

Abstract
Connector enhancer of KSR (CNK) is a multidomain protein required for RAS signaling. Its C-terminal portion (CNKC-term) directly binds to RAF. Herein, we show that the N-terminal portion of CNK (CNKN-term) strongly cooperates with RAS, whereas CNKC-term efficiently blocks RAS- and RAF-dependent signaling when overexpressed in the Drosophila eye. Two effector loop mutants of RASV12, S35 and C40, which selectively activate the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase pathways, respectively, do not cooperate with CNK. However, a strong cooperation is observed between CNK and RASV12G37, an effector loop mutant known in mammals to activate specifically the RAL pathway. We have identified two domains in CNKN-term that are critical for cooperation with RAS. Our results suggest that CNK functions in more than one pathway downstream of RAS. CNKc-term seems to regulate RAF, a component of the MAPK pathway, whereas CNKN-term seems to be involved in a MAPK-independent pathway.
Keywords: RAF
The small GTPase RAS plays a central role in integrating and transmitting proliferation, differentiation, and survival signals elicited by membrane receptors to downstream effector pathways (1, 2). RAS controls these pathways by binding its effector loop region to the so-called RAS effector proteins, which in turn is thought to activate specific signaling cascades. The relatively large number of putative RAS effectors identified suggests that RAS controls multiple pathways. Evidence supporting this hypothesis has been provided by the use of specific RAS effector loop mutants that abrogate RAS’s ability to modulate particular downstream pathways (3). Biochemical and genetic studies have now confirmed the functional relevance of three types of RAS effectors (1, 2). The first type of effector is the RAF protein kinase family, which controls the activation of the mitogen-activated protein kinase (MAPK) pathway. This pathway includes two additional kinases, MAPK kinase and MAPK, and plays a major role in controlling both proliferation and differentiation. The second type of effector is phosphatidylinositol-3-kinase (PI3-K), which seems to mediate some of the RAS-dependent actin cytoskeleton remodeling and protection against apoptosis. Finally, the third bona fide RAS effector is RAL-GDS, an exchange factor for the small GTPase RAL, which seems to regulate multiple processes including receptor endocytosis, cytoskeletal changes, and DNA synthesis.
Although the MAPK pathway has been extensively studied, our understanding of how signal transmission through this kinase cascade is regulated is still far from complete. A number of proteins of unknown biochemical function have been shown, through genetic analysis, to play a role in this pathway (for example, see ref. 4). One of those proteins is kinase suppressor of RAS (KSR), which is a putative kinase that is structurally related to RAF and was identified originally in RAS-dependent genetic screens in Drosophila melanogaster and Caenorhabditis elegans (5–7). Genetic data indicate that KSR is essential for cell proliferation and cell differentiation in Drosophila and that KSR seems to function upstream or in parallel to RAF (7). Biochemical studies have established that KSR directly interacts with components of the MAPK pathway and suggest that KSR has a scaffolding function (8–14). We recently completed a genetic screen based on a KSR-dependent phenotype to identify genes that might help to elucidate the function of KSR. Several complementation groups were established, and the cloning and preliminary characterization of one of them, named connector enhancer of KSR (cnk), was recently reported (15). The cnk gene encodes a protein composed of several putative protein–protein interaction domains, which suggests a multiadaptor function. The biochemical relationship of CNK to KSR is unknown. Nonetheless, like KSR, CNK seems to be required in a step between RAS and RAF or in parallel to RAF, and its function is required for normal cell proliferation and differentiation. Interestingly, CNK was found to cooperate very strongly with activated RAS when coexpressed in the Drosophila eye. However, CNK completely antagonized RAF activity in the same assay. These results lead us to propose that CNK function might be RAS-dependent, such that, in the absence of a RAS-dependent signal, CNK might titrate a signaling component of the MAPK pathway, which could explain its ability to suppress RAF activity. The finding that the C-terminal portion of CNK (CNKC-term) physically interacts with RAF is consistent with this hypothesis and suggests that CNK regulates an aspect of RAF function.
In this paper, we show that the ability of CNK to enhance RAS signaling and its ability to suppress RAF activity map to different parts of the CNK protein. The cooperation with RAS is mediated by two N-terminal domains, whereas the ability to suppress RAF activity maps to CNKC-term, the portion that binds RAF. Although the expression of the N-terminal portion of CNK (CNKN-term) in the Drosophila eye strongly enhanced RAS signaling, we detected no effect on MAPK activation in cultured Schneider cells. Moreover, CNK strongly enhanced signaling by RAS1V12G37, an effector loop mutant known to activate the RAL pathway in mammals, but did not cooperate with two other effector mutants that predominantly activate the MAPK or the PI3-K pathways. Taken together, these results suggest that CNK functions in at least two pathways downstream of RAS: the MAPK pathway by means of CNKC-term and a MAPK-independent pathway through CNKN-term.
Materials and Methods
Plasmids.
psE-CNKN-term and psE-CNKC-term were constructed by cloning a KpnI–NotI fragment from pBluescript-CNKN-term (15) and pBluescript-CNKC-term (15) into the psE vector (16).
pMet-CNKN-term has been described (15). pMet-RAS1V12 was generated by transferring a KpnI–BamHI fragment encompassing the activated RAS1 gene from pBluescript-RAS1V12 (17) into pMet, a vector containing the metallothionein promoter that is inducible by heavy metals.
pUAS-CNKN-term was generated by transferring a KpnI–NotI fragment from pBluescript-CNKN-term into a pUAST vector (18) that was modified to include a NotI site downstream of the KpnI site, allowing the directed insertion of the CNKN-term KpnI–NotI fragment in the sense orientation. The three pUAS-CNKN-term mutant domain constructs were generated in two steps. First, three independent site-directed mutageneses were performed on pBluescript-CNKN-term by using the QuickChange kit (Stratagene) to alter specific amino acids in the SAM, CRIC, and PDZ domains, respectively (see Fig. 1). Then, a KpnI–NotI fragment corresponding to each CNKN-term mutant was moved into the modified pUAST vector.
Figure 1.

Schematic representation of the CNK constructs used in this study. CNK is 1,554 amino acids long and contains several domains that probably recognize other proteins: a sterile alpha motif (SAM) domain; a conserved region in CNK (CRIC) domain; a PSD-95, ZO-1/2, Dlg-1 (PDZ) domain; a pleckstrin homology (PH) domain; and two proline-rich (Pro) stretches that have consensus binding sites for SH3 domains. The regions present in the truncated CNK variants are denoted by a line, which corresponds to amino acids 2–384 for CNKN-term constructs (X schematically indicates the position of the mutant domains) and to amino acids 381–1,554 for CNKC-term. To disrupt the normal function of CNKN-term domains, conserved residues in the SAM and PDZ domains were changed by site-directed mutagenesis, whereas a mutation analogous to the one found in the cnkS-726 loss-of-function was introduced to generate a mutant CRIC domain. CNKN-termSAMmut has amino acids WI at positions 17 and 18 changed to SS. CNKN-termCRICmut has a deletion of amino acids AHR at position 162. CNKN-termPDZmut has amino acids GF at positions 217 and 218 changed to SS. All CNK constructs have an N-terminal FLAG epitope (MDYKDDDDK).
Genetics, Germ-Line Transformation, Histology, and Fly Stocks.
Fly culture and crosses were performed according to standard procedures. P element-mediated germ-line transformation and scanning electron microscopy were carried out as described in refs. 19 and 20, respectively.
sev-RAS1V12 flies are described in ref. 17. sE-RAFTor4021 flies are described in ref. 16. UAS-RAS1V12S35, UAS-RAS1V12G37, and UAS-RAS1V12C40 flies are described in ref. 21 and were kindly provided by Felix Karim (Exelixis Pharmaceuticals, South San Francisco, CA). The sev-GAL4 and sE-GAL4 lines were kindly provided by Adina Bailey (University of California, Berkeley) and Barry Dickson (Research Institute of Molecular Pathology, Vienna), respectively.
Cell Transfection and Protein Analysis.
For transient transfection experiments, 107 Schneider-2 (S2) cells were transfected with pMet-RAS1V12 alone or in combination with pMet-CNKN-term. Protein expression was induced 24 h after transfection by adding 0.7 mM CuSO4. Cells were harvested in Nonidet P-40 lysis buffer (8) at different time points. Equal amounts of proteins were loaded onto SDS/12% PAGE and analyzed by immunoblotting by using anti-Drosophila RAS1 monoclonal antibody (I. Rebay and G.M.R., unpublished work) and anti-FLAG M5 monoclonal antibody (Sigma) to monitor protein expression and by using anti-phospho ERK-1&2 monoclonal antibody (Sigma) to detect the levels of activated MAPK.
Results
CNK Contains Domains with Opposite Effects on RAS-Dependent Signaling.
We previously showed that overexpression of full-length CNK (Fig. 1) in the developing Drosophila eye under the control of the sevenless enhancer and HSP70 proximal promoter (sE-CNK) ablated photoreceptor cells and resulted in the roughening of the adult eye surface (ref. 15; Fig. 2, compare A and B). We proposed that the mild antagonistic effect of overexpression of CNK on eye development was due to the titration of endogenous signaling components required for RAS signaling. We also showed that overexpression of CNK strongly enhanced the activated RAS1 (RAS1V12) rough-eye phenotype (ref. 15; Fig. 2, compare E and F) but completely suppressed the activated RAF (RAFTor4021) rough eye (ref. 15; Fig. 2, compare I and J). Together, these results suggested that CNK function is RAS-dependent.
Figure 2.

Expression of CNKN-term enhances the activated RAS1 rough-eye phenotype, whereas expression of CNKC-term suppresses activated RAS1 and activated RAF rough-eye phenotypes. Scanning electron micrographs of adult eyes of the following genotypes: (A) wild type, (B) sE-CNK/sE-CNK, (C) sE-CNKN-term/sE-CNKN-term, (D) sE-CNKC-term/sE-CNKC-term, (E) sev-RAS1V12/+, (F) sE-CNK/sev-RAS1V12, (G) sE-CNKN-term/sev-RAS1V12, (H) sE-CNKC-term/sev-RAS1V12, (I) sE-RAFTor4021/+, (J) sE-RAFTor4021/sE-CNK, (K) sE-RAFTor4021/sE-CNKN-term, and (L) sE-RAFTor4021/sE-CNKC-term. Anterior is to the right.
To determine whether CNK’s ability to cooperate with RAS during eye development can be assigned to a particular region of CNK, we generated transgenic lines overexpressing either CNKN-term (amino acids 1–384) or CNKC-term (amino acids 381–1,554) in the Drosophila eye by using the sE expression system. We tested the effects of these transgenes on eye development either by themselves or in the presence of activated RAS1 or activated RAF. Overexpression of CNKN-term in the eye has no effect on its own. sE-CNKN-term flies have a normal eye surface (Fig. 2C) and wild-type arrays of ommatidia, as determined by examination of sections of fixed eyes (data not shown). However, overexpression of CNKN-term strongly enhanced the activated RAS1 rough-eye phenotype (Fig. 2, compare E and G) but had no effect on the activated RAF rough-eye phenotype (Fig. 2, compare I and K). In contrast, overexpression of CNKC-term produces a rough eye (Fig. 2, compare A and D) because of its ability to block photoreceptor differentiation, as evidenced by the missing photoreceptor rhabdomeres in a large proportion of adult ommatidia (data not shown). In addition, overexpression of CNKC-term was able to suppress the rough-eye phenotypes produced by either activated RAS1 or activated RAF (Fig. 2, compare E and H as well as I and L, respectively). Together, these results indicate that different domains of the protein mediate the abilities of full-length CNK to cooperate with RAS1 and to block photoreceptor cell differentiation. CNKN-term contains the domain(s) required for the cooperation with RAS1, whereas CNKN-term seems to contain the domain(s) responsible for the ability of full-length CNK, when expressed at high levels, to block photoreceptor cell differentiation and to suppress the rough-eye phenotype produced by activated RAF.
A MAPK-Independent Function for CNK?
The strong enhancement of activated RAS1 by CNK or CNKN-term suggests that CNK facilitates RAS1-dependent signaling. Because the MAPK pathway is an important mediator of RAS1 signals during eye development, we tested whether CNK could stimulate RAS1-dependent activation of MAPK in a cell culture system. We transiently expressed RAS1V12 alone or with CNKN-term in Drosophila S2 cells and then measured the levels of endogenous activated MAPK. As shown in Fig. 3, expression of RAS1V12 alone stimulated MAPK activation in S2 cells. However, coexpression of CNKN-term with RAS1V12 did not result in significantly higher levels of activated MAPK. We obtained comparable results when various amounts of RAS1V12 and/or CNKN-term (or full-length CNK) expression constructs were transfected and also when they were assayed at different time points (data not shown). Therefore, these data suggest either that the effect of CNK is mediated by a MAPK-independent pathway or that S2 cells lack or have limiting amounts of a component(s) required to mediate the effect of CNK on RAS1V12 signaling.
Figure 3.

Expression of CNKN-term does not influence MAPK activation in S2 cells. Lysates from plain S2 cells (−) or S2 cells transfected with pMet-RAS1V12 alone or in combination with pMet-CNKN-term (amounts are indicated) were analyzed by immunoblotting by using an anti-phospho MAPK monoclonal antibody to detect the levels of activated MAPK.
CNK Specifically Cooperates with the RAS1V12G37 Effector Loop Mutant.
Distinct RAS effector loop mutations have been shown to impair signaling selectively through different RAS-dependent pathways (3, 22, 23). For instance, changing Thr-35 to Ser (S35) disables signaling via the RAL-GDS and the PI3-K pathways but does not affect signaling via the RAF/MAPK pathway. Changing Glu-37 to Gly (G37) perturbs signaling via the RAF/MAPK and the PI3-K pathways but does not affect the RAL-GDS pathway. Finally, changing Tyr-40 to Cys (C40) disrupts signaling via the RAF/MAPK and the RAL-GDS pathways but does not affect the PI3-K pathway. Therefore, combining these mutations with the V12 activating mutation predominantly leads to the activation of particular effector pathways. Although it remains to be established, it is likely that RAS1 in Drosophila also controls other pathways, which probably include a PI3-K and a RAL pathway. Effector loop mutations in Drosophila RAS1 previously have been used to show that, like RAS1V12, ectopic expression of RAS1V12S35 induced cell proliferation, whereas the two other effector loop mutants were unable to stimulate cell proliferation. This result suggested that, unlike mammalian cells, activation of the MAPK pathway alone is sufficient for the control of cell proliferation in Drosophila (21).
We examined the ability of CNK to cooperate with the different RAS1V12 effector loop mutants in an attempt to determine which signaling pathway downstream of RAS might be enhanced by CNK. We separately overexpressed the three double-mutant variants of RAS1 (V12S35, V12G37, and V12C40) in the eye by using the UAS/GAL4 expression system and tested the effect of coexpressing full-length CNK. Expression of all three double mutants produced a rough eye when tested alone (Fig. 4 A, C, and E). Strikingly, when CNK is coexpressed (Fig. 4 B, D, and F), a massive cooperation is observed with RAS1V12G37 (Fig. 4D) but not with the other two effector loop mutations. Similar results were observed with CNKN-term (Fig. 5, compare A and B and data not shown). These results indicate that the cooperation observed between CNK and RAS1 is mediated by a pathway unaffected by the G37 mutation, implying that CNK does not cooperate with RAS1 through an effect on the MAPK pathway.
Figure 4.
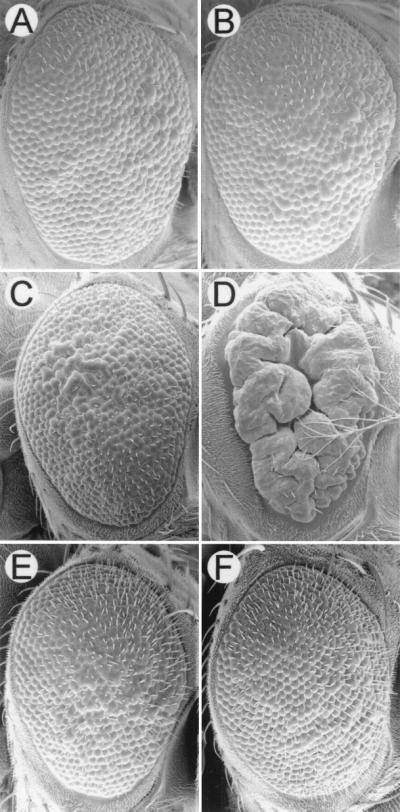
CNK cooperates only with RAS1V12G37. Scanning electron micrographs of adult eyes of the following genotypes: (A) UAS-RAS1V12S35/sev-GAL4, (B) UAS-RAS1V12S35/sev-GAL4; sE-CNK/+, (C) UAS-RAS1V12G37/+; sE-GAL4/+, (D) UAS-RAS1V12G37/+; sE-GAL4/sE-CNK, (E) UAS-RAS1V12C40/+; sE-GAL4/+, and (F) UAS-RAS1V12C40/+; sE-GAL4/sE-CNK. sev-GAL4 expresses lower levels of GAL4 than sE-GAL4; however, both constructs express GAL4 in the same subset of cells in the developing eye. To obtain similar eye roughness, RAS1V12S35 was expressed by the sev-GAL4 driver, whereas RAS1V12G37 and RAS1V12C40 were expressed by the sE-GAL4 driver. Anterior is to the right.
Figure 5.

The SAM and the CRIC domains of CNK are required for the cooperation with RAS1. Scanning electron micrographs of adult eyes of the following genotypes: (A) UAS-RAS1V12G37/+; sE-GAL4/+, (B) UAS-RAS1V12G37/+; UAS-CNKN-term/+; sE-GAL4/+, (C) UAS-RAS1V12G37/+; UAS-CNKN-termSAMmut/+; sE-GAL4/+, (D) UAS-RAS1V12G37/+; UAS-CNKN-termCRICmut/+; sE-GAL4/+, and (E) UAS-RAS1V12G37/+; UAS-CNKN-termPDZmut/+; sE-GAL4/+. At least three independent transformants for each CNKN-term construct were tested. The phenotypes were uniform in all cases. Anterior is to the right.
Two Domains in CNK Are Required for the Enhancement of RAS1 Signaling.
Three domains (SAM, CRIC, and PDZ) have been identified in CNKN-term and account for most of the amino acid sequence of this region (ref. 15; see Fig. 1). To determine which of these domains is relevant for the cooperation with RAS1, we generated three separate CNKN-term constructs, each containing a mutation impairing a different domain (Fig. 1), and then tested the ability of these mutants to enhance RAS1V12G37 activity. When coexpressed in the eye, CNKN-termSAMmut had a much reduced ability to enhance RAS1V12G37 (Fig. 5C), and CNKN-termCRICmut did not enhance RAS1V12G37 at all (Fig. 5D); however, CNKN-termPDZmut strongly enhanced RAS1V12G37 activity (Fig. 5E). Therefore, these results indicate that the SAM and the CRIC domains are involved in the cooperation between CNK and RAS1, whereas the PDZ domain seems dispensable.
Discussion
CNK is a RAF-binding protein required for RAS signaling. In this paper, we present evidence suggesting that CNK has multiple functions downstream of RAS. The ability of CNKC-term to associate physically with RAF and to suppress RAS or RAF-dependent signaling when overexpressed in the Drosophila eye suggests that CNK regulates some aspects of RAF function, thereby affecting the MAPK pathway. In addition, the ability of CNKN-term to cooperate strongly with RAS1V12G37 and its failure to stimulate MAPK activation in S2 cells suggest that CNK also plays a role in a MAPK-independent pathway.
Previously, we have shown that full-length CNK strongly cooperates with activated RAS but suppresses activated RAF function (ref. 15; Fig. 2 F and J), suggesting a RAS-dependent function for CNK. However, it was not clear whether the same domains of CNK mediated the opposite effects of CNK on RAS and RAF signaling and whether the effects were functionally related. In this report, we show, by overexpressing CNKN-term and CNKC-term in the developing Drosophila eye, that the effects of CNK on RAS and RAF signaling depend on different regions of the protein and seem to be functionally distinct. The ability of CNK to cooperate with RAS is mediated by two domains localized in CNKN-term (Fig. 5 C and D), which do not modulate activated RAF-dependent signaling (Fig. 2K). In contrast, CNKC-term antagonizes RAS and RAF signaling when overexpressed (Fig. 2 H and L), suggesting that the ability of full-length CNK to suppress activated RAF is mediated by CNKC-term. The domain(s) in CNKC-term responsible for this negative effect have not yet been characterized. However, under physiological conditions, they seem to play a positive role during RAS signaling, because several of the cnk loss-of-function alleles identified in a KSR-dependent screen have a truncated C-terminal domain (15). It may be that overexpressed CNKC-term has a negative effect on RAS and RAF signaling, because it sequesters and/or mislocalizes a signaling component. The fact that RAF physically associates with CNKC-term is consistent with this possibility.
The ability of CNK or CNKN-term to enhance activated RAS but not activated RAF suggests that this effect occurs upstream of RAF or in a pathway parallel to RAF. Two independent sets of results presented in this paper are consistent with the idea that the CNK/RAS cooperation is mediated by a RAF/MAPK-independent pathway. The first is the fact that the coexpression of CNKN-term and RAS1V12 did not produce higher levels of activated MAPK (Fig. 3). The second set of evidence is the striking observation that CNK strongly cooperates with RAS1V12G37, a RAS effector loop mutant known to stimulate the RAL pathway in mammals (22), but not with two other RAS effector loop mutants known to stimulate the MAPK and PI3-K pathways (Fig. 4). Our data cannot rule out the alternative hypothesis that the enhancement of RAS1V12G37 by CNK results from CNK-mediated stimulation of the MAPK pathway acting synergistically with the RAS1V12G37-stimulated pathway. This possibility would be analogous to the strong cooperation observed in mammalian cells between the MAPK pathway and the RAL pathway to transform cells (22). Consistent with this possibility, we found that a loss-of-function mutation in the rolled/mapk locus did not suppress the RAS1V12G37 mild rough-eye phenotype but suppressed the CNK/RAS1V12G37 cooperation (data not shown). Moreover, we found that the RAS1V12G37 mutant is leaky and stimulates MAPK when expressed in S2 cells to approximately 10% of the levels obtained with RAS1V12 or RAS1V12S35 (data not shown). It is thus formally possible that CNK functions in the MAPK pathway, which collaborates efficiently with another pathway also stimulated by RAS1V12G37. A strong argument against this model is the observation that CNK strongly cooperates with RAS1V12 but not with RAS1V12S35, a RAS effector loop mutant known to stimulate the RAF/MAPK pathway (ref. 3; Figs. 2L and 4B). It would be difficult to reconcile this observation with a model in which CNK is merely enhancing signaling through the MAPK pathway. More likely, the phenotypic effect of suppressing MAPK signaling might be due to the fact that MAPK is involved in secondary developmental defects resulting from the strong stimulation of the RAS1V12G37-specific pathway. Alternatively, it might reflect the fact that the CNK/RAS1V12G37-stimulated pathway also requires a basal level of MAPK signaling to mediate its effects. A similar dependency in basal MAPK activity has been suggested recently for the RAL pathway to induce the differentiation of F9 embryonal carcinoma cells (24).
We expect that RAS1, like its mammalian homologues, controls the RAL pathway in Drosophila. Because the effector loop regions of Drosophila RAS1 and mammalian RAS proteins are identical in sequence and because the mammalian RALA and Drosophila RAL are nearly identical (M.T. and G.M.R., unpublished observation), it is likely that RAS1V12G37 also stimulates the RAL pathway in Drosophila. Although little is known regarding the RAL pathway in Drosophila, it has been reported recently that overexpression of activated RAL in the Drosophila eye disrupts the normal actin cytoskeleton assembly but does not interfere with cell differentiation (25). This result is consistent with studies conducted in mammalian cells that suggested that RAL controls the organization of the actin cytoskeleton (26). Based on these findings, it will be interesting to determine whether RAS1V12G37 has a similar effect on the actin cytoskeleton and whether CNK enhances this effect.
The CNK/RAS cooperation clearly depends on the integrity of the SAM and the CRIC domains. SAM domains have been found in various types of proteins and seem to mediate homodimerization and/or heterodimerization with other SAM domain-containing proteins (27). The CRIC domain is a unique region shared by all CNK homologues identified thus far (ref. 15; M.T. and G.M.R., unpublished results). The boundaries of this domain (≈80 amino acids) have been arbitrarily defined based on sequence homology. Its functional relevance was initially suggested by a cnk loss-of-function allele, cnkXE-726, which has a 3-amino acid in-frame deletion within this region (15). The elucidation of the functions of the SAM and CRIC domains of CNK awaits the molecular characterization of their effect on RAS signaling and the identification of the proteins that interact with them.
Acknowledgments
We are grateful to Iswar Hariharan and Felix Karim for critical reading of the manuscript. We thank Susan Mullaney for Drosophila embryo injections. This work was supported by funds from the Medical Research Council of Canada (to M.T.) and the Howard Hughes Medical Institute (to G.M.R.).
Abbreviations
- MAPK
mitogen-activated protein kinase
- PI3-K
phosphatidylinositol-3-kinase
- CNKN-term/CNKC-term
N-/C-terminal portion of CNK
- S2
Schneider-2
- SAM
sterile alpha motif
- CRIC
conserved region in CNK
- PDZ
PSD-95, ZO-1/2, Dlg-1
References
- 1.Katz M E, McCormick F. Curr Opin Genet Dev. 1997;7:75–79. doi: 10.1016/s0959-437x(97)80112-8. [DOI] [PubMed] [Google Scholar]
- 2.Bos J L. EMBO J. 1998;17:6776–6782. doi: 10.1093/emboj/17.23.6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White M A, Nicolette C, Minden A, Polverino A, Van Aelst L, Karin M, Wigler M H. Cell. 1995;80:533–541. doi: 10.1016/0092-8674(95)90507-3. [DOI] [PubMed] [Google Scholar]
- 4.Sternberg P W, Alberola-Ila J. Cell. 1998;95:447–450. doi: 10.1016/s0092-8674(00)81612-8. [DOI] [PubMed] [Google Scholar]
- 5.Kornfeld K, Hom D B, Horvitz H R. Cell. 1995;83:903–913. doi: 10.1016/0092-8674(95)90206-6. [DOI] [PubMed] [Google Scholar]
- 6.Sundaram M, Han M. Cell. 1995;83:889–901. doi: 10.1016/0092-8674(95)90205-8. [DOI] [PubMed] [Google Scholar]
- 7.Therrien M, Chang H C, Solomon N M, Karim F D, Wassarman D A, Rubin G M. Cell. 1995;83:879–888. doi: 10.1016/0092-8674(95)90204-x. [DOI] [PubMed] [Google Scholar]
- 8.Therrien M, Michaud N R, Rubin G M, Morrison D K. Genes Dev. 1996;10:2684–2695. doi: 10.1101/gad.10.21.2684. [DOI] [PubMed] [Google Scholar]
- 9.Denouel-Galy A, Douville E M, Warne P H, Papin C, Laugier D, Calothy G, Downward J, Eychene A. Curr Biol. 1997;8:46–55. doi: 10.1016/s0960-9822(98)70019-3. [DOI] [PubMed] [Google Scholar]
- 10.Michaud N R, Therrien M, Cacace A, Edsall L C, Spiegel S, Rubin G M, Morrison D K. Proc Natl Acad Sci USA. 1997;94:12792–12796. doi: 10.1073/pnas.94.24.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xing H, Kornfeld K, Muslin A J. Curr Biol. 1997;7:294–300. doi: 10.1016/s0960-9822(06)00152-7. [DOI] [PubMed] [Google Scholar]
- 12.Yu W, Fantl W J, Harrowe G, Williams L T. Curr Biol. 1997;8:56–64. doi: 10.1016/s0960-9822(98)70020-x. [DOI] [PubMed] [Google Scholar]
- 13.Cacace A M, Michaud N R, Therrien M, Mathes K, Copeland T, Rubin G M, Morrison D K. Mol Cell Biol. 1999;19:229–240. doi: 10.1128/mcb.19.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stewart S, Sundaram M, Zhang Y, Lee J, Han M, Guan K L. Mol Cell Biol. 1999;19:5523–5534. doi: 10.1128/mcb.19.8.5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Therrien M, Wong A M, Rubin G M. Cell. 1998;95:343–353. doi: 10.1016/s0092-8674(00)81766-3. [DOI] [PubMed] [Google Scholar]
- 16.Dickson B, Sprenger F, Morrison D, Hafen E. Nature (London) 1992;360:600–603. doi: 10.1038/360600a0. [DOI] [PubMed] [Google Scholar]
- 17.Fortini M E, Simon M A, Rubin G M. Nature (London) 1992;355:559–561. doi: 10.1038/355559a0. [DOI] [PubMed] [Google Scholar]
- 18.Brand A H, Perrimon N. Development (Cambridge, UK) 1994;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 19.Rubin G M, Spradling A C. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- 20.Kimmel B E, Heberlein U, Rubin G M. Genes Dev. 1990;4:712–727. doi: 10.1101/gad.4.5.712. [DOI] [PubMed] [Google Scholar]
- 21.Karim F D, Rubin G M. Development (Cambridge, UK) 1998;125:1–9. doi: 10.1242/dev.125.1.1. [DOI] [PubMed] [Google Scholar]
- 22.White M A, Vale T, Camonis J H, Schaefer E, Wigler M H. J Biol Chem. 1996;271:16439–16442. doi: 10.1074/jbc.271.28.16439. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez-Viciana P, Warne P H, Khwaja A, Marte B M, Pappin D, Das P, Waterfield M D, Ridley A, Downward J. Cell. 1997;89:457–467. doi: 10.1016/s0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 24.Verheijen M H, Wolthuis R M, Defize L H, den Hertog J, Bos J L. Oncogene. 1999;18:4435–4439. doi: 10.1038/sj.onc.1202834. [DOI] [PubMed] [Google Scholar]
- 25.Sawamoto K, Yamada C, Kishida S, Hirota Y, Taguchi A, Kikuchi A, Okano H. Oncogene. 1999;18:1967–1974. doi: 10.1038/sj.onc.1202522. [DOI] [PubMed] [Google Scholar]
- 26.Feig L A, Urano T, Cantor S. Trends Biochem Sci. 1996;21:438–441. doi: 10.1016/s0968-0004(96)10058-x. [DOI] [PubMed] [Google Scholar]
- 27.Stapleton D, Balan I, Pawson T, Sicheri F. Nat Struct Biol. 1999;6:44–49. doi: 10.1038/4917. [DOI] [PubMed] [Google Scholar]