (Livermore, CA). The peptide was conjugated to KLH and injected into rabbits by Cocalico Biologicals, Inc. (Reamstown, PA). 500 μg of antigen peptide was combined with Complete Freund’s Adjuvant for initial inoculation then animals received boost inoculations of 250 μg of antigen peptide on days 14, 21, 49, 56, and monthly thereafter. Bleeds (6–7 ml) were collected once per month.
Affinity Purification
Columns were prepared by activating Sepharose 4B beads with 0.5 g of CNBr then conjugating 2–5 μmol of pY12-Kir3.1 peptide. Sera (3 ml) were added to the Sepharose beads and incubated overnight at 4 °C with gentle rocking. Unbound protein was washed off resin in a 0.7 × 15 cm column using 20 ml of Tris (pH 7.4, 0.05 M) and NaCl (0.15 M), then antibody was eluted with 5 ml MgCl2 (5 M) in Tris (pH 7.4, 0.05 M) and NaCl (0.15 M) and concentrated using a Centriprep-30. Antibody aliquots were stored at −20 °C in 50% glycerol until use.
ELISA Assay
96-well plates were incubated for 3 h in a 10 μg/ml solution (in 50 mM sodium carbonate buffer, pH 9.6) of a peptide synthesized to correspond to either the first 17 residues of the native Kir3.1 N-terminal tail or the first 17 residues of Kir3.1 N-terminal tail, phos-phorylated at tyrosine, position 12 (pY12-Kir3.1). Unbound antigen was rinsed from wells with 1×PBS/Tween 20 (0.05%). Wells were incubated at room temperature for 1 h in blocking solution (1× PBS/Tween 20 (0.05%) and 3% bovine serum albumin), then overnight in increasing concentrations (triplicate) of purified pY12-Kir3.1 antibody. Wells were incubated for 3 h at 37 °C in IgG/alkaline-phosphatase-conjugated secondary antibody (Promega, Madison, WI) diluted 1:1000 in 1× PBS-Tween 20 (0.05%). Alkaline phosphatase was detected by 10 mg/ml p-nitrophenyl phosphate in 0.05 M sodium carbonate buffer (pH 9.8, with 1 M MgCl2). The reaction was stopped by the addition of 2 M NaOH. Absorbance was measured by spectrophotometer at 405 nm, and serum antibody titer was estimated based on absorbance reading plotted against dilution for each triplicate.
Immunoblots
Seven days after nerve ligation or 40 min after formalin or saline injections, mice were killed by decapitation, and the spinal cord was dissected and placed on dry ice, then cut by scalpel into four longitudinal sections corresponding to dorsal/ventral horns, ipsilateral and contralateral to injection. Tissue was placed into 250 μl of ice-cold lysis buffer (12) and homogenized by Dounce homogenizer. Lysates were boiled for 2 min, sonicated 20 s to shear genomic DNA, and then centrifuged 10 min (14,000 × g, 4 °C). Protein concentration was determined by Pierce bicinchroninic assay (BCA) before loading 20 μg onto non-denaturing 10% bisacrylamide precast gels (Invitrogen) and running at 150 V for 1.5 h. Blots were transferred to nitrocellulose for 1.5 h at 30 V, blocked in 1× TBS/5% nonfat dry milk/0.1% Tween 20 for 2 h, incubated 8 h room temperature in 0.03 mg/ml pY12-Kir3.1 antibody and washed. Blots were incubated for 1 h in horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Promega) before treating with chemiluminescence reagents (PerkinElmer Life Sciences) and developing by Kodak Biomax x-ray film (Rochester, NY). Band intensity was quantified by scanning film and comparing average pixel intensity of each band to vehicle controls using NIH Image v1.61 software (Research Service Support Branch of the NIMH, National Institutes of Health, Bethesda, MD).
Immunocytochemistry and Confocal Microscopy
NIH3t3-trkB or SH-SY5Y neuroblastoma cells were plated onto coverslips and incubated in a 37 °C (5% CO2) incubator. After drug treatments, cells were washed once in PBS, then fixed in 4% paraformaldehyde for 20 min, washed, blocked 2 h in0.3% gelatin/0.025% Triton X-100 in PBS at room temperature, incubated 1–3 days at 4 °C in 1.5 μg/ml primary affinity-purified pY12-Kir3.1 antibody or 1:100 phospho-ERK1/2 (Cell Signaling, Beverly, MA), then incubated 2 h at room temperature in goat anti-rabbit rhodamine-conjugated secondary antibody at 1:1000. Coverslips were viewed by Leica confocal microscopy. All quantitations were performed using Metamorph Imaging System software (Universal Imaging Corporation, Downington, PA), in which plasma membranes were outlined, and the average pixel intensity of the defined region was determined. Quantifications were performed blind.
Primary Mouse Cardiac Myocyte Cultures
Atria or ventricles were dissected from postnatal day 1–5 mouse pups, cut coarsely, digested in pancreatin (Sigma) and collagenase (Worthington, Lakewood, NJ) for 30 min at 37 °C then collected by centrifugation in fetal bovine serum (Sigma). After 2 h of incubation at 37 °C to allow unwanted fibroblasts to adhere to the plates, supernatant was collected, and cells were plated on coverslips coated with 50 μg/ml poly-D-lysine. Cells were used within 48 h after culture for atrial myoctyes and 5–6 days after culture for ventricular myocytes. ICC was conducted as described above for NIH3t3 cell lines.
Animals and Housing
Male C57Bl/6 mice (Charles River Laboratories, Wilmington, MA) weighing 25–35 g, housed in the Animal Core Facility at the University of Washington in groups of two to four in plastic cages bedded with Bed-A-Cob, were used in these experiments. Kir3.1 knock-out mice, generated as described (4), were backcrossed for nine generations against the C57BL/6 mouse strain. PCR of tail DNA samples was used to confirm null-mutation genotype. Animals were maintained in pathogen-free housing units illuminated on a 12 h light-dark cycle with artificial lights on at 7 AM. Food and water were available ad libitum. Protocols were approved by the University of Washington Animal Care and Use Committee in accordance with the 1996 National Institutes of Health Guide for the Care and Use of Laboratory Animals, and guidelines for the International Association for the Study of Pain (13). Veterinary staff inspected mice regularly to ensure compliance.
Partial Sciatic Nerve Ligation (pSNL)
pSNL was used as a model of neuropathic pain (14). The animals were anesthetized with pentobarbital sodium (intraperitoneal, 80 mg/kg). The right hind legs were cleaned with 70% ethanol. Using autoclaved/sterilized instruments, the right sciatic nerve was exposed, and ~one-third to one-half of its diameter was tightly ligated using 7-0 silk suture (Surgical Specialties Corporation, Reading, PA). After checking hemostasis, the muscle and the adjacent fascia were closed with sutures, and the skin was closed with clips. The mice were euthanized with halothane when experiments were completed.
Hindpaw Formalin Injection
The left hindpaw of wild-type C57Bl/6 mice was gently held and injected intraplantarly with saline or 4% formalin in pH 7.4 phosphate buffer using a 26.5-gauge needle. The mouse was returned to a clear cage for observation. Paw flinches and licks were counted during the acute (0–5 min) and tonic phases (10–40 min) of response to formalin treatment. Mice were deeply anesthetized by halothane inhalation and cardioperfused with 4% paraformaldehyde 40 min after injection for immunohistochemistry experiments, or killed by decapitation for Western blot analyses.
Spinal Cord Immunohistochemistry
After cardioperfusion with paraformaldehyde, spinal cords were dissected and placed in 4% paraformaldehyde for an additional 2 h, then transferred to a 30% sucrose solution and incubated for 1–2 days at 4 °C for cryoprotection. A microtome (Leica Microsystems, Wetzlar, Germany) was used to cut 40-μm floating sections stored in phosphate buffer. Sections were rinsed three times in PBS then blocked in 4% goat serum and 0.3% Triton X-100. After three more PBS rinses, sections were incubated 2 days in primary antibodies (1:100 pY12-Kir3.1, 1:1000 GFAP (Advanced Immunochemical, Inc., Long Beach, CA), 1:250 NeuN (Chemicon, Temecula, CA) diluted in blocking buffer, rinsed again in PBS, and then incubated in secondary antibodies (1:250 Rhodamine-conjugated goat anti-rabbit, 1:1000 donkey anti-guinea pig, 1:500 fluorescein isothiocyanate-conjugated goat anti-mouse (Jackson Laboratories, West Grove, PA). Slices were mounted onto glass slides and dried overnight, then mounted with Vectashield mounting media (Vector Laboratories, Burlingame, CA) and coverslipped. Images were taken using an SP1 confocal microscope and image capturing software (Leica Microsystems, Wetzlar, Germany). Quantifications were performed blind using Metamorph Imaging System software (Universal Imaging Corporation) or NIH Image v1.61 software. Average pixel intensities within a region approximating laminae I–II of the spinal cord dorsal horn were calculated and normalized to a region of comparable area in the ventral horn.
Forced Swim Stress
C57Bl/6 mice were exposed to a modified Porsolt forced swim stress as described previously (15–16). Briefly, mice swam in a 30 °C water bath in a 2-day procedure with no opportunity to escape. On day 1, animals swam for 15 min. On day 2, mice swam for four intervals of 6 min separated by 7–12-min rest periods. Following each swim, mice were towel-dried and returned to their home cages. Exclusion criteria included difficulty in staying afloat; no mice met these criteria in this study.
Statistics
Student’s t tests were performed to determine statistical significance of results. p values <0.05 were used to define a significant difference between conditions.
RESULTS
Generation of pY12-Kir3.1, an Antibody Recognizing Kir3.1, Phosphorylated at Tyrosine, Position 12
An antibody selective for Kir3.1 phosphorylated at tyrosine 12 within the N-terminal domain (pY12-Kir3.1) was generated in rabbits and affinity-purified. A peptide for the first 17 residues of Kir3.1 phosphorylated at position 12 was synthesized and conjugated to KLH (Fig. 1A), and rabbits were immunized to the conjugate by a schedule of injections as detailed under “Experimental Procedures.” Following monthly boosts with the peptide, antibodies to pY12-Kir3.1 were affinity-purified from the raw sera by a CNBr column conjugated to the pY12-Kir3.1 peptide. Antibody bound to the column was isolated, and its specificity for phosphorylated over unmodified peptide was verified by ELISA assay. The affinity-purified antisera preferentially bound to the phosphorylated pY12-Kir3.1 peptide (first 17 amino acids) over the peptide in the native state (Fig. 1B). These results suggest specificity of affinity-purified pY12-Kir3.1 for Kir3.1 phosphorylated at tyrosine 12.
FIGURE 1. pY12-Kir3.1 is phosphoselective and specific for Kir3.1.
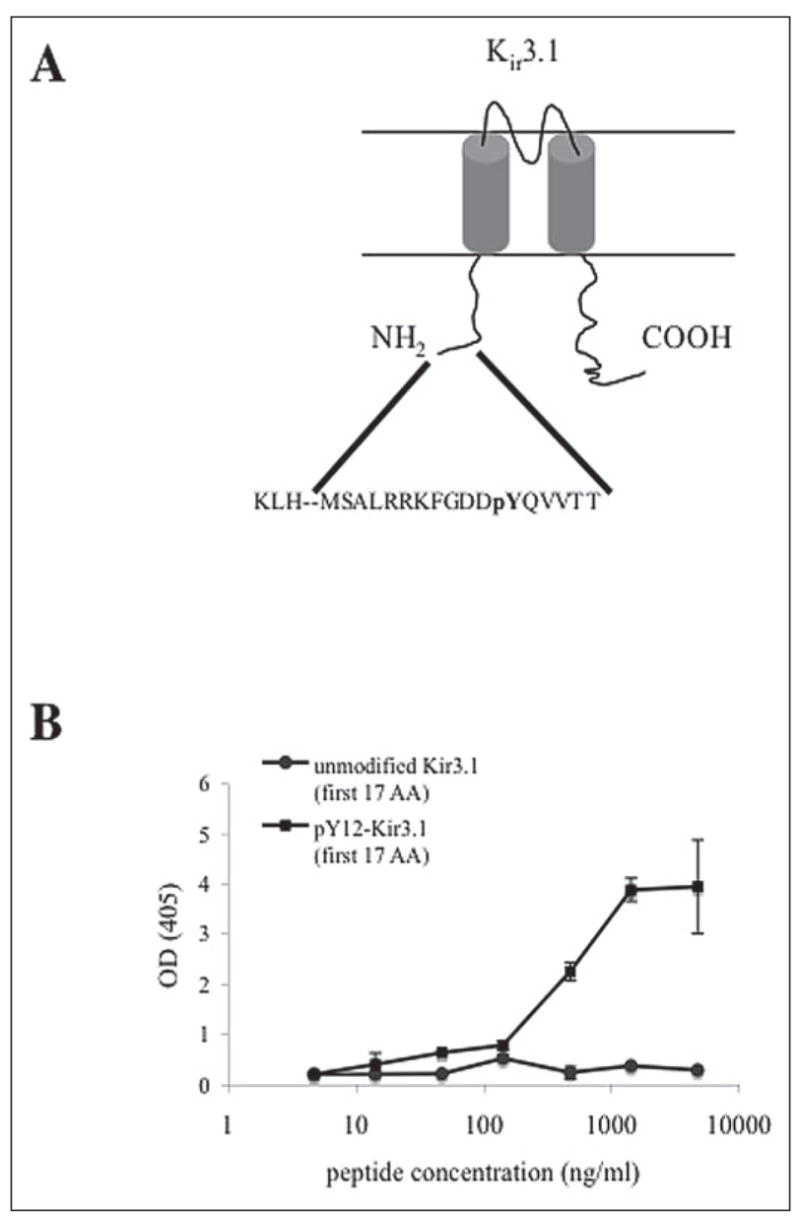
A, pY12-Kir3.1 was generated by immunizing rabbits with a KLH-conjugated synthetic peptide recognizing the first 17 amino acids of Kir3.1 N-terminal tail. Sera were purified over a CNBr column conjugated to phospho-Y12-Kir3.1. B, ELISA assays were performed in which affinity-purified antisera were coincubated with synthetic peptides of either unmodified Kir3.1 or pY12-Kir3.1 (first 17 amino acids).
We confirmed the expression of Kir3.1 in cultured SH-SY5Y neuroblastoma cells differentiated 10 days with 10 μM all-trans-retinoic acid (ATRA) by immunoblot analysis using a commercially available, non-phosphoselective antibody to Kir3.1 (data not shown). Treatment of cultures for 15 min with 4.5 mM hydrogen peroxide, a tyrosine phosphatase inhibitor and activator of non-receptor tyrosine kinases such as Src (17), resulted in enhanced pY12-Kir3.1 immunoreactivity (IR) (Fig. 2,A and B), suggesting both phosphoselectivity and specificity for K ir3.1. The peroxide-enhanced pY12-Kir3.1-IR in SH-SY5Y cells was prolonged by subsequent incubation of SH-SY5Y cells in the tyrosine phosphatase inhibitor phenylarsine oxide (PAO, 20 μM) (TABLE ONE), again suggesting antibody phosphosensitivity.
FIGURE 2. pY12-Kir3.1-IR is increased following stimulation of cells expressing Kir3.1.
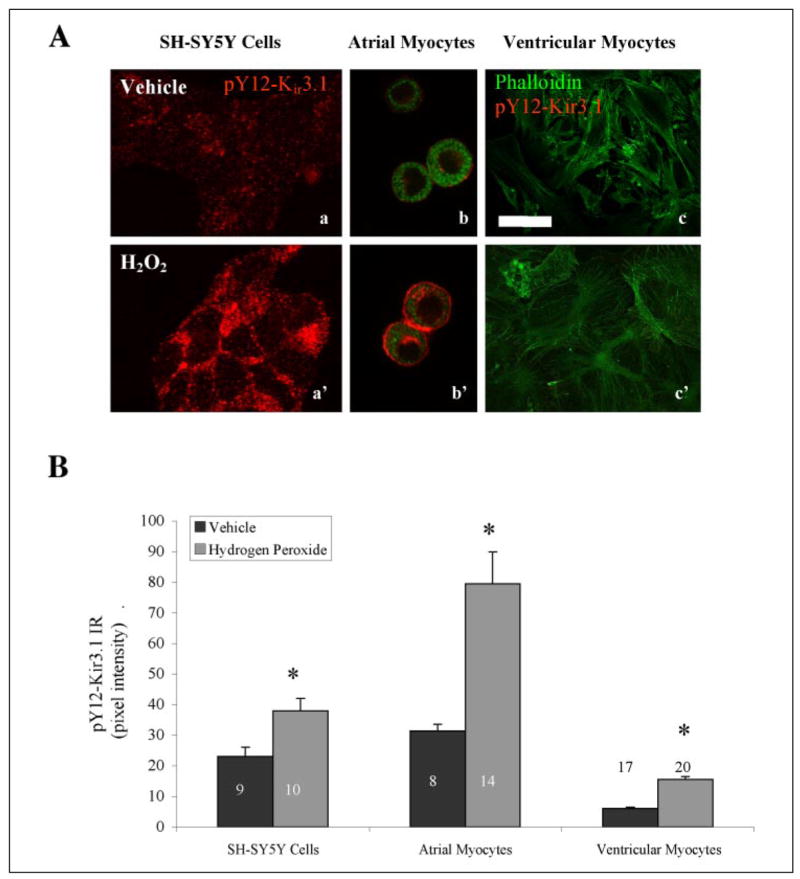
A, hydrogen peroxide elevates pY12-Kir3.1-IR in SH-SY5Y cells and atrial myocytes but not in ventricular myocytes. Scale bar, 100 μm. B, quantification of peroxide-induced increase in pY12-Kir3.1-IR. Black bars, vehicle; gray bars, hydrogen peroxide-treated. *, p < 0.05 by Student’s t test.
TABLE ONE.
Tyrosine phosphatase inhibition by phenylarsine oxide (20 μM) prolongs hydrogen peroxide-mediated enhancement of pY12-Kir3.1-IR in SH-SY5Ycells
| Time after hydrogen peroxide treatment | Treatment after hydrogen peroxide removeda | |
|---|---|---|
| Vehicle | PAO | |
| min | ||
| −15 | 22 ± 2 (10)b | |
| 0 | 38 ± 2 (10) | |
| 5 | 34 ± 5 (8) | 34 ± 6 (6) |
| 15 | 37 ± 2 (6) | 42 ± 10 (3) |
| 30 | 20 ± 1 (5) | 42 ± 12 (3) |
Data representative of two independent experiments.
Mean pixel intensity ± S.E. (n).
These results were confirmed in AtT20 cells, a mouse pituitary tumor cell line endogenously expressing Kir3.1 (Ref. 18, and data not shown). Further, peroxide treatment had no effect on pY12-Kir3.1-IR in CHO cells (data not shown), a cell line that does not endogenously express Kir3.1 (19).
To extend these results to native tissues, we cultured atrial and ventricular cardiac myocytes from day 1–5 mouse pups and examined pY12-Kir3.1-IR (Fig. 2, A and B). Atria endogenously express Kir3.1, whereas ventricular myocytes express substantially less Kir3.1 protein (1). pY12-Kir3.1-IR was very low in vehicle-treated cells (Fig. 2, A and B). Treating atrial myocytes with increasing doses of hydrogen peroxide resulted in enhanced pY12-Kir3.1-IR as determined by confocal microscopy and image analysis using Metamorph software (TABLE TWO).
TABLE TWO.
pY12-Kir3.1-IR in hydrogen peroxide-treated atrial myocytes
| Dose hydrogen peroxide | pY12-Kir3.1 IR |
|---|---|
| mM | |
| 0.0 | 0 ± 0 (10)a |
| 0.2 | 8 ± 2 (12) |
| 1.0 | 7 ± 1 (13) |
| 4.5 | 49 ± 13 (5) |
Mean pixel intensity ± S.E. (n).
To exclude the possibility that the hydrogen peroxide-induced enhancement pY12-Kir3.1-IR was a result of apoptosis, we preincubated SH-SY5Y cells or atrial myocytes in 50–500 μM anisomycin for 15 min prior to fixation and immunostaining with pY12-Kir3.1. Both hydrogen peroxide and anisomycin activate comparable pro-apoptotic signaling cascades at 15-min post-treatment, including p38 MAPK and JNK (c-Jun N-terminal kinase), but anisomycin is not associated with Src activation and tyrosine phosphatase inhibition (20–22). Supporting the antibody specificity for tyrosine kinase-mediated signaling events, anisomycin pretreatment did not increase pY12-Kir3-IR in ATRA-differentiated SH-SY5Y cells or atrial myocytes (TABLE THREE).
TABLE THREE.
pY12-Kir3.1-IR in SHSY5Y cells and a trial myocytes after anisomycin treatment
| Cell type | Vehicle | Anisomycin | |
|---|---|---|---|
| pY12-Kir3.1 IR | Dose | pY12-Kir3.1 IR | |
| μM | |||
| SH-SY5Y | 20 ± 2 (9)a | 50 | 25 ± 6 (13) |
| Atrial myocytes | 21 ± 2 (5) | 100 | 22 ± 6 (4) |
| 500 | 24 ± 2 (6) | ||
Mean pixel intensity ± S.E. (n).
Brain-derived neurotrophic factor (BDNF) activation of trkB receptor is a more physiologically relevant stimulus, and previous results suggested that trkB activation by BDNF could modulate Kir3 signaling (7, 8). Thus, we investigated whether pY12-Kir3.1-IR was elevated following trkB activation in NIH-3t3 fibroblasts stably transfected with full-length trkB, then transiently transfected with YFP-tagged constructs of Kir3.1[F137S] or Kir3.1[F137S/Y12F]. The single point mutation in the pore region allows the channel to traffic normally and function as a homomeric channel (23). Expression of the YFP-tagged channels occurred throughout the cytoplasm (Fig. 3A), in concurrence with previously published literature indicating Golgi and endoplasmic reticulum as well as plasma membrane expression of these channels (24). To verify functional trkB in these NIH-3t3 cells, we fixed cells at 2-, 5-, 15-, and 30-min post-BDNF treatment (0.2 μg/ml, 15 min) and immunostained for phospho-ERK1/2, one of the first signaling events observed following BDNF activation of trkB (25). In accordance with previously published reports (25), the peak of phospho-ERK1/2 immunoreactivity occurred between 5 and 15 min (TABLE FOUR).
FIGURE 3. TrkB activation elevates pY12-Kir3.1-IR in NIH-3t3 cells stably transfected with trkB.
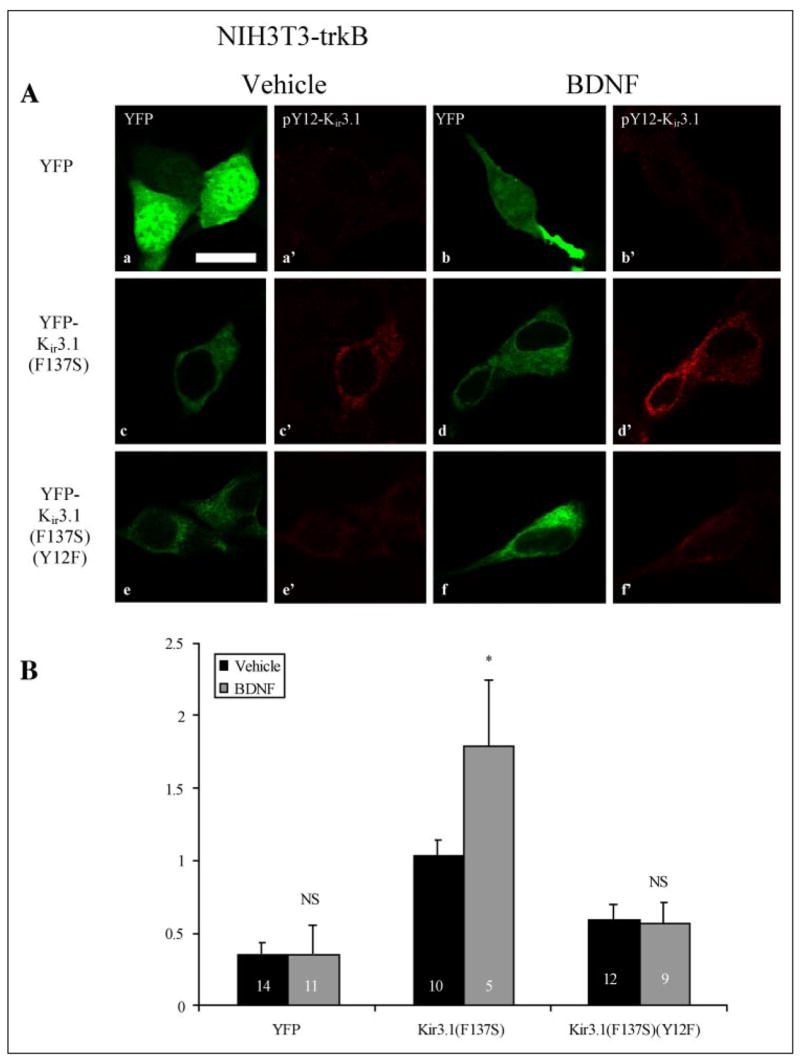
A, NIH3t3 cells were transfected with YFP (upper panels), YFP-K ir3.1[F137S] (middle panels), or YFP-Kir3.1[F137S/Y12F] and treated in vehicle (panels a, a′, c, c′, e, e′) or 0.2 μg/ml BDNF (panels b, b′, d, d′, f, f ′) for 15 min. Images taken with a 488-nm laser (panels a, b, c, d, e, ) excited the YFP f or the YFP-tagged channel, indicating transfection efficacy, whereas images taken with a 568-nm laser (panels a′, b′, c′, d′, e′, f′) excited a rhodamine-conjugated secondary antibody recognizing pY12-Kir3.1. Scale bar, 100 μm. B, metamorph software was used to quantify pY12-Kir3.1-IR as ratio of YFP signal intensity in vehicle- (black bars) and BDNF-treated (gray bars) cells. n equals the number of cells analyzed.
TABLE FOUR.
Phospho-ERK1/2-IR in NIH3t3 fibroblasts stably expressing trkB after 0.2 μg/ml BDNF treatment
| Time after BDNF addition | Phospho-ERK1/2 IR |
|---|---|
| min | |
| 0 | 65 ± 1 (26)a |
| 2 | 80 ± 2 (9) |
| 5 | 187 ± 5 (8) |
| 15 | 172 ± 3 (8) |
| 30 | 152 ± 1 (20) |
| 45 | 122 ± 2 (9) |
Mean pixel intensity ± S.E. (n).
NIH3t3 cells transfected with YFP-Kir3.1[F137S] and treated with 0.2 μg/ml DNF for 15 min (i.e. near the peak of the ERK1/2 activation response reported in TABLE FOUR) showed enhanced pY12-Kir3.1-IR. In contrast, cells transfected with either YFP-containing vector lacking the Kir3.1 coding region or YFP-Kir3.1[F137S/Y12F] did not exhibit a comparable increase (Fig. 3A). To correct for differences in transfection efficiency, results of confocal imaging experiments were expressed as ratios of pY12-Kir3.1-IR to YFP signal as computed by Metamorph software analysis of mean pixel intensities (Fig. 3B). Vehicle treatment resulted in ratios of 0.36 ± 0.08 (n = 14), 1.04 ± 0.1(n = 10) and 0.6 ± 0.1 (n = 12) whereas BDNF treatment yielded ratios of 0.36 ± 0.2 (n = 11), 1.79 ± 0.45 (n = 5), and 0.57 ± 0.14 (n = 9) in NIH3t3-trkB cells expressing YFP, YFP-Kir3.1[F137S], and YFP-Kir3.1[F137S/Y12F], respectively (Fig. 3B). These results were replicated in two independent experiments. Similar results were obtained when data were not normalized for transfection efficacy (TABLE FIVE). The lack of staining of the Y12F mutant following BDNF treatment provides an important specificity control.
TABLE FIVE.
pY12-Kir3.1-IR in BDNF-treated NIH3t3 fibroblasts without correcting for transfection efficacy
| Construct transfected | Vehicle | BDNF |
|---|---|---|
| YFP | 16 ± 1 (14)a | 16 ± 1 (11) |
| YFP-Kir3.1[F137S] | 46 ± 6 (10) | 83 ± 15 (5) |
| YFP-Kir3.1[F137S/Y12F] | 28 ± 2 (12) | 33 ± 3 (9) |
Mean pixel intensity ± S.E. (n)
pY12-Kir3.1 in Neuropathic Pain
Earlier work in heterologous expression systems suggests that trkB activation decreases Kir3 amplitude (7) and accelerates muopioid peptide receptor (MOP-R)-dependent channel deactivation (8). To determine whether tyrosine phosphorylation of Kir3.1 could be triggered in vivo, we investigated whether stimuli thought to activate tyrosine kinase cascades in vivo could enhance pY12-Kir3.1-IR. Because the clinically relevant phenomenon of neuropathic pain is associated with elevated receptor and non-receptor tyrosine kinase activity in the spinal cord dorsal horn (26), we investigated pY12-Kir3.1-IR in this region in mice subjected to pSNL, an animal model for neuropathic pain (14). Seven days after one-third to one-half of the sciatic nerve was ligated, pY12 Kir3.1-IR was elevated unilaterally in the superficial layers of the dorsal horn, as determined by confocal microscopy (Fig. 4, A and B). In contrast, shamligation did not significantly elevate pY12 Kir3.1-IR. In accordance with previously published data on the anatomical location of Kir3.1 (5), we observed increased pY12-Kir3.1-IR in the outer regions of lamina II (lamina IIo) in the dorsal horn. As previously observed for Kir3.1, pY12-Kir3.1-IR did not co-localize with immunoreactivity of an antibody directed against calcitonin gene-related peptide (CGRP, Ref. 6 and data not shown), nor was co-localization observed with an antibody recognizing glutamic acid decarboxylase (GAD), a marker for GABA-ergic neurons (data not shown). The spinal cord cell type expressing the increased pY12-Kir3.1-IR was not further characterized.
FIGURE 4. Partial sciatic nerve ligation ipsilaterally increases pY12-Kir3.1-IR in mouse dorsal horn.
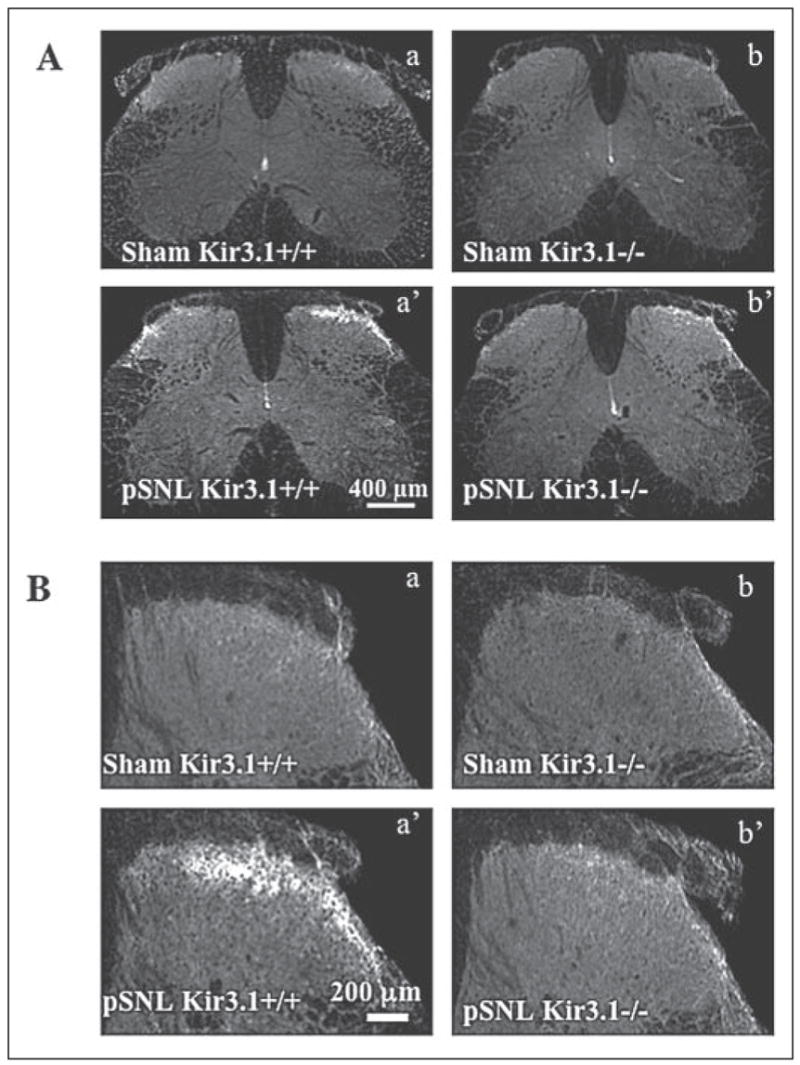
A, confocal microscopy of the dorsal horn of spinal cord sections from wild-type (letters) or Kir3.1−/− (primed letters) mice 7 days subsequent to sham operation (upper panels) or nerve partial sciatic nerve ligation (middle panels). Left panels, ipsilateral to nerve injury; right panels, contralateral to nerve injury. Scale bars, 400 μm. B, higher magnification of side ipsilateral to nerve injury. Scale bars, 200 μm.
Consistent with the specificity of the phosphoantibody demonstrated in vitro, partial sciatic nerve ligation of Kir3.1 knock-out mice (4) did not increase pY12-Kir3.1-IR in the dorsal horn (Fig. 4, A and B). pY12-Kir3.1-IR in lamina I–II of the dorsal horn was 127 ± 17% (n = 6) and 124 ± 12% (n = 6) of ventral horn-IR in sham-operated wild-type and Kir3.1 knock-out mice, respectively, and 262 ± 22% (n = 16) and 142 ± 15% (n = 7) in pSNL wild-type and Kir3.1 knock-out mice. Western blot analysis confirmed these results in wild-type animals (Fig. 5). Furthermore, pSNL caused no change in unmodified Kir3.1 expression between the sides of the dorsal horn ipsilateral and contralateral to the nerve injury (Fig. 5). pSNL of Kir3.1 knock-out mice failed to increase pY12-Kir3.1-IR as detected by Western blot analysis of dorsal horn homogenates (data not shown).
FIGURE 5. Western analysis of dorsal horn spinal cord from mice subjected to pSNL.
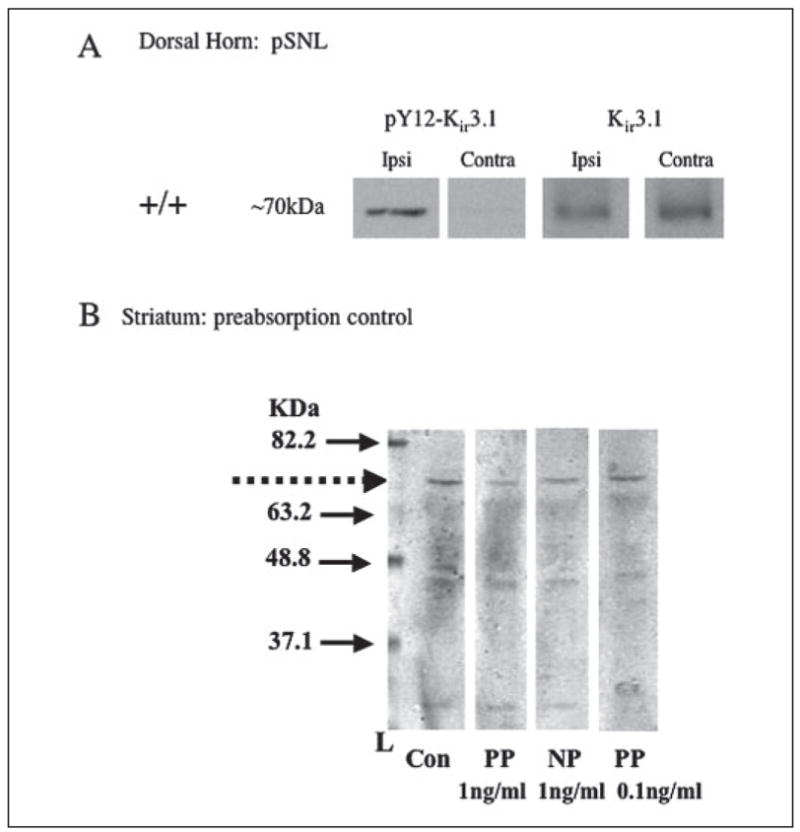
A, left panels, dorsal horn homogenates from ipsilateral to surgery; right panels, contralateral side. Left two columns of panels, +/+ mice; right two columns of panels, Kir3.1−/− mice. Upper panels, pY12-Kir3.1 antibody; lower panels, Kir3.1 (unmodified). B, preabsorption of pY12-Kir3.1 antibody with the phosphopeptide immunogen was more effective in reducing signal intensity detected in Western blot analysis. Mouse striatal protein from mice subjected to pSNL was resolved by electrophoresis and transferred to nitro-cellulose as described under “Experimental Procedures.” Replicate lanes separated by a prestained ladder (Invitrogen) were individually exposed to pY12-Kir3.1 antibody (0.03 mg/ml) that had either been preabsorbed for 1 h at room temp with phosphopeptide (PP), the equivalent non-phosphopeptide (NP), or saline (Con). As indicated in this representative Western blot, 1 ng/ml phosphopeptide effectively decreased the pY12-Kir3.1 band at the approximate molecular mass of ~70 kDa (marked by the dashed arrow). Data are representative of two independent experiments.
Because the pY12-Kir3.1 antibody was affinity purified using a phosphopeptide-conjugated column protocol, preabsorption of the antibody with phosphopeptide (1 ng/ml) was expected to block immunostaining as shown (Fig. 5B). Significantly, preabsorption of the antibody with 1 ng/ml of the non-phosphorylated peptide did not block staining. These results indicate that the antibody also showed phosphoselectivity under conditions used for Western blot analysis; however the antibody was not absolutely phosphospecific as higher concentrations of non-phosphorylated peptide (300 ng/ml) did block staining (data not shown). Thus, the basal staining evident in unstimulated tissue may be a mixture of unphosphorylated and phosphorylated Kir3.1, and the increase in pY12-Kir3.1-IR in the absence of an increase in Kir3.1 expression was likely caused by Tyr12 phosphorylation.
pY12-Kir3.1 in Formalin-induced Inflammation
As a chronic pain stimulus, sciatic nerve ligation produces neurochemical changes distinct from those observed during acute inflammatory pain (11). To investigate whether acute inflammatory pain also elevated pY12-Kir3.1-IR, we injected the left hindpaw of mice with saline or 4% formalin (27). In accordance with previously published literature (28–29), effectiveness of formalin treatment was confirmed by observing more paw flinches and licks in the 40-min interval post-injection (120 ± 22, n = 5) compared with saline-injected controls (0.67 ± 0.54, n = 6). Formalin treatment induced a more pronounced elevation in pY12-Kir3.1-IR than saline-injected control mice (Fig. 6, A and B) in one side of the dorsal horn. Mean pixel intensities for pY12-Kir3.1-IR for the two sides of the dorsal horn was 98 ± 6 (n = 19) and 126 ± 5(n = 19) for formalin-injected animals and 55 ± 4 (n = 20) and 66 ± 18 (n = 20) for saline-injected mice (Fig. 6B).
FIGURE 6. Hindpaw formalin induces unilateral elevation in pY12-Kir3.1-IR in spinal cord dorsal horn.
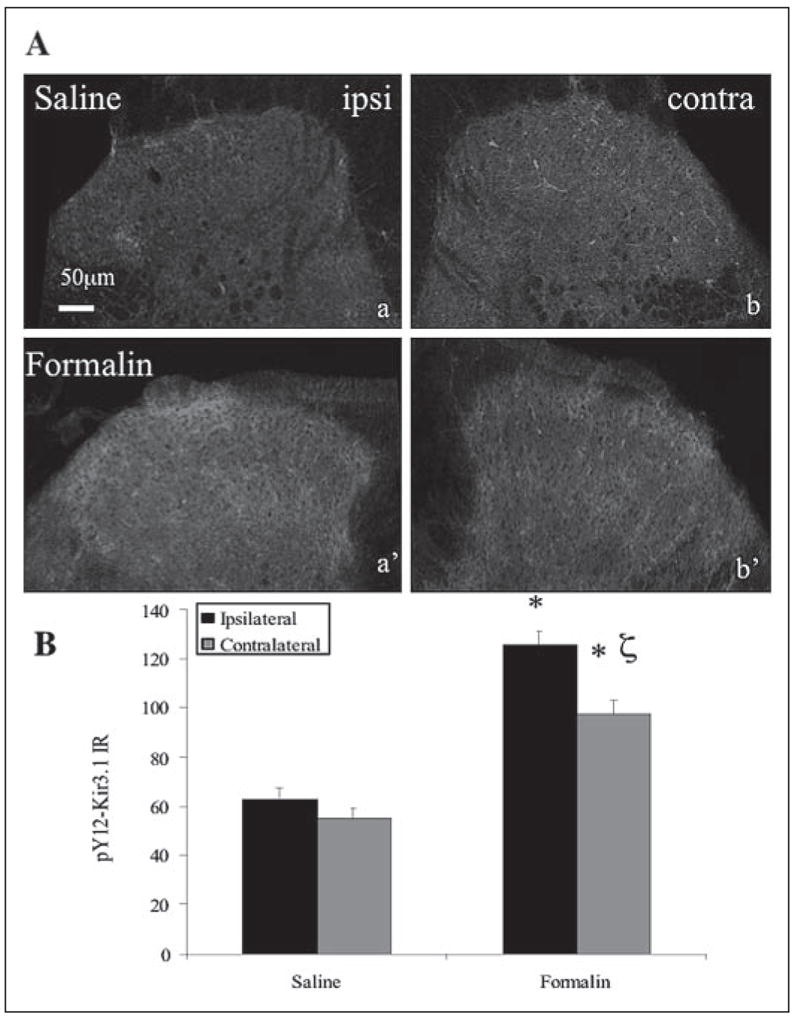
A, confocal images show dorsal horn sections from spinal cord 40 min after saline (letters) or formalin (primed letters) injection into the left hindpaw. Scale bar, 50 μm. B, quantification of pY12-K ir3.1-IR using Metamorph software. *, p < 0.05 compared with saline-injected paw;ζ, p < 0.05 compared with side 1. n equals the number of tissue sections with 3–4 animals per treatment group.
High power images show punctuate staining of pY12-Kir3.1-IR in regions surrounding the nucleus of neurons in both mice injected with formalin and pSNL animals, as identified by double labeling with NeuN, a neuronal nuclear marker (Fig. 7).
FIGURE 7. High power confocal images from formalin and nerve-ligated spinal cord dorsal horn sections.
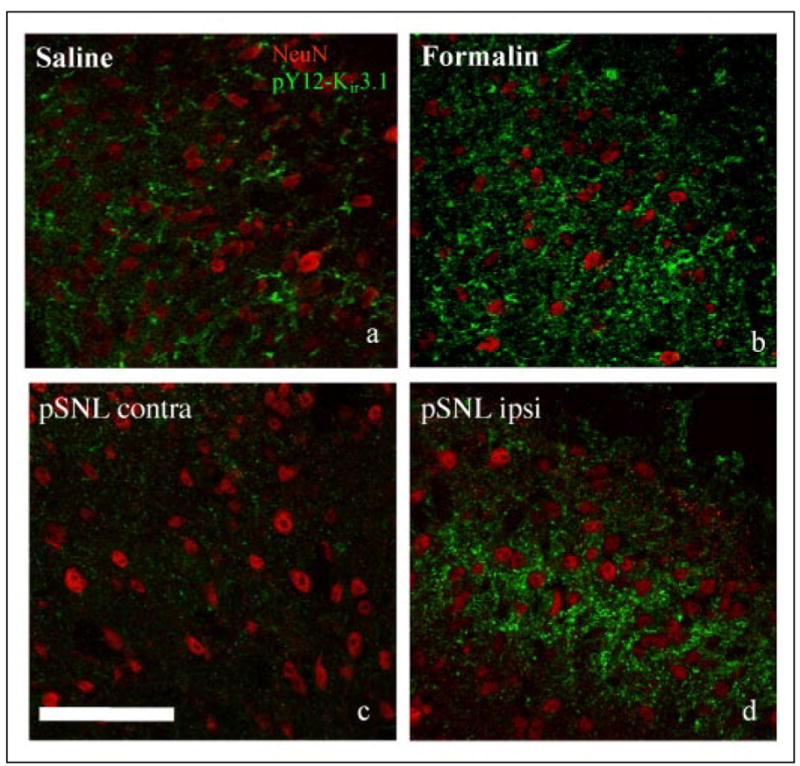
pY12-Kir3.1 appears as punctate staining around neuronal nuclei in sections from mice 40-min postsaline (panel a) or formalin (panel b) injection, or mice 7 days post-pSNL (panel c, contralateral; panel d, ipsilateral to partial nerve ligation). Red, NeuN neuronal nuclei marker; green, pY12-K ir3.1. Scale bars, 100 μm.
Western blot analyses were used to confirm that the pY12-Kir3.1-IR was elevated on the side of the dorsal horn ipsilateral to formalin injection (Fig. 8). To demonstrate pY12-Kir3.1 phosphoselectivity in the hindpaw formalin assay, we injected 28–30 g mice with 80 μg of K252A (tyrosine kinase inhibitor) both 12 and 2 h prior to hindpaw injection. The K252A dosing protocol was a modified version of previously published studies in guinea pigs (30). Vehicle (Me2SO) delivery did not exceed 40 μl. Results of immunoblotting experiments confirmed that formalin-enhanced pY12-Kir3.1-IR was inhibited by K252A on the side ipsilateral to the formalin compared with saline injections (Fig. 8, A and B). As quantified by NIH Image software, formalin injection elevated pY12-Kir3.1-IR to 142 ± 15% (n = 11) of saline-treated controls in the dorsal horn ipsilateral to injection, an effect inhibited by K252A (86 ± 17% (n = 4) of saline injection) (Fig. 8B). The dorsal horn tissue contralateral to injection site was not significantly elevated over saline injection in either formalin-injected (95 ± 23, n = 7) or K252A-pretreated, formalin-injected mice (84 ± 18, n = 3) (Fig. 8B).
FIGURE 8. Intraperitoneal injection of K252A blocks enhances pY12-Kir3.1-IR enhancement in spinal cord dorsal horn.
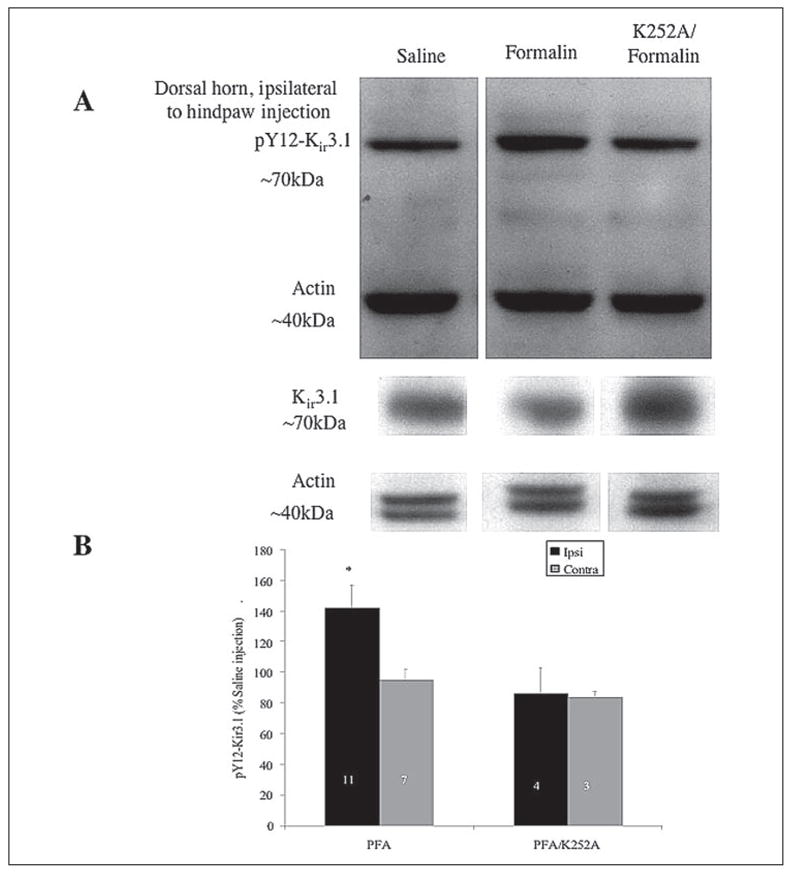
A, homogenates from mice injected intraplantarly with saline (left lanes, 40 min) or formalin (middle lanes, 40 min) or pre-treated intraperitoneal with K252A 12 h prior to formalin intraplantar injection (right lanes) were immunoblotted with pY12-Kir3.1 or an antibody recognizing unmodified Kir3.1 (lower panels). An antibody recognizing actin served as the loading control. B, quantification of gel analyses using NIH Image analysis software. n equals the number of tissue sections from two animals.
To evaluate the effect of vehicle (Me2SO) treatment on our experimental results, we treated mice with Me2SO or K252A and measured pY12-Kir3.1-IR postsaline or formalin injections. When band intensities were normalized to actin-loading controls, formalin injection elevated pY12-Kir3.1-IR to 176 ± 61% of Me2SO-treated, saline-injected mice (n = 3), an effect inhibited by K252A treatment (116 ± 32% of Me2SO-treated, saline-injected controls, n = 3). K252A treatment alone had little effect on Kir3.1-IR, as evidenced by K252A-pretreated, saline-injected mice yielding pY12-Kir3.1-IR of 122 ± 24% (n = 3) for Me2SO-pretreated, saline-injected animals.
pY12-Kir3.1-IR in Forced Swim Stress
Biochemical changes in the dorsal horn induced by stimuli unrelated to pain also elevate tyrosine kinases. Mice subjected to chronic swim stress, for example, exhibit an elevation of kinase activity in brain (29). To determine whether Kir3.1 phosphorylation occurred in response to stimuli minimally involved in nociception, we exposed mice to a modified Porsolt swim-stress protocol (see “Experimental Procedures”) and performed Western immunoblotting (Fig. 9A and data not shown) and immunohistochemistry (Fig. 9B) on lumbar spinal cord sections from swim-stressed mice. Unstressed mice served as a negative control. Swim stress bilaterally elevated pY12-Kir3.1-IR in spinal cord slices from swim-stressed compared with unstressed mice (Fig. 9B and data not shown). pY12-Kir3.1 staining was more diffuse than in mice subjected to pSNL, and closely resembled that observed in formalin-injected mice. These results were consistent with Western blot experiments (Fig. 9A). Co-labeling with an antibody recognizing a glial marker (GFAP) resulted in some co-localization of pY12-Kir3.1-IR and GFAP-IR, suggesting that pY12-Kir3.1-IR up-regulation may be occurring in some cells of glial morphology following swim stress as compared with pSNL (Fig. 10).
FIGURE 9. Swim stress bilaterally elevates pY12-Kir3.1 in spinal cord dorsal horn.
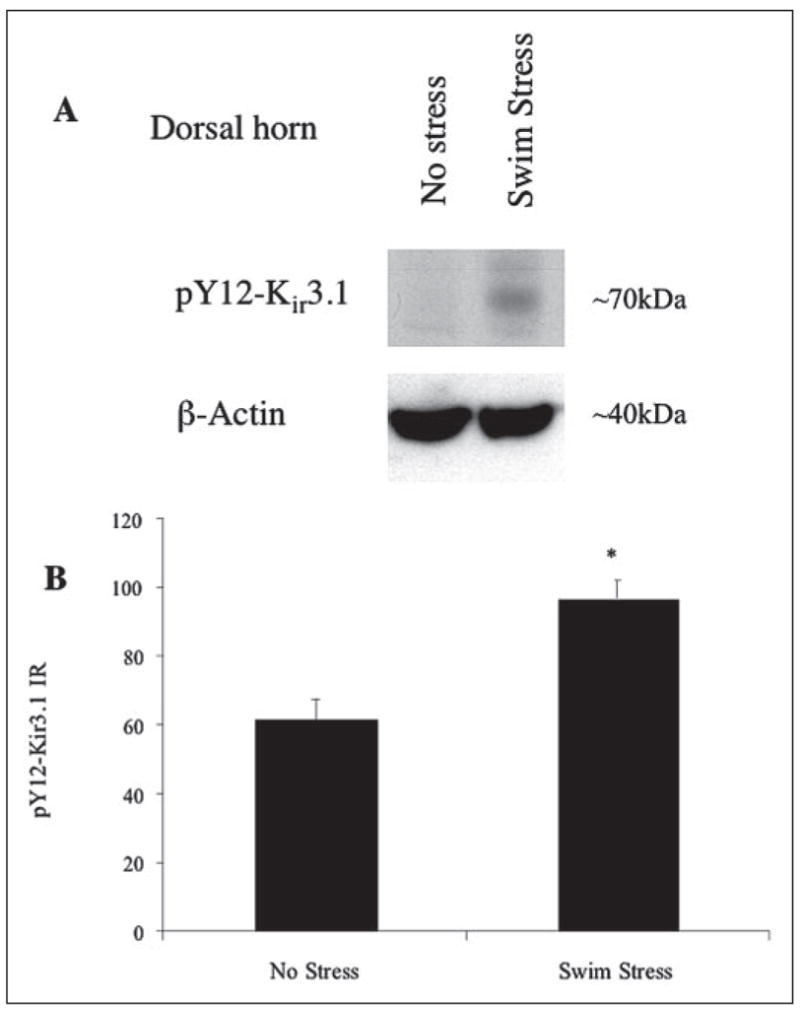
A, Western analysis of pY12-Kir3.1 in unstressed (left lanes) or swim-stressed mice (right lanes) using an antibody recognizing actin as a loading control. B, Metamorph software was used to quantify pixel intensities in laminae I–II of the dorsal horn from spinal cord sections taken from unstressed (left bars) or swim-stressed mice (right bars). n equals the number of tissue sections with 3–4 animals per treatment group.
FIGURE 10. High power confocal images comparing spinal cord sections from sham-operated (panels a–c), pSNL (panels d–f), and swim-stressed (panels g–i) mice.
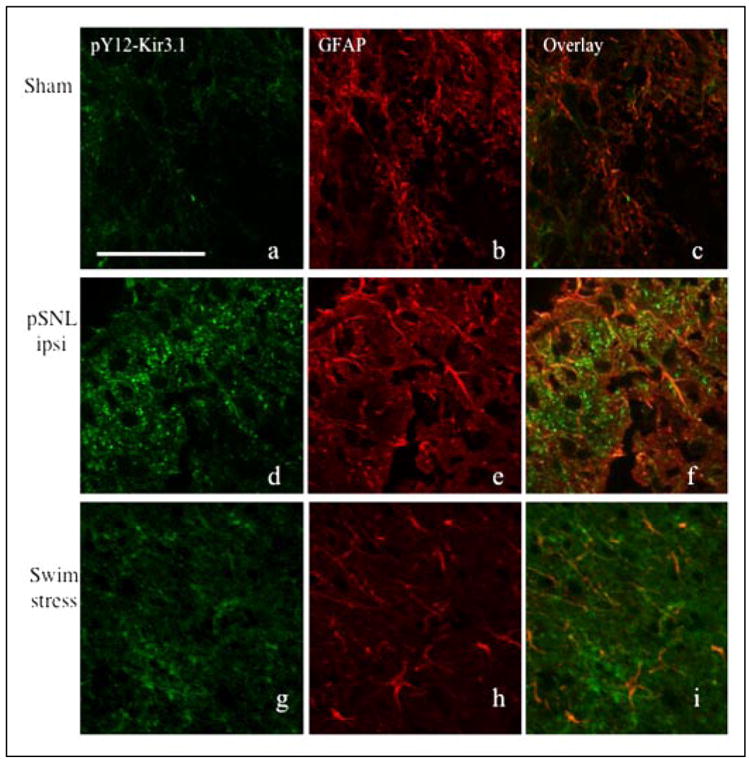
Spinal cord sections were double-labeled with pY12-Kir3.1 (green, panels a, d, and g) and GFAP (red, panels b, e, and h). Overlays are shown in panels c, f, and i Scale bar, 100 μm.
DISCUSSION
In this study, we asked whether a tyrosine phosphorylation event previously shown to alter channel gating occurs in vivo following physiologically appropriate stimuli. To address that question we generated and characterized a phosphoselective antibody able to distinguish pY12-Kir3.1 from native, unphosphorylated channel expressed by cells within the dorsal horn of the mouse spinal cord. We found that Kir3.1 can be phosphorylated at tyrosine 12 in neuronal cell lines and primary atrial myocyte cultures. Further, we provide evidence that tyro-sine phosphorylation is elevated in lamina IIo of the spinal cord dorsal horn following behavioral activation using physiological stimuli associated with spinally mediated nociception and chronic stress, including sciatic nerve ligation, formalin injection, and forced swim stress. The generality of the response suggests that this mode of channel regulation occurs following a wide variety of stimuli capable of tyrosine kinase activation.
Generation of a Probe to Detect Phosphorylation of Kir3.1 at Tyrosine 12
Based on homology of the K ir3.1 sequence to known recognition motifs for tyrosine kinases, a computer-generated algorithm predicts that the tyrosine at position 12 in Kir3.1 has a greater than 90% probability of being phosphorylated by tyrosine kinases in response to appropriate stimuli (31). Recent crystal structure data indicate that, like the Gβγ-binding domain, the N-terminal tail of Kir3.1 is located outside the central pore (32). Further, tyrosine 12 lies within the N-terminal domain of the channel that binds to the Gα subunit and gates the channel in the absence of GPCR activation (33).
The conclusion that the affinity-purified polyclonal antibody generated for this study was able to selectively detect the tyrosine 12 phosphorylated form of Kir3.1 rests on a series of control experiments that all provided consistent results. As described in this report, the pY12-Kir3.1 antibody reacted more strongly with the synthetic phosphopeptide than the non-phosphorylated version in ELISA assay. Second, pY12-Kir3.1-IR was detected in cells expressing Kir3.1 (NIH3t3-trkB, SH-SY5Y and primary atrial myocytes) but not in cells not expressing channel (untransfected cells and primary ventricular myocytes). Third, treatment of cells with reagents known to activate tyrosine kinase cascades (peroxide and BDNF) significantly increased pY12-Kir3.1-IR. Fourth, the increased IR was blocked by tyrosine kinase inhibition, prolonged by tyrosine phosphatase inhibition and not evident in cells expressing the Y12F mutant form of Kir3.1. Fifth, mice lacking Kir3.1 following gene disruption did not show the increased pY12-Kir3.1-IR. Sixth, the antibody detected a band on gels of the appropriate molecular mass, and preabsorption of the antibody with the phosphopeptide was more effective in blocking the IR detected than preabsorption with the non-phosphorylated peptide. These results are consistent with the conclusion that the antibody can be used to detect pY12-Kir3.1. The importance of this new tool is that it enables the extension of molecular observations of Kir3.1 regulation by tyrosine phosphorylation previously made in Xenopus oocytes and transfected cells to mammalian systems.
Hydrogen Peroxide- and trkB-mediated Phosphorylation of Kir3.1
Hydrogen peroxide (4.5 mM) and BDNF (0.2 μg/ml), but not anisomycin (50–500 μM) treatment resulted in increased IR of Kir3.1 at tyrosine position 12 in neuronal cells and atrial myocytes (Figs. 2 and 3). In addition to inhibiting tyrosine phosphatases by oxidizing residues within their catalytic binding pocket (34), hydrogen peroxide directly activates Src tyrosine kinase (17). Although hydrogen peroxide is a byproduct of cellular metabolism and signal transduction events (34–35), the concentrations (4.5 mM) required to see enhanced pY12-Kir3.1-IR in this study are probably not physiologically relevant. Nevertheless, peroxide is a useful pharmacological tool. Hydrogen peroxide treatment subjects the cell to oxidative stress, a phenomenon observed during pathologies including ischemic stress subsequent to myocardial infarction or stroke (36). If such ischemic events were to induce local elevation of hydrogen peroxide to millimolar levels, tyrosine phosphorylation of Kir3 might occur in these regions. The effects could include increased probability of cardiac arrhythmias subsequent to (a) elevated resting membrane potential of atrial myocytes or (b) atrial myocytes sub-responsiveness to vagal stimulation since Kir3 channels in the atria contribute to the resting membrane potential in heart (4). Right ventricular infarction in dogs, for example, leads to an apparent vagal denervation (37). The physiological consequences of Kir3.1 tyrosine phosphorylation in atrial myocytes remain to be established.
Phosphorylation of Kir3.1 in Models of Inflammatory and Neuropathic Pain
In the spinal cord, tyrosine kinases play a pivotal role in modulating an organism response to pain by inducing central sensitization to normally innocuous stimuli (38). Protein kinases act to post-translationally regulate proteins during inflammatory pain, including the suppression of potassium channels (11). TrkB receptor activation in the dorsal horn, for instance, induces Src-mediated tyrosine phosphorylation of N-methyl-D-aspartate (NMDA) receptors, thereby enhancing the activity of these excitatory ion channels and promoting central sensitization through a mechanism resembling hippocampal long term potentiation (39–40). Previous investigators have shown the susceptibility of receptor tyrosine kinases in promoting long-term potentiation in the spinal cord and hippocampus (41). Kir3.1 has been documented to play a role in pain circuitry, particularly in the response of neurons to analgesic drugs (5–6). The results presented in this study suggest that Kir3.1 becomes tyrosine-phosphorylated following acute and chronic nociceptive stimuli, but how that event contributes to the pain response is not yet clear.
We used a third behavioral stimuli known to activate tyrosine kinase cascades to assess the generality of the phosphorylation response. Kir3.1 phosphorylation also occurred bilaterally in the superficial laminae of the spinal cord dorsal horn following repeated swim stress in mice. Thus, tyrosine phosphorylation Kir3.1 may contribute more broadly to the stress response.
Our results further suggest that tyrosine phosphorylation occurs in regions of the spinal cord where Kir3.1 and Kir3.2 are enriched (5–6). As suggested by pY12-Kir3.1 labeling of cells not immunoreactive for NeuN and its colocalization with GFAP-IR, tyrosine phosphorylation of Kir3.1 may occur in glial as well as neuronal cell types (Fig. 10). Similar results have been observed for serine-phosphorylated kappa opioid receptors in the dorsal horn of the spinal cord following pSNL (43). Although the significance of our finding of phosphorylated Kir3 channels in spinal cord glia remains to be established, Kir channels have previously been reported to play a role in potassium flux in glial cells (44). It is possible that glial Kir3.1 channels may play a role in potassium buffering following the repeated glutamatergic activation occurring in response to painful stimuli, allowing removal of synaptic potassium to facilitate the maintenance of neuronal equilibrium potential. If the results observed in our oocyte expression systems are applicable to the physiology of neurons (7–8), Kir3.1 tyrosine phosphorylation could attenuate this buffering capacity, thereby providing a negative feedback mechanism to ultimately promote spinal analgesia.
CONCLUSIONS
In summary, in this study we generated and characterized a new tool for probing for Kir3 tyrosine phosphorylation in mammalian systems. Our data suggest that Kir3.1 is phosphorylated at position Tyr12 in the dorsal horn of the spinal cord in response to painful and stressful stimuli. These results suggest a role for tyrosine phosphorylation in spinally mediated nociception. Experiments are underway to understand the electrophysiological consequences of Kir3.1 tyrosine phosphorylation following pSNL, formalin-induced inflammation, and swim stress.
Acknowledgments
We thank AMGEN Corporation for supplying the BDNF used in this study.
Footnotes
This work was supported by the United States Public Health Service Grant DA11672 from NIDA, National Institutes of Health and National Institutes of Health Grants MH61933 and DA11806.
The abbreviations used are: Kir3, G-protein-gated inwardly rectifying potassium channel; trkB, tyrosine kinase receptor, ligand BDNF; BDNF, brain-derived neurotrophic factor; IR, immunoreactivity; ERK, extracellular signal-regulated kinase; pY, phosphorylated tyrosine; PBS, phosphate-buffered saline; ELISA, enzyme-linked immunosorbent assay; YFP, yellow fluorescent protein; pSNL, partial sciatic nerve ligation; PAO, phenylarsine oxide; KLH, keyhole limpet hemocyanin; GFAP, glial fibrillary acidic protein.
References
- 1.Dascal N. Cell Signal. 1997;9:551–573. doi: 10.1016/s0898-6568(97)00095-8. [DOI] [PubMed] [Google Scholar]
- 2.Torrecilla M, Marker CL, Cintora SC, Stoffel M, Williams JT, Wickman K. J Neurosci. 2002;22:4328–4334. doi: 10.1523/JNEUROSCI.22-11-04328.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wickman K, Nemec J, Gendler SJ, Clapham DE. Neuron. 1998;20:103–114. doi: 10.1016/s0896-6273(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 4.Bettahi I, Marker CL, Roman MI, Wickman K. J Biol Chem. 2002;277:48282–48288. doi: 10.1074/jbc.M209599200. [DOI] [PubMed] [Google Scholar]
- 5.Marker CL, Stoffel M, Wickman K. J Neurosci. 2004;24:2806–2812. doi: 10.1523/JNEUROSCI.5251-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marker CL, Lujan R, Loh HH, Wickman K. J Neurosci. 2005;25:3551–3559. doi: 10.1523/JNEUROSCI.4899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogalski SL, Appleyard SM, Pattillo A, Terman GW, Chavkin C. J Biol Chem. 2000;275:25082–25088. doi: 10.1074/jbc.M000183200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ippolito DL, Temkin PA, Rogalski SL, Chavkin C. J Biol Chem. 2002;277:32692–32696. doi: 10.1074/jbc.M204407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tucker K, Fadool DA. J Physiol. 2002;542:413–429. doi: 10.1113/jphysiol.2002.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong Y, Brandt GS, Li M, Shapovalov G, Slimko E, Karschin A, Dougherty DA, Lester HA. J Gen Physiol. 2001;117:103–118. doi: 10.1085/jgp.117.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ji RR, Strichartz G. Sci STKE. 2004;2004:reE14. doi: 10.1126/stke.2522004re14. [DOI] [PubMed] [Google Scholar]
- 12.Belcheva MM, Szucs M, Wang D, Sadee W, Coscia CJ. J Biol Chem. 2001;276:33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- 13.Zimmermann M. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- 14.Seltzer Z, Dubner R, Shir Y. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- 15.McLaughlin JP, Xu M, Mackie K, Chavkin C. J Biol Chem. 2003;278:34631–34640. doi: 10.1074/jbc.M304022200. [DOI] [PubMed] [Google Scholar]
- 16.Porsolt RD, Le Pichon M, Jalfre M. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- 17.Takano T, Sada K, Yamamura H. Antioxid Redox Signal. 2002;4:533–541. doi: 10.1089/15230860260196335. [DOI] [PubMed] [Google Scholar]
- 18.Celver J, Xu M, Jin W, Lowe J, Chavkin C. Mol Pharmacol. 2004;65:528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- 19.Doupnik CA, Davidson N, Lester HA, Kofuji P. Proc Natl Acad Sci U S A. 1997;94:10461–10466. doi: 10.1073/pnas.94.19.10461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang X, Gorospe M, Holbrook NJ. J Biol Chem. 1999;274:29599–29602. doi: 10.1074/jbc.274.42.29599. [DOI] [PubMed] [Google Scholar]
- 21.Roux PP, Blenis J. Microbiol Mol Biol Rev. 2004;68:320–344. doi: 10.1128/MMBR.68.2.320-344.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu SC, Gavrilin MA, Tsai MH, Han J, Lai MZ. J Biol Chem. 1999;274:25769–25776. doi: 10.1074/jbc.274.36.25769. [DOI] [PubMed] [Google Scholar]
- 23.Chan KW, Sui JL, Vivaudou M, Logothetis DE. Proc Natl Acad Sci U S A. 1996;93:14193–14198. doi: 10.1073/pnas.93.24.14193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma D, Zerangue N, Raab-Graham K, Fried SR, Jan YN, Jan LY. Neuron. 2002;33:715–729. doi: 10.1016/s0896-6273(02)00614-1. [DOI] [PubMed] [Google Scholar]
- 25.Huang EJ, Reichardt LF. Annu Rev Biochem. 2003;72:609–642. doi: 10.1146/annurev.biochem.72.121801.161629. [DOI] [PubMed] [Google Scholar]
- 26.Woolf CJ, Costigan M. Proc Natl Acad Sci. 1999;96:7723–7730. doi: 10.1073/pnas.96.14.7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berrino L, Oliva P, Massimo F, Aurilio C, Maione S, Grella A, Rossi F. Eur J Pain. 2003;7:131–137. doi: 10.1016/S1090-3801(02)00086-1. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Lighthall G, Liang DY, Clark JD. J Neurosci Res. 2004;78:533–541. doi: 10.1002/jnr.20274. [DOI] [PubMed] [Google Scholar]
- 29.Liu YF, Bertram K, Perides G, McEwen BS, Wang D. J Neurochem. 2004;89:1034–1043. doi: 10.1111/j.1471-4159.2004.02391.x. [DOI] [PubMed] [Google Scholar]
- 30.Paz O, Ashkenazy Y, Moshonov S, Fischer G, Feigel D, Kusniec F, Geltner D, Zor U. J Basic Clin Physiol Pharmacol. 1991;2:287–295. doi: 10.1515/jbcpp.1991.2.4.287. [DOI] [PubMed] [Google Scholar]
- 31.Blom N, Gammeltoft S, Brunak S. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 32.Nishida M, MacKinnon R. Cell. 2002;111:957–965. doi: 10.1016/s0092-8674(02)01227-8. [DOI] [PubMed] [Google Scholar]
- 33.Peleg S, Varon D, Ivanina T, Dessauer CW, Dascal N. Neuron. 2002;33:87–99. doi: 10.1016/s0896-6273(01)00567-0. [DOI] [PubMed] [Google Scholar]
- 34.Rhee SG, Bae YS, Lee SR, Kwon J. Sci STKE. 2000;2000:PE1. doi: 10.1126/stke.2000.53.pe1. [DOI] [PubMed] [Google Scholar]
- 35.Rhee SG. Exp Mol Med. 1999;31:53–59. doi: 10.1038/emm.1999.9. [DOI] [PubMed] [Google Scholar]
- 36.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 3. Oxford Science Publications, Oxford University Press; Oxford, UK: 1999. [Google Scholar]
- 37.Elvan A, Zipes DP. Circulation. 1998;97:484–492. doi: 10.1161/01.cir.97.5.484. [DOI] [PubMed] [Google Scholar]
- 38.Garraway SM, Petruska JC, Mendell LM. Eur J Neurosci. 2003;18:2467–2476. doi: 10.1046/j.1460-9568.2003.02982.x. [DOI] [PubMed] [Google Scholar]
- 39.Woolf CJ, Salter MW. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 40.Salter MW, Kalia LV. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- 41.Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, Taghibiglou C, Wang Y, Lu J, Wong TP, Sheng M, Wang YT. EMBO J. 2004;23:1040–1050. doi: 10.1038/sj.emboj.7600126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor BK, Peterson MA, Roderick RE, Tate J, Green PG, Levine JO, Basbaum AI. Pain. 2000;84:263–270. doi: 10.1016/s0304-3959(99)00212-2. [DOI] [PubMed] [Google Scholar]
- 43.Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kofuji P, Newman EA. Neuroscience. 2004;129:1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]