

Summary
We report two unrelated boys with the X-linked creatine transporter defect (CRTR) and clinical features more severe than those previously described with this disorder. These two boys presented at ages 12 and 30 months with severe mental retardation, absent speech development, hypotonia, myopathy and extra-pyramidal movement disorder. One boy has seizures and some dysmorphic features; he also has evidence of an oxidative phosphorylation defect. They both had classical absence of creatine peak on brain magnetic resonance spectroscopy (MRS). In one, however, this critical finding was overlooked in the initial interpretation and was discovered upon subsequent review of the MRS.
Molecular studies showed large genomic deletions of a large part of the 3′end of the complete open reading frame of the SLC6A8 gene. This report emphasizes the importance of MRS in evaluating neurological symptoms, broadens the phenotypic spectrum of CRTR and adds knowledge about the pathogenesis of creatine depletion in the brain and retina.
Creatine provides a source of high-energy phosphate for normal cellular function through intramitochondrial conversion to phosphocreatine. Approximately 50% of the required creatine is derived from the diet (mainly animal protein), with the remainder provided by de novo synthesis, primarily in the liver, pancreas and kidneys (Wyss and Kaddurah-Daouk 2000). The first step in synthesis is the rate-limiting intramitochondrial transfer of the amidino group from arginine to glycine by arginine:glycine amindinotransferase (AGAT) to form guanidinoacetate, which is then methylated in the cytosol by guanidinoacetate methyltransferase (GAMT) to form creatine. The creatine transporter is expressed primarily in skeletal and cardiac muscle and kidney, with lesser expression in the CNS, placenta and lungs (Salomons et al 2003).
The role of creatine and phosphocreatine in energy metabolism has long been recognized, but disorders of creatine deficiency have only recently been discovered. Two of these disorders are autosomal recessive GAMT deficiency (Stockler et al 1996) and AGAT deficiency (Item et al 2001), which are characterized by mild to severe developmental delay, particularly expressive language delay, seizures and, in some cases, severe truncal hypotonia and dystonia (Schulze 2003; Stromberger et al 2003). In 2001, Salomons and colleagues described an X-linked deficiency of creatine, presumed to be due to lack of transport of creatine into brain, later shown to be due to a mutation in the creatine transporter gene, SLC6A8 (Salomons et al 2001). The diagnostic hallmarks in affected males are reduced or absent creatine signal in magnetic resonance spectroscopy (MRS) of brain, impaired creatine uptake in fibroblasts, increased creatine/creatinine ratio in urine and a hemizygous mutation in the creatine transporter gene SLC6A8 mapped to Xq28. The phenotype of creatine transporter defect (CRTR) reported so far has been a mild–moderate mental retardation, expressive speech and language delay, easily controlled seizures and autistic behaviour (Hahn et al 2002; Salomons et al 2001), although severe mental retardation occasionally occunrs. To date, 15 families have been diagnosed with a mutation in the SLC6A8 gene. However, clinical descriptions have been published only of the first nine families (Mancini et al 2005; Salomons et al 2003).
We report two unrelated cases of CRTR, due to large genomic deletions in SLC6A8, with symptoms more severe than those previously reported.
Clinical presentation
Case 1
S.M. was referred to our Metabolic Service at age 30 months for evaluation of severe developmental delay, hypotonia and dystonia. He was the fourth child of his consanguineous Middle Eastern parents (third-degree cousins), born following an uncomplicated pregnancy and delivery. Poor feeding and severe failure to thrive were noted from birth due to poor suck, difficulty swallowing and vomiting. He never achieved head control. Dystonia was diagnosed at 3 months of age when he developed abnormal posturing in his arms and legs. These abnormal movements progressed into prolonged episodes of dystonia during acute illness, such as upper respiratory tract infections. An extensive evaluation, which included brain MRI and lysosomal studies, was unrevealing. A presumptive diagnosis of dopa-responsive dystonia was made, but a trial of carbidopa/levodopa combination was unsuccessful. Transient hyperammonaemia and lactic acidosis were noted and the possibility of mitochondrial disease was suggested but not evaluated. Family history was unremarkable; two older sisters and a brother were clinically normal.
At the time of our initial evaluation, his weight was 8.2 kg, height 78.5 cm and head circumference 46 cm, all below the 3rd centile. He was profoundly hypotonic, with severe oromandibular dystonia and bruxism. Dystonia was also evident in his upper and lower extremities. Deep tendon reflexes were slightly depressed throughout. Speech and language development were severely delayed. He was nondysmorphic and the remainder of his physical examination was unremarkable. Ophthalmic evaluation revealed intermittent esotropia. Grating acuities by preferential looking were at the lower limits of normal for age, 20/130 in each eye, and 20/94 with binocular viewing. Fundoscopy showed no evidence of retinal disease.
Laboratory values are summarized in Table 1. Plasma creatinine was persistently low, and urine organic acids analysis was interpreted as reflecting generalized mitochondrial dysfunction because of increased excretion of the tricarboxylic acid (TCA) cycle intermediates 2-ketoglutaric acid and citric acid, as well as mildly increased excretion of ethyl-malonic acid and 3-methylglutaconic acid. MRI showed abnormal increased signal in the periatrial white matter on the T2-weighted images with associated thinning of the splenium of the corpus callosum. Muscle biopsy histology showed focal predominance of type I fibers and mildly increased amounts of lipid in all fibers, but PAS, NADH and AT-Pase stains were normal. Electron microscopy showed normal number and morphology of mitochondria and OXPHOS studies revealed normal electron transport chain enzyme activities. Magnetic resonance spectroscopy (MRS) imaging showed almost complete absence of the creatine signal (Fig. 1a).
Table 1.
Laboratory results
| Biochemical markera | Normal range | Patient 1 | Patient 2 |
|---|---|---|---|
| Lactate (mmol/L) | 0.5–2.2 | 2.9–3.6 | 4 |
| Pyruvate (mmol/L) | 0.03–0.08 | 0.09–0.18 | 0.06–0.14 |
| Lactate (CSF) (mmol/L) | 1–1.5 | 1.5 | 5.1 |
| CK (u/L) | <175 | 239 | 91 |
| NH3 (μg/L) | 40–80 | 25–209b | 40–217b |
| Creatinine (P) (mg/dl) | 0.3–0.7 | 0.1–0.3 | 0.2 |
| GAA (P) (μmol/L) | 1.04 ± 0.4 | 1.41 | 1.64 |
| GAA (U)(mmol/mol creatinine) | 54 ± 26 | 121.3c | 168d |
| Creatine/creatinine (U) | 0.39 ± 0.29 | 3.67 | 2.00 |
CK, creatine kinase; GAA, guanidinoacetic acid; P, plasma; U, urine
Elevated NH3 values were encountered during febrile episodes
Urine creatinine was 39 mg/dl
Urine creatinine was 28.1 mg/dl
Fig. 1.
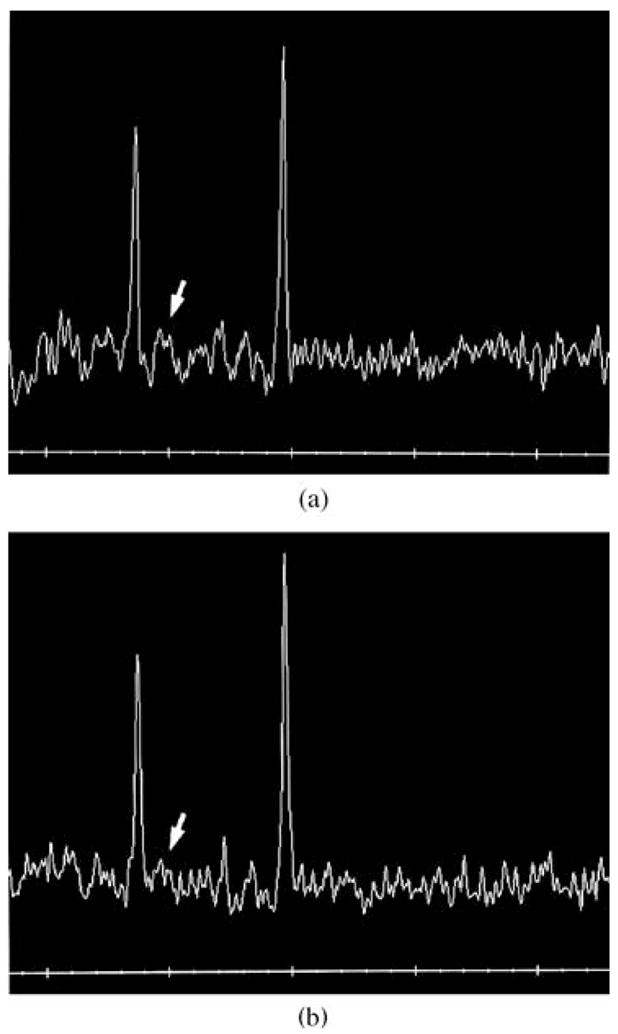
MRS images showing classical absence of creatine peak (arrow) in case 1 (a) and case 2 (b)
The elevated creatine/creatinine ratio in urine and absent creatine peak on MRS prompted further evaluation of the creatine transporter. Impaired creatine uptake in fibroblasts and deletion of exons 8 to 13 in the SLC6A8 gene were detected (see below). Creatine supplementation was initiated (250 mg/kg per day increasing to 750 mg/kg per day). Tri-hexyphenidyl and lorazepam were started for the movement disorder and a gastro-stomy tube was inserted to improve his poor nutritional status. Follow-up 1 year later showed that he had improved weight gain and general condition, but hypotonia and poor head control persisted. Dystonia improved somewhat, but at the age of about 33 months he developed choreiform movements, which were not observed on the initial presentation. Treatment with risperidone resulted in disappearance of the movements.
Case 2
A.R. was referred to our Metabolic Service at age 12 months for evaluation of global developmental delay, severe head and truncal hypotonia, dystonia, weakness, and failure to thrive. Mitochondrial dysfunction had been suspected because of marked elevation of the TCA cycle intermediates in the urine. He was the second-born child of healthy non-consanguineous Hispanic parents. He was delivered by normal vaginal delivery following a pregnancy complicated by oligohydramnios. He required initial resuscitation with bagging. Poor feeding and poor weight gain were noted early in infancy and were attributed to gastro-oesophageal reflux. During an evaluation for failure to thrive at 4 months of age, he was noted to have mild dysmorphic facial features. A renal ultrasound demonstrated bilateral hydronephrosis and a vesicocystourethrogram (VCUG) showed left vesicoureteric reflux with mild obstruction. Karyotype and fluorescent in situ hybridzation studies of 22q11 deletion were normal. Despite adequate energy intake after gastrostomy tube placement at 9 months of age, he continued to have poor weight gain and minimal developmental progress. An ophthalmic examination at 11 months of age was unremarkable apart from intermittent esotropia. Family history was unremarkable; his mother and older sister were clinically normal.
At the time of referral he was 12 months old but still had no head control. His language was limited to cooing. Physical examination was significant for microcephaly and small size. Head circumference, height and weight were significantly below the 5th centile (at the 50th centile for a 3-month-old). He was dysmorphic, with thin, cupped ear, deep sunken eyes, prominent nasal bridge, high-arched palate with submucous cleft, and fifth finger clinodactyly. Neurological examination showed severe axial and truncal hypotonia and weakness. There was an intermittent esotropia with variable angle.
Further laboratory evaluation is summarized in Table 1. MRI of the brain showed areas of hyperintensity within the globus pallidus bilaterally and delayed myelination of white matter with thinning of the entire corpus callosum. MRS study of the brain was initially reported as normal (see below). Bilateral sensorineural hearing loss was diagnosed at age 15 months. A fresh muscle biopsy specimen at age 16 months showed mild variability in myofibre size and increased myofibre lipid in type I fibres, consistent with a defect of oxidative phosphorylation (OXPHOS). Enzymatic assay showed diminished activity of the mitochondrial respiratory chain complex I (zero activity; control 106 ± 46 nmol substrate per mg mitochondrial protein) and, to a milder extent, complet III (641 nmol substrate/min per mg mitochondrial protein; control 1377 ± 367) and complex IV (681 nmol substrate/min per mg mitochondrial protein; control 1583 ± 370). The assay was repeated three times on the muscle biopsy specimen, with consistently significantly lower values compared to the concurrently run control mitochondria, which showed normal respiratory chain enzyme activity. No mitochondrial mutations were found. A presumptive diagnosis of mitochondrial disease was made and treatment with coenzyme Q10, carnitine and vitamin supplementation (B1, B2, C, E and K) was started, with no significant improvement.
After the diagnosis of creatine transporter deficiency was made in case 1, this diagnostic possibility was entertained in case 2, given the similarity in their clinical profiles. Consequently, his MRS was reviewed and the missing creatine peak, initially overlooked, was recognized as very similar to that seen in case 1 (Fig. 1b). Subsequent testing revealed normal plasma guanidinoacetic acid and a markedly increased creatine/creatinine ratio (Table 1), consistent with CRTR. Impaired creatine uptake studies in fibroblast culture confirmed the diagnosis. Molecular testing demonstrated a large deletion of SLC6A8 (see below). Following diagnosis, he was treated with supplemental creatine but had no appreciable clinical improvement. He has shown no progress in his development over several years and has no expressive language. He has had several hospitalizations for hyperpyrexia (up to 40.5°C) without any recognizable infectious aetiology. At the age of 3 years he developed choreoathetoid movements, which initially were intermittent and easily controlled with clonazepam but later became more pronounced. At 3 years 10 months he had his first seizure that progressed to status epilepticus and required ICU admission for management. Treatment with phenobarbital has so far been successful in controlling seizure activity. At the age of 4 years, he still has difficulty with head control, is unable to roll over and has no significant expressive language. Repeat MRS study showed persistent absence of creatine peak despite oral creatine therapy. Interestingly, his ophthalmological examination evolved as follows: At age 2 years, grating acuities by preferential looking were near the lower limits of normal for age, 20/190 in each eye and 20/94 with binocular viewing. At age 3 years 8 months, a pigmentary retinopathy became apparent (Fig. 2). ERG responses to full-field stimuli, when compared to normal values (Fulton and Hansen 2000; Fulton et al 2003; Hansen and Fulton 2005), demonstrated deficits in both scotopic and photopic conditions. In scotopic conditions the saturated b-wave amplitudes were 43% and 30% of the normal mean for age; both of these values are below the 99% prediction interval for normal. A sensitivity parameter, the stimulus evoking a half-maximum response, was near the lower limits of normal for age. Photopic response amplitudes, at 14% and 22% of the normal mean, were also significantly attenuated.
Fig. 2.
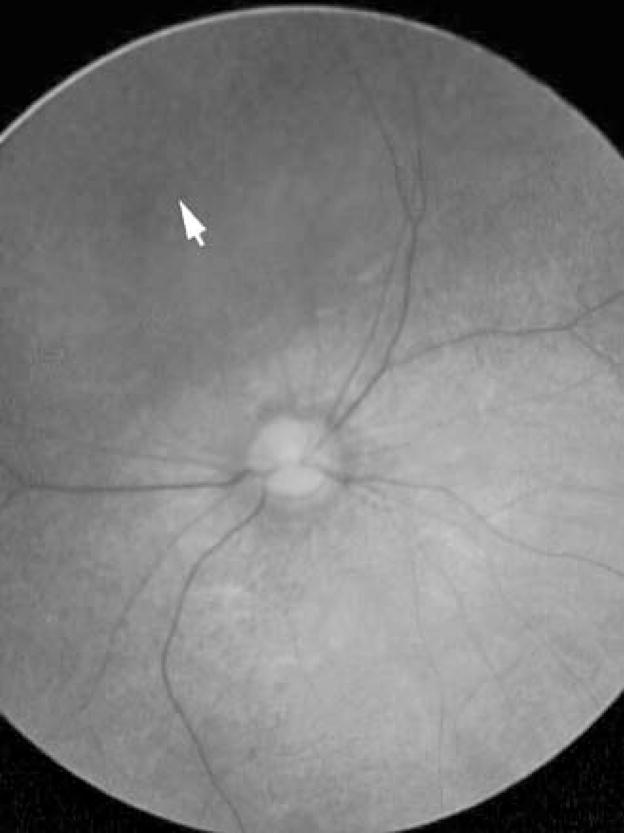
Fundus photograph, right eye, patient 2. At the equator, 360°, was a band of diffuse granular pigmentation (arrow). This pigmentation and mild attenuation of the retinal arterioles and mild pallor of the optic nerve head were consistent with early retinal degeneration. Features of the left fundus were similar
Molecular and creatine uptake studies
Amplification by PCR of all 13 exons and adjacent splice sites of SLC6A8 at the genomic DNA level was performed as described previously (Rosenberg et al 2004). In addition, three amplicons of autosomal gene regions were generated of approximately 1 kb, which confirmed proper quality of the genomic DNAs of both patients. In the DNA of patient 1, we successfully amplified exons 1, 2, 3, 4 and 5–7, but the single exons 8, 9, 10, 11, 12 and 13 could not be amplified using exon-specific primers. In the genomic DNA of patient 2, none of these combined or single exons could be amplified. Furthermore, mRNA was isolated from cultured fibroblasts of both patients. Indeed, in contrast to mRNAs in controls, the SLC6A8 mRNA could not be detected by RT-PCR (Salomons et al 2001). Specific amplification of gene sequences in the upstream and downstream genes, specifically exon 4 of the centromeric DUSP9 gene, and exon 1 of the telomeric DXS1357/BCAP31 and ABCD1 genes, was carried out successfully. These data are consistent with large genomic deletions of exons 8–13 of SLC6A8 in patient 1 and of the complete coding region in patient 2. These findings were confirmed by multiplex ligation-dependent probe amplification test. The de novo nature of these deletions was confirmed by the normal results on the mothers of the two patients. Creatine uptake in both patients showed impaired creatine uptake profiles both at physiological (25 μmol/L) and at supraphysiological levels (500 μmol/L): the creatine concentrations in the fibroblasts at 25 μmol/L substrate concentration were 0.8 and 1.0 pmol/μg protein, respectively (controls, n = 19, 25 ± 5.2; CRTR patients, n = 14, 0.94 ± 0.96). Incubations with 500 μmol/L creatine resulted in 7.6 and 9.9 pmol/μg protein, respectively (control, n = 19, 36 ± 5.1; CRTR patients, n = 14, 8.37 ± 2.0).
Discussion
CRTR belongs to a novel group of inborn errors of metabolism, the creatine deficiency syndromes (Rosenberg et al 2004; Salomons et al 2001; Schulze 2003; Stromberger 2003). A number of reviews have been published to further characterize the clinical, laboratory, molecular and imaging profiles of CRTR (deGrauw et al 2002, 2003; Salomons et al 2003). Dystonia and choreoathetoid movements, prominent features of our cases, are common in GAMT but were only reported once in CRTR (Hahn et al 2002). Similarly, the seizure disorder seen in our cases contrasts with the relatively milder form reported previously in CRTR. That the guanidinoacetate (GAA) level was normal in both cases raises doubt about the suggestion that the movement disorder seen in GAMT deficiency is due to high levels of GAA (Leuzzi 2002). It appears that creatine deficiency plays a more important role in the pathogenesis of the movement disorder seen in these conditions.
The various indicators of mitochondrial dysfunction clouded the clinical picture in both patients whose work-up initially focused on a primary mitochondrial pathology (see below). It was only after MRS studies revealed the absence of creatine peak, the classical sign shared by all three disorders of creation metabolism, that the possibility of CRTR was entertained. As seen in the example of the second case, that classical sign was overlooked initially. Realization of the powerful potential of MRS to detect this group of disorders, and that it is not as rare as initially thought (see below), should improve the detection rate.
The lack of biochemical or clinical response in our two patients to creatine supplementation, even at high doses, is not surprising. Trials of treatment of males affected with CRTR deficiency with creatine monohydrate only have not proved to be successful (Bizzi et al 2002; deGrauw et al 2002). Supplementation with high doses of arginine and glycine, which are the primary substrates for creatine biosynthesis, combined with high doses of creatine monohydrate, is being investigatedv (unpublished data, Mancini GMS, van der Knaap N, Salomons GS).
Both of our patients showed evidence of disturbance in mitochondrial energy metabolism, with transient hyperammonaemia and lactic acidaemia, along with specific mitochondrial respiratory chain enzyme deficiencies in one patient and evidence of mild myopathy (increased CPK and/or abnormal muscle histology) in both. This suggests that some of the underlying pathogenesis of the creatine disorders is due to mitochondrial energy depletion, at least in these two patients. This is of interest given the well-described role of creatine in mitochondrial metabolism, which includes a protective and beneficial role of creatine for mitochondria (Brustovetsky et al 2001; O’Gorman et al 1997;Tarnopolsky et al 1997). In addition, the specific importance of creatine to the mitochondrial respiratory chain and the detrimental effect of blocking the uptake of creatine with specific analogues have been demonstrated (O’Gorman et al 1996). Moreover, there is some evidence, at least in fibroblasts of patients with GAMT deficiency, that different components of respiratory chain are upregulated (Das et al 2000), suggesting that in the absence of the short-term energy buffering of creatine and phosphocreatine the cells rely almost exclusively on mitochondrially generated ATP, which puts extra stress on the respiratory chain. While some tissues with low energy requirement can compensate for this by upregulating their respiratory chain, as demonstrated in fibroblasts, in other tissues with high energy requirement, such as brain, retinal photoreceptors and muscle, the compensatory upregulation may be overwhelmed with the resulting deficiency in respiratory chain components.
Interestingly, patient 2 also had evidence of pigmentary retinopathy and significant deficits in retinal function, as do some children with mitochondrial disorders (Cooper et al 2002). Although the retinopathy and ERG deficits in mitochondrial disorders have a broad spectrum, the most subtle ERG signature of a mitochondrial disorder is low scotopic b-wave sensitivity with good preservation of the amplitude of the scotopic b-wave (Cooper et al 2002). The mechanism appears to be energy depletion and depressed ionic pumps in the rod photoreceptors. In contrast, patient 2 had attenuated amplitudes, while sensitivity was relatively better preserved. Thus, mechanisms other than energy depletion warrant consideration.
The clinical description of previously reported patients with CRTR does not allow for a definitive conclusion about evidence of mitochondrial disturbance in those cases. Nevertheless, our report suggests that CRTR should be considered in the differential diagnosis of patients with evidence of mitochondrial pathology. Likewise, clinical description of patients with CRTR should include mitochondrial studies to better characterize this association.
Patient 2 also had mild dysmorphic features, as did some previously reported patients (Hahn et al 2002). It is possible that this or other features encountered in our patients may be due to (partial) deletion of other genes flanking SLC6A8. However, gene sequences in the upstream and downstream genes could be properly amplified, suggesting that this is not the case for those currently known to be flanking SLC6A8. Work is under way to characterize the breakpoints. Even if we fail to show direct involvement of other genes, one should remember that the deleted regions may contain long-range control sequences affecting other genes (Kleinjan and van Heyningen 2005). So far, all other patients with CRTR have had small base changes leading to nonsense mutations, single amino acid deletions, missense mutations and splice site errors (Salomons et al 2003). Therefore, the presence of large deletions in our patients may also suggest genotype–phenotype correlation, although this cannot be clearly established because of the small number of cases reported. It is of note that a contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E, with features of seizures and hypotonia that are also seen in our patients, has been described (Corzo et al 2002); this locus is telomeric to the SLC6A8 locus. Very long-chain fatty acids were checked in both patients and the results were unremarkable.
Recent studies in a panel of X-linked mental retardation males from the European consortium showed that approximately 2.1% of males had CRTR (Rosenberg et al 2004), suggesting that this disorder is a relatively frequent cause of mental retardation. Accordingly, in the absence of a confirmed alternative diagnosis, we recommend that all mentally retarded males should be tested for CRTR.
Footnotes
Competing interests: None declared
References
- Bizzi A, Bugiani M, Salomons GS, et al. X-linked creatine deficiency syndrome: a novel mutation in creatine transporter gene SLC6A8. Ann Neurol. 2002;52:227–231. doi: 10.1002/ana.10246. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Dubinsky JM. On the mechanisms of neuroprotection by creatine and phosphocreatine. J Neurochem. 2001;76:425–434. doi: 10.1046/j.1471-4159.2001.00052.x. [DOI] [PubMed] [Google Scholar]
- Cooper LL, Hansen RM, Darras BT, et al. Rod photoreceptor function in children with mitochondrial disorders. Arch Ophthalmol. 2002;120:1055–1062. doi: 10.1001/archopht.120.8.1055. [DOI] [PubMed] [Google Scholar]
- Corzo D, Gibson W, Johnson K, et al. Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am J Hum Genet. 2002;70:1520–1531. doi: 10.1086/340849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das AM, Ullrich K, Isbrandt D. Upregulation of respiratory chain enzymes in guanidinoacetate methyltransferase deficiency. J Inherit Metab Dis. 2000;23:375–377. doi: 10.1023/a:1005643617251. [DOI] [PubMed] [Google Scholar]
- deGrauw TJ, Salomons GS, Cecil KM, et al. Congenital creatine transporter deficiency. Neuropediatrics. 2002;33:232–238. doi: 10.1055/s-2002-36743. [DOI] [PubMed] [Google Scholar]
- deGrauw TJ, Cecil KM, Byars AW, Salomons GS, Ball WS, Jakobs C. The clinical syndrome of creatine transporter deficiency. Mol Cell Biochem. 2003;244:45–48. [PubMed] [Google Scholar]
- Fulton AB, Hansen RM. The development of scotopic sensitivity. Invest Ophthalmol Vis Sci. 2000;41:1588–1596. [PubMed] [Google Scholar]
- Fulton AB, Hansen RM, Westall CA. Development of ERG responses: the ISCEV rod, maximal and cone responses in normal subjects. Doc Ophthalmol. 2003;107:235–241. doi: 10.1023/b:doop.0000005332.88367.b8. [DOI] [PubMed] [Google Scholar]
- Hahn KA, Salomons GS, Tackels-Horne D, et al. X-linked mental retardation with seizures and carrier manifestations is caused by a mutation in the creatine-transporter gene (SLC6A8) located in Xq28. Am J Hum Genet. 2002;70:1349–1356. doi: 10.1086/340092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen RM, Fulton AB. Development of the cone ERG in infants. Invest Ophthalmol Vis Sci. 2005;46:3458–3462. doi: 10.1167/iovs.05-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Item CB, Stockler-Ipsiroglu S, Stromberger C, et al. Arginine:glycine amidinotransferase deficiency: the third inborn error of creatine metabolism in humans. Am J Hum Genet. 2001;69:1127–1133. doi: 10.1086/323765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinjan DA, van Heyningen V. Long-range control of gene expression: emerging mechanisms and disruption in disease. Am J Hum Genet. 2005;76:8–32. doi: 10.1086/426833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuzzi V. Inborn errors of creatine metabolism and epilepsy: clinical features, diagnosis, and treatment. J Child Neurol. 2002;17(supplement 3):3S89–97. [PubMed] [Google Scholar]
- Mancini GM, Catsman-Berrevoets CE, de Coo IF, et al. Two novel mutations in SLC6A8 cause creatine transporter defect and distinctive X-linked mental retardation in two unrelated Dutch families. Am J Med Genet A. 2005;132:288–295. doi: 10.1002/ajmg.a.30473. [DOI] [PubMed] [Google Scholar]
- O’Gorman E, Beutner G, Wallimann T, Brdiczka D. Differential effects of creatine depletion on the regulation of enzyme activities and on creatine-stimulated mitochondrial respiration in skeletal muscle, heart, and brain. Biochim Biophys Acta. 1996;1276:161–170. doi: 10.1016/0005-2728(96)00074-6. [DOI] [PubMed] [Google Scholar]
- O’Gorman E, Fuchs KH, Tittmann P, Gross H, Wallimann T. Crystalline mitochondrial inclusion bodies isolated from creatine depleted rat soleus muscle. J Cell Sci. 1997;110(12):1403–1411. doi: 10.1242/jcs.110.12.1403. [DOI] [PubMed] [Google Scholar]
- Rosenberg EH, Almeida LS, Kleefstra T, et al. High prevalence of SLC6A8 deficiency in X-linked mental retardation. Am J Hum Genet. 2004;75:97–105. doi: 10.1086/422102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomons GS, van Dooren SJ, Verhoeven NM, Cecil KM, Ball WS, Degrauw TJ, Jakobs C. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am J Hum Genet. 2001;68:1497–1500. doi: 10.1086/320595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomons GS, van Dooren SJ, Verhoeven NM, et al. X-linked creatine transporter defect: an overview. J Inherit Metab Dis. 2003;26:309–318. doi: 10.1023/a:1024405821638. [DOI] [PubMed] [Google Scholar]
- Schulze A. Creatine deficiency syndromes. Mol Cell Biochem. 2003;244:143–150. [PubMed] [Google Scholar]
- Stockler S, Isbrandt D, Hanefeld F, Schmidt B, von Figura K. Guanidinoacetate methyltransferase deficiency: the first inborn error of creatine metabolism in man. Am J Hum Genet. 1996;58:914–922. [PMC free article] [PubMed] [Google Scholar]
- Stromberger C, Bodamer OA, Stockler-Ipsiroglu S. Clinical characteristics and diagnostic clues in inborn errors of creatine metabolism. J Inherit Metab Dis. 2003;26:299–308. doi: 10.1023/a:1024453704800. [DOI] [PubMed] [Google Scholar]
- Tarnopolsky MA, Roy BD, MacDonald JR. A randomized, controlled trial of creatine monohydrate in patients with mitochondrial cytopathies. Muscle Nerve. 1997;20:1502–1509. doi: 10.1002/(sici)1097-4598(199712)20:12<1502::aid-mus4>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. 2000;80:1107–1213. doi: 10.1152/physrev.2000.80.3.1107. [DOI] [PubMed] [Google Scholar]