

Abstract
Retinoic acid (RA) is a critical regulator of gene expression during embryonic development and in the maintenance of adult epithelial tissues. Genetic polymorphisms of CYP26A1 could cause inter-individual variation in the metabolism of retinoic acid, thus altering signaling during embryonic development. A total of 13 single nucleotide polymorphisms (SNPs) were identified in CYP26A1 in 92 racially diverse individuals (24 Caucasians, 24 African-Americans, 24 Asians and 20 individuals of unknown racial origin). Three of the 13 SNPS produced coding changes: R173S, F186L and C358R. These alleles were termed CYP26A1*2, CYP26A1*3, and CYP26A1*4, respectively, by the Human Cytochrome P450 (CYP) Allele Nomenclature Committee at http://www.cypalleles.ki.se/. cDNA constructs for wild-type and mutant alleles of CYP26A1 were constructed in a pcDNA3.1 expression vector containing a FLAG tag at the C-terminal end, which was used to identify and quantitate the CYP26A1 allelic proteins when expressed in COS-1 cells. Wild type CYP26A1 protein metabolized all-trans-retinoic acid (at-RA) to 4-oxo-RA, 4-OH-RA and 18-OH-RA as well as water-soluble metabolites. CYP26A1.3 (F186L) and CYP26A1.4 (C358R) allelic proteins exhibited significantly lower metabolism (40-80%) of at-RA than wild-type CYP26A1.1 protein. This study identifies two CYP26A1 coding alleles, CYP26A1*3 and CYP26A1*4, which are predicted to be defective in retinoic acid metabolism based on the metabolism of at-RA by the recombinant proteins. This is the first study to identify coding alleles of CYP26A1. The in vitro characterization of the recombinant allelic proteins suggests the need for future clinical studies of genotype/phenotype relationships of CYP26A1 in embryonic development.
Keywords: P450, CYP26A1, retinoic acid, polymorphisms, retinoic acid metabolism
Introduction
Retinoid derivatives, including retinol, retinal, retinoic acid (RA) and other related compounds, are important regulators of gene expression [1, 2]. They are essential for embryonic development, differentiation of adult epithelial cells, regulation of cell growth and other physiological processes [2, 3]. Among various retinoid compounds, at-RA generated from all-trans retinaldehyde is suggested to be one of the biologically most important retinoids [4, 5]. at-RA exerts its biological effects via retinoic acid receptors (RARs) thus regulating gene expression during cell proliferation, embryonic development, and differentiation of adult epithelial tissues. Steady-state concentrations of at-RA are tightly controlled by the synthesis and catabolism of at-RA. CYP26A1 has been reported to be the principal enzyme responsible for the catabolism of at-RA to the inactive polar metabolites, 4-hydroxy-RA (4-OH-RA) , 4-oxo-RA, and 18-OH-RA [6-8]. Although some studies have suggested that 4-OH-RA and 4-oxo-RA have biological activity [9-11], other recent studies have suggested that these metabolites are principally inactive breakdown products of at-RA [1, 5]. Cyp26a1 null mice die during mid-late gestation [7, 12] . These mice exhibit spina bidfida, and a number of developmental abnormalities including truncation of the tail, malformation of the lumbrosacral region, and abnormal development of the hindbrain, hindgut and kidneys. A recent study showed that the lethal phenotype of Cyp26a1-/- mice was rescued by the heterozygous disruption of Aldh1a2 (the enzyme responsible for synthesis of at-RA) [5]. This study suggests that the principal function of CYP26A1 in the embryo is to protect tissues from excess exposure to at-RA.
Thus polymorphisms in human CYP26A1 that affect enzyme activity could be important in regulating the cellular concentrations of RA, particularly in the embryo. Therefore, we sequenced 150 bp upstream of the translation start site, the coding regions, and intron-exon junctions of the CYP26A1 gene in 92 racially diverse individuals. A nucleotide sequence analysis program was used to predict possible new splice sites introduced by mutations. Newly identified coding variants were constructed by site-directed mutagenesis and evaluated in a recombinant system in COS-1 cells. Catalytic activities of the mutant and wild-type recombinant alleles (RefSeq NM_000783) were compared using at-RA as a substrate. Alterations in the catalytic activity of CYP26A1 could affect cellular concentrations of at-RA, potentially affecting gene regulation in embryonic development and in the maintenance of adult epithelial tissues.
Materials and Methods
Chemicals and reagents
Restriction enzymes were obtained from New England Biolabs (Beverly, MA, USA). E. coli DH5α competent cells and antibiotics were purchased from Invitrogen (Carlsbad, CA, USA). Oligonucleotide primers were synthesized by Sigma Genosys (Woodlands, TX, USA). Polymerase chain reaction (PCR) was performed by using a proofreading Pfu DNA polymerase from Stratagene (La Jolla, CA, USA). at-RA, 9-cis-RA, 13-cis-RA, and reduced β-nicotinamide adenine dinucleotide phosphate (NADPH) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). 4-oxo-RA and 5,6-epoxy-RA were a gift from Hoffmann-La Roche (Nutley, NJ, USA) [13]. at-[3H] RA was purchased from PerkinElmer Life Sciences (Boston, MA, USA). All other chemicals and organic solvents for HPLC were of the highest grade from commercial sources.
DNA samples
Genomic DNA was obtained from lymphoblastoid cell lines isolated from 92 different human subjects. These samples were from the Human Genetic Cell Repository sponsored by the National Institutes of Health, housed at the Coriell Institute, Camden, NJ and represented all three major racial/ethnic groups (Caucasians [US and European], Asians, and Africans). Samples originated from the following ethnic ancestries: 24 Africans (15 African-Americans, 9 African Pygmies), 24 Asians (4 Indo-Pakistani, 5 native Taiwanese, 5 mainland Chinese [Beijing], 3 Cambodian, 3 Japanese, 4 Melanesian), 24 Caucasians (7 Utah, 5 Druze [Lebanon], 7 eastern European [Adygei], 5 Russian [Moscow]) and 20 individuals of unspecified ancestry, representing the ethnic diversity of the U.S. population, from the NIH DNA Polymorphism Discovery Resource [14]. Since the samples are commercially available cell lines from anonymous healthy donors, the protocol was considered exempt by the Lawrence Livermore National Laboratory Review Board under 10CRF745.101(b) 4.
Direct DNA sequencing and variant identification
All eight exons, intron-exon junctions, 150 bp upstream from the translation start site, and 293 bp downstream of the stop codon were amplified using CYP26A1 specific primers (Table 1) and sequenced. PCR amplification of the genomic sequence was initiated approximately 50-100 nucleotides from each intron-exon boundary. Appended oligonucleotide sequences were added to the 5’-end of the PCR primers for annealing of the forward or reverse energy transfer (ET) DNA sequencing primers (Amersham Biosciences, Piscataway, NJ, USA). The amplification products were directly sequenced using a DYEnamic Direct cycle sequencing kit with DYEnamic ET primers (Amersham Biosciences, Piscataway, NJ, USA). The reaction products were loaded onto ABI Prism 377 stretch DNA sequencers (Foster City, CA, USA). Identification of variants with single nucleotide substitutions in heterozygous and homozygous individuals was performed using “PolyPhred” (version 2.1) which includes a software package that utilizes the output from Phred, Phrap and Consed. A nucleotide sequence analysis program at http://www.fruitfly.org/seq_tools/splice.html was used to predict possible new splice sites introduced by mutations.
Table 1.
PCR Primers used for the identification of CYP26A1 variants
| SNP ID | Forward primer | Reverse primer |
|---|---|---|
| 1671 -1673 | CCAATCCGCAATTAAAGATGA | CCTGCTCCTAGCCGCAATAG |
| 1669 -1670 | CTTCTGCTGAAGTCGGGGTAG | GGGAAAGCTAACCTGGAGCTG |
| 1720 -1721 | GATGGAGGCTTTTAACGCTGT | GATCTTGGCGCGAATGTTCT |
| 1722 -1723 | GAAATGACCCGCAATCTCTT | TTCGGGATTCCATAGCTGTAGT |
| 1724 | AAGGGACGTTGCATTTTGTTT | CAAAGCTAATTCTGCCCACTG |
| 1725 -1726 | GGCTGATTTTATTGGAGCACA | GGGCTGACAAACTGGAATTTA |
| 1746 | CCTCTCCCTTGCTTTTAGCTC | CAAATAAGTCCAGGTAGGCATTG |
Oligonucleotide sequence, GTTTTCCCAGTCACGACG, was attached to the 5’-end of the forward primer and AGGAAACAGCTATGACCAT was attached to the 5’-end of the reverse primer for DNA sequencing.
Construction of expression vectors for CYP26A1 alleles and site-directed mutagenesis
Full-length CYP26A1 wild type cDNA was amplified by PCR using human universal QUICK clone cDNA as a PCR template (Clonetech Laboratories Inc., Mountain View, CA, USA) and cloned into pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA, USA). PCR primers used for human CYP26A1 cDNA cloning were: first forward primer, 5’-cggcacgagtggcgcgggaggtcg-3’; second nested forward primer, 5’-gagtggcgcgggagg tcgcggcgc-3’; first reverse primer, 5’-atatgttacacttccaataagtctcagg-3’; second nested reverse primer, 5’-acacttccaataagtctcaggtttgaac-3’. After cloning CYP26A1 cDNA into pCR2.1-TOPO vector, another round of PCR amplification was performed to add a FLAG tag and restriction enzyme sites: forward primer, 5’-ggtggtaagcttccatggggctcccagcgctgctggccagtg-3’ and reverse primer, 5’-ggtggtctcgagtcatcacttgtcatcgtcatccttgtagtcgatttccccatggaaatgggtg-3’. The PCR product containing a CYP26A1-FLAG was digested with HindIII and XhoI and sub-cloned into the pcDNA3.1 expression vector (Invitrogen). The pcDNA3.1-CYP26A1-FLAG was sequenced and used as a wild-type template for site-directed mutagenesis using a QuickChange kit (Stratagene, La Jolla, CA, USA). PCR primers with N-terminal modification of CYP26A1 cDNA for E.coli expression [15-17] were: forward primer, 5’-ggtggtcatatggctctgttattagcagtttttctcctcaccttcgtgctgccg-3’ and reverse primer, 5’-gctgccaagctttcagtgatggtgatggtggatttccccatggaaatgg-3’. A second possible shorter CYP26A1 variant predicted in the NCBI (RefSeq NM_057157) was also constructed. Primers used for the construction of an alternative N-terminal truncated CYP26A1 protein based on a putative alternative start site (RefSeq NM_057157) were: forward primer, 5’-ggtggtaagcttatgaagcgcaggaaatacgg-3’ and reverse primer, 5’-ggtggtctcgagtcatcacttgtcatcgtcatc-3’. Primers used for the mutagenesis are listed in Table 2. After construction of recombinant plasmids, the entire cDNA was verified by sequencing in both directions before expression.
Table 2.
PCR Primers for site-directed mutagenesis of CYP26A1
| Mutation | Primer sequence (5’→3’) |
|---|---|
| Arg173Ser | CTGAGCTGCGGCGAGAGCGGCCTCCTGGTCTAC |
| Phe186Leu | GTGAAGCGCCTCATGTTACGAATCGCCATGCGCATC |
| Cys358Arg | CAACTTAAATACATCGGGCGTGTTATTAAGGAGACC |
Underlined and bold nucleotides indicate the mutated base from wild type CYP26A1 (cDNA Accession number: NM_000783).
Cell culture and transient transfection of COS-1 cells
COS-1 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen) supplemented with 10% FBS, 100 U/ml penicillin, and 100 ug/ml streptomycin at 37° in 5% CO2. Exponentially growing cells were plated into six-well plates in triplicate at 2 × 105 cells/well 12 hr before transfection. Cells were transfected with 1 μg of pcDNA3.1 empty vector, pcDNA3.1-CYP26A1-FLAG tagged wild-type, or pcDNA3.1-CYP26-FLAG-variants using FuGENE 6 transfection reagent and conditions recommended by the manufacturer (Roche Applied Science, Indianapolis, IN, USA).
Analysis of retinoic acid metabolism
Media was removed 48 hr after transfection with pcDNA3.1 control vector or pcDNA3.1-FLAG CYP26A1 constructs. COS-1 cells were washed once with DMEM, and incubated with 0.5 ml of DMEM containing 100 nM at-[3H] RA (1 mCi/ml, 44.5 Ci/mmol, PerkinElmer Life Sciences, USA), 100 μM cold at-RA, and 0.2 mM NADPH. After various incubation times (2 min - 3 hrs) at 37°C, medium was removed and mixed with an equal volume of dichloromethane (DCM) /methanol (2:1, v/v) as described [13]. All experimental procedures with retinoids were conducted with protection from light. The aqueous phase and the organic phase were separated by centrifugation at 13000 rpm for 10 min. The organic phase, containing hydrophobic metabolites and retinoic acids, was dried using a Savant Speed Vac Concentrator (GMI Inc., MN, USA) and then resuspended in ethanol for HPLC injection. The radioactivity in the reaction was measured using a β-scintillation counter (Beckman LS6500 Multi-Purpose Scintillation Counter, Beckman Coulter Inc., Fullerton, California, USA). Cellular retinoids were extracted using methanol, dried as above, and residues were resuspended in ethanol for HPLC analysis. Metabolites were analyzed by HPLC using a reverse phase analytical column (Ultrasphere C18 5 μm, 4.6 × 250 mm, Beckman Instruments, Fullerton, CA, USA), and Waters Alliance 2695 HPLC system (Milford, MA, USA). The radiolabelled at-RA metabolites were identified by comparison of theirs elution times with the UV peaks of unlabelled standard metabolites. Metabolites were separated using the method of Marill et. al [18], consisting of a linear gradient of 1% ammonium acetate in water (solvent A):methanol (solvent B) at a starting ratio of 35: 65, increasing to 100% methanol over 35 minutes at a flow rate of 1 ml/min.
Western blot analysis
Transfected COS-1 cells were harvested, resuspended with cell lysis buffer (Cell Signalling Technology, Danvers, MA, USA), and sonicated. Total cellular protein (1.25 μg/lane) was heated at 70°C in sodium dodecyl sulfate (SDS) sample loading buffer for 10 min before electrophoresis on a 10% SDS-polyacrylamide gel (Criterion XT Bis-Tris precast gel, Bio-Rad Laboratories, Hercules, CA, USA). Amino-terminal Met-FLAG-BAP-fusion protein (Sigma-Aldrich, St. Louis, MO, USA) was used as a positive control for FLAG detection. After electrophoresis, proteins were transferred to a nitrocellulose membrane (Bio-Rad Laboratories, CA, USA). The membrane was blocked with 3% BSA in Tris buffered saline (TBS) buffer. It was then incubated with monoclonal anti-FLAG M2-horseradish peroxidase antibody (Sigma-Aldrich, St. Louis, MO, USA) at a 1: 4000 dilution. FLAG epitope tagged proteins were visualized with Super Signal West Pico Chemiluminescent Substrate (Pierce Biotechnology, Inc., Rockford, IL, USA). Protein bands containing the FLAG tag were visualized by autoradiography on Kodak Biomax MR Film (Eastman Kodak Company, Rochester, NY, USA). The density of the bands was quantified using a Fluorskan 8900 (Alpha Innotech, San Leandro, CA, USA).
Statistical Analysis
All data represent the mean ± S.E of triplicates. Experiments shown represent one of two independent experiments. Statistical significance (p<0.05) was determined for each metabolite between wild-type and variant using analysis of variance followed by a two-tailed Student’s t-test.
Putative model structure of CYP26A
There are four X-ray crystal structures of mammalian cytochrome P450 proteins available in the protein database: CYP2C5 [19], CYP2C8 [20], CYP2C9 [21] and CYP3A4 [22]. The corresponding sequences along with the sequence of CYP26A1 were aligned using Clustal-X and T-coffee alignment programs. The alignments of the X-ray crystal structures using Syby17.1 (Tripos, Inc.) show that all the structurally important regions (helices and beta sheets) aligned well against each other. Therefore, a possible structure template for CYP26A1 was created based on the X-ray crystal structure of CYP2C5 [19] using Sybyl 7.1 (Tripos, Inc.).
Results
CYP26A1 SNP discovery
A total of 13 CYP26A1 SNPs were identified in 92 racially diverse individuals (Table 3). Two variants were found upstream of the translation start site, seven in introns, and four in the coding regions of CYP26A1. All SNPs are numbered from the start site of translation and are named according the the recommendations of the Nomenclature Working group [23]. Three newly identified SNPs produced amino acid changes: a 558C>A in the cDNA (numbering from the start codon) coding for F186L (one heterozygous mutation found in Whites), a 517.C>A coding for R173S (three heterozygous mutations found in African-Americans, and a 1072T>C coding for C358R (one heterozygous mutation found in Asians). The C358R coding change was found as a heterozygous mutation in an individual who was homozygous for a g.199G>C change in intron 1 (IVS1+10) and heterozygous for two changes in intron 4 (IVS4+7 and IVS4+8)(g.1380A>G and g.1381G>A), suggesting the C358R is linked to the inron 1 change and perhaps to the intron 4 changes. The other two coding SNPs were not linked with any other SNPs. Coding alleles were submitted to the CYPalleles nomenclature committee at http://www.imm.ki.se/CYPalleles for official nomenclature. The R173S allele was designated CYP26A1*2, the F186L allele designated CYP26A1*3, and the C358R allele designated CYP26A1*4 by this committee. We also found a silent mutation in exon 7 (1314 C>G of the cDNA) coding for G438G. The change in intron 1 (IVS+10) was frequent in individuals of African descent (8/30 alleles in African-Americans, 4/18 alleles in African pygmies) and also found in 2/46 alleles in Asians, and 4/40 alleles of unknown ancestry). Two SNPs identified in intron 4, an IVS4+7 and IVS4+8 (GA>AG) were found as a heterozygous mutation in eight of nine individuals who were homozygous for the IVS+10 change. We found a total of three changes in intron 1 (IVS1+10, IVS1-44, IVS1-5, three changes in intron 4 (IVS4+7, IVS44+8, and IVS 4-8, and 1 change in intron 6 (IVS6+69. None of the intron/exon SNPs appeared to create or delete acceptor or donor splice sites using a splice site prediction program http://www.fruitfly.org/seq_tools/splice.html. Neither of the two mutations found in the promoter appeared to affect any known transcription factor binding sites, including a RA response element [24], as analyzed by MatInspector http://www.genomatix.de/products/MatInspector/index.html).
Table 3.
Single nucleotide polymorphisms (SNP) found in CYP26A1
| SNP IDa | Site | Nucleotide change | Amino acid change | Number of allelese |
|||||
|---|---|---|---|---|---|---|---|---|---|
| Africans | Caucasians | Asians | Unknown | Positiond | |||||
| AAb | APc | ||||||||
| 1671 | Promoter | ccgtg(G>A)ggttt | Non coding | 0/30 | 0/18 | 3/46 | 1/48 | 0/40 | -59 |
| 1673 | Promoter | ctggc(G>T)gcggc | Non coding | 1/30 | 0/18 | 0/46 | 0/46 | 0/40 | -36 |
| 1672 | IVS1+10 | gggag(G>C)gtggg | Non coding | 8/30 | 4/18 | 0/48 | 2/46 | 4/40 | +199 |
| 1669 | IVS1-44 | ccccg(G>A)cgccc | Non coding | 3/30 | 0/18 | 0/48 | 0/48 | 0/40 | +330 |
| 1670 | IVS1-5 | cctcc(C>T)acagc | Non coding | 0/30 | 0/18 | 0/48 | 1/48 | 0/40 | +369 |
| 1721 | Exon3 | gcgag(C>A)gcggc | Arg173Ser | 3/30 | 0/18 | 0/48 | 0/48 | 0/40 | +947 |
| 1720 | Exon3 | atgtt(C>A)cgaat | Phe186Leu | 0/30 | 0/18 | 1/48 | 0/48 | 0/40 | +988 |
| 1723 | IVS4+7 | tgagt (A>G)gcagc | Non coding | 4/30 | 1/16 | 0/48 | 1/46 | 2/40 | +1380 |
| 1722 | IVS4+8 | gagta(G>A)cagct | Non coding | 4/30 | 1/16 | 0/48 | 1/46 | 2/40 | +1381 |
| 1724 | IVS4-8 | actct(A>G)ttctt | Non coding | 0/30 | 0/18 | 1/48 | 0/48 | 0/40 | +1884 |
| 1726 | Exon 6 | tcggg(T>C)gtgtt | Cys358Arg | 0/30 | 0/18 | 0/48 | 1/46 | 0/40 | +2682 |
| 1725 | IVS6+69 | tgctg(T>G)ggctg | Non coding | 0/30 | 1/18 | 0/48 | 0/46 | 1/40 | +2901 |
| 1746 | Exon 7 | ggagg(C>G)cttag | Gly438Gly | 0/30 | 0/16 | 0/46 | 2/48 | 0/40 | +3190 |
The SNP ID numbers are from the NIEHS SNP database.
AA, African-American,
AP, African Pygmies.
Position number was based on the reference CYP26A1 gene sequence obtained from NCBI accession number, NT_030059.12 with the A of the start codon being +1, and negative numbers representing the upstream region. Intron changes are also denoted IVS followed the position relative to the exon . CYP26A1 was resequenced in DNA from 15 African-Americans (30 alleles), 9 African Pygmies (18 alleles), 24 Caucasians (48 alleles), 24 Asians (48 alleles) and 20 unknown racial group (40 alleles).
Expression of CYP26A1
Initially, CYP26A1 wild-type cDNA was introduced into the pCW vector after N-terminal modification and transfected into E. coli as described previously for CYP3A4 and CYP3A5 [15-17]. Although CYP3A4 and CYP3A5 proteins were successfully expressed in the E. coli cDNA expression system as positive controls, no reduced CO-difference spectrum indicative of cytochrome P450 (450nm) was observed for CYP26A1 in more than three separate experiments. Therefore, COS-1 cells were subsequently used for transient transfection for CYP26A1 protein production, since this system was used previously for expression of recombinant CYP26A1 and CYP26C1 [25, 26]. Since no antibody for CYP26A1 is available, a FLAG peptide was added to the CYP26A1 C-terminus and utilized to monitor the amount of protein expressed in transfections (Fig. 1A) and to quantitate CYP26A1 protein by Western blot analysis by comparison with a linear curve for the amino-terminal Met-FLAG-BAP-fusion protein (Fig. 1B). Western blotting showed that the expression of CYP26A1 protein (50 kD band) was similar in all transfected COS-1 cells.
Fig. 1.
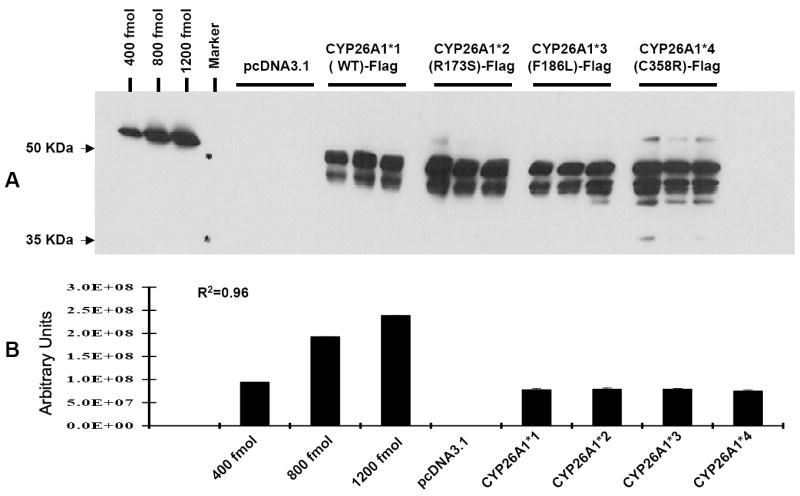
Western blot analysis of the expression of CYP26A1 wild-type and CYP26A1 variants in COS-1 cells. A. COS-1 cells were transiently transfected with pcDNA3.1 alone (EV, empty vector), pcDNA3.1-CYP26A1*1( WT)-FLAG, pcDNA3.1-CYP26A1*2 (R173S)-FLAG, pcDNA3.1-CYP26A1*3( F186L)-FLAG, and pcDNA3.1-CYP26A1*4( C358R)-FLAG. After 48 hr transfection, cells were washed with medium and incubated with 100 μM at-RA and 0.2 mM NADPH. After 2 hr at 37°C, COS-1 cells were harvested and extracted as described and subjected to electrophoresis on a 10% SDS-polyacrylamide gel as described in Materials and Methods. Proteins were detected with a monoclonal anti-FLAG M2- horseradish peroxidase antibody as described under Materials and Methods. B. The image density of the immunoreactive bands on Kodak Biomax MR Film was quantitated using a Fluorskan 8900. Values represent the means ± S.E of triplicate transfections. Differing amounts of amino-terminal Met-FLAG-BAP-fusion protein (400-1200 fmol) were used as an internal standard for FLAG quantitation (R2=0.98 in the linear regression analysis).
at-RA metabolism
Expression of metabolically active CYP26A1 was verified by the production of specific nonpolar metabolites of at-RA in the medium. Figure 2 shows the HPLC profile of the organic phase extracted from the medium after cells were transfected with empty vector (A), CYP26A1 wild-type cDNA (B), and standards for at-RA metabolites (C). The identity of at-RA, 9-cis-RA, 13-cis-RA, 4-oxo-RA, 4-OH-RA, 18-OH-RA and 5, 6-epoxy-RA was verified by comparing the retention times and UV spectra with those of the available standard compounds. The elution order and retention times of metabolites were the same as those reported by Marill et al [17]. The time course for metabolite formation was examined in cells transfected with wild-type CYP26A1*1 after 2 min (to allow for cellular uptake), 1 hr, 2 hr, and 3 hr. The production of metabolites in the organic phase doubled between one and two hrs. Therefore, the 2 hr time point was chosen to compare catalytic differences between the wild-type and variant alleles. Figure 3 summarizes the results of one of two independent transfections of all allelic constructs, performed in triplicate. Results of both experiments showed that cells transfected the R173S (CYP26A1*2) allelic variant metabolize at-RA to a profile of metabolites (4-oxo-RA, 4-OH-RA, and 18-OH-RA) similar to those transfected with wild-type CYP26A1*1. The profile of metabolites in the cells and organic phase is identical to those reported by Kim et al [13]. In contrast, cells transfected with the F186L (CYP26A1*3) and C358R (CYP2C6A1*4) variants exhibited significantly lower production of at-metabolites compared to those transfected with wild-type CYP26A1*1 in both the organic phase from the media (Fig. 3A) and cellular fractions (Fig. 3B). Analysis of metabolites obtained from the organic phase (Fig. 3A) showed a 43% decrease in 4-OH-RA (p<0.05) production by cells transfected with CYP26A1*3, a 50% decrease in 4-oxo-RA (p<0.03), and a 73% decrease in 18-OH-RA (p<0.002) compared to that of cells transfected with wild-type CYP26A1*1. Similarly in the cellular fractions from this experiment (Fig. 3B), there was 58% decrease in 4-OH-RA (p<0.003) and an 89% decrease in 4-oxo-RA (p<0.005) compared to cells transfected with CYP26A1*1. 18-OH-RA was not detectable in the cellular fractions obtained from the CYP26A1*3 transfections, although it was detected in cells transfected with wild-type CYP26A1*1. Therefore, synthesis of the 18-OH-RA metabolite appears to be more dramatically affected by the amino acid change F186L than that of other metabolites. In the organic phase extracted from the medium from cells transfected with the CYP26A1*4(C358R) variant (Fig 3A), there was a 50% decrease in the production of 4-OH-RA (p<0.03), a 68% decrease in 4-oxo-RA (p<0.01), and a 46% decrease in 18-OH-RA (p<0.16) compared to that produced by cells transfected with wild-type CYP26A1*1. In the cellular fraction (Fig. 3B), the CYP26A1*4 variant exhibited a 39% decrease in production of 4-OH-RA (p<0.005), a 43% decrease in 4-oxo-RA (p<0.18), and a 52% decrease in 18-OH-RA production (p<0.02) compared to wild-type CYP26A1. The low production of metabolites by COS-1 cells transfected with CYP26A1 protein in COS-1 cells prevented the determination of kinetic parameters. Therefore, it is possible we underestimate the effects of the amino acid mutations.
Fig. 2. HPLC analysis of at-RA metabolites produced in COS-1 cells.

COS-1 cells were transfected with pcDNA3.1 control vector and pcDNA3.1- CYP26A1 wild-type vector for 48 hr. COS-1 cells were then washed with medium and incubated with 0.5 ml of DMEM containing 100 nM at-[3H] RA (1 mCi/ml, 44.5 Ci/mmol, PerkinElmer Life Sciences, USA), 100 μM cold at-RA and 0.2 mM NADPH. After 2 hr at 37°C, medium was removed and mixed with an equal volume of dichloromethane (DCM) /methanol (2:1, v/v) as described in Materials and Methods. A. The profile of the organic phase extracted from the pcDNA3.1 control vector transfection. B. The profile of the organic phase extracted from pcDNA3.1-CYP26A1 wild-type transfection. C. Separation of standard metabolites. 1, 4-oxo-RA; 2, 4-OH-RA; 3, 18-OH-RA, 4, 5,6-epoxy-RA; 5, 13-cis-RA; 6, 9-cis-RA; 7, at-RA; 8, unknown metabolite. Identity of the at-RA metabolite was verified by comparing retention times and UV spectra of the compound with the standard. Authentic standards for two of the metabolites (4-OH-RA or 18-OH-RA) were not available but identified based on the profile of metabolites reported previously [13, 18].
Fig. 3. at-RA metabolism by CYP26A1 wild-type and CYP26A1-variants in COS-1 cells.
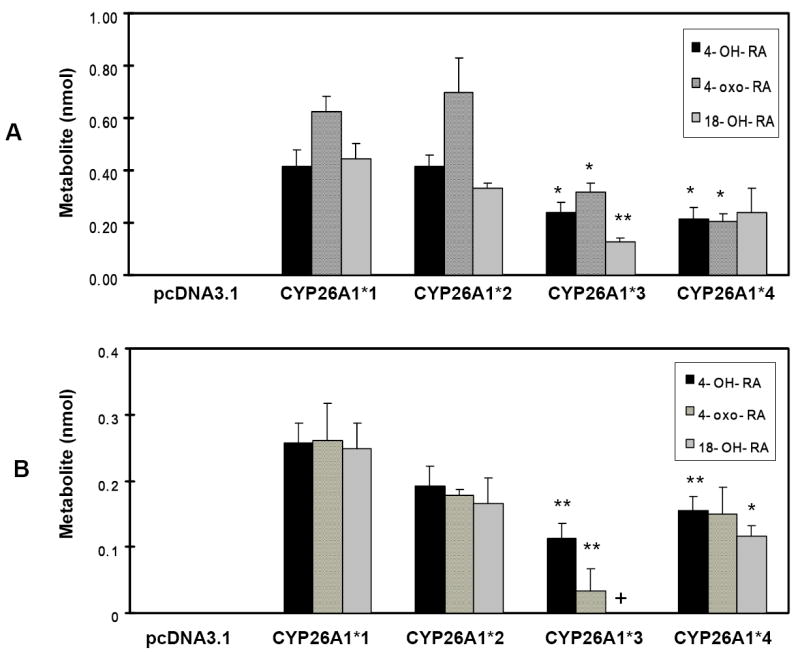
The pcDNA3.1, pcDNA3.1-CYP26A1*1 wt, and pcDNA3.1-CYP26A*2(R173S), CYP26A*3(F186L), CYP26A*4(C358R) variants were transfected into COS-1 cells in triplicate. After 48 hr transfection, cells were washed with medium and incubated with 0.5 ml of buffer containing 100 nM at-[3H] RA (1 mCi/ml, 44.5 Ci/mmol and 100 μM cold at-RA and 0.2 mM NADPH. After 2 hr at 37°C, the organic phase and cellular fractions were extracted as described in Materials and Methods. A. nmol at-RA metabolites obtained from organic phase. B. nmol at-RA metabolites obtained from cellular fraction. CYP26A*3 (F186L) and CYP26A*4 (C358R) allelic variants exhibited a significantly decreased activity compared with the pcDNA3.1-CYP26A1*1 wt vector control *, p < 0.05 and **, p<0.005. EV, empty pcDNA3.1 vector; +, not detected. Values represent the means ± S.E. of three independent transfections.
A second possible shorter transcriptional variant of human CYP26A1 mRNA had been predicted in the NCBI Reference Sequences (RefSeq).The full length CYP26A1 (RefSeq NM_000783) codes for a total of 497 amino acids. The shorter predicted variant (RefSeq NM_057157) would lack 69 amino acid in the N-terminus, resulting in a truncated protein of 428 amino acids which would still contain the heme binding region. Although functional studies for the full length CYP26A1 protein (isoform 1) have been reported [26], there are no functional studies reported for the shorter variant. In the present study, we constructed the two cDNA variants and transfected both into COS-1 cells. When cells transfected with the truncated variant were incubated with radioactive at-RA for a 2 hour period, there was no change in radioactivity in the aqueous phase compared to cells transfected with the empty vector alone (data not shown). In contrast, there was a marked increase in radioactivity in the aqueous phase in with the longer variant. This data suggests that the shorter variant may not be functional.
Discussion
Inherited genetic polymorphisms in cytochrome P450 genes are one of the major reasons for inter-individual variation in the metabolism of clinical drugs, environmental chemicals, and endogenous compounds [27, 28]. In humans, the CYP26A gene family metabolizes retinoids [5-7]. Among the three members of this subfamily, CYP26A1appears to be essential for maintaining retinoic acid homeostasis in the embryo [7, 12, 29]. The present study identifies genetic variants of CYP26A1 including three new coding changes in the protein. Importantly, our results indicate that new F186L (CYP26A1*3) and C358R (CYP26A1*4) coding variants of CYP26A1 are significantly defective in metabolizing at-RA compared to wild-type in recombinant studies. If these differences occur in vivo, these defects in CYP26A1 could have an important impact on embryonic development. Knockouts of Cyp26a1 in mice are known to be embryonic lethal and to cause caudal agenesis and malformation of the lumbosacral elements [7, 12]. These abnormalities resemble the teratogenic effects of excess retinoic acid in mice [30].
In humans, a rare congenital defect known as caudal regression syndrome involving severe malformations of the spine and spinal cord occurs in 1/7,500 births [31, 32]. A recent study by De Marco and coworkers [33] reported a number of SNPS of CYP26A1 in 49 patients with caudal regression syndrome and 131 controls; however, the incidence of these SNPs did not differ between patients and controls. Three of the SNPS were also found in our study. These included the IVS1+10 change, two SNPs at IVS4+8AG>GA, and a silent base pair change in the cDNA coding for G438G. DeMarco and coworkers [33] reported that the SNP at IVS1+10 affected splicing of a CYP26A1 minigene fragment using an in vitro expression system in COS-1 cells [33]. They suggested this change might cause initiation downstream at the second initiation site. We found the change in intron 10 to be frequent in African-Americans (8/30 alleles). The high incidence of the intron 1 SNP in African-Americans would seem surprising if this were a null defect. However, if this SNP affects splicing in vivo, it could cause decreased expression of the C358R allelic protein. De Marco and coworkers [33] also reported that the IVS4+8 change appeared to produce a moderate decrease in splicing of a CYP26A1 minigene transcript in vitro. Both the coding and intron mutations found in CYP26A1 will require further study in developmental disorders.
No crystal structure if available for CYP26A1 protein. To gain insight on possible structural and functional effects of the amino acid changes observed in the new CYP26A1 alleles found in the present study, homology modeling and sequence alignment were used. Currently, four x-ray crystal structures have been determined for mammalian CYP450s: CYP3A4 [22], CYP2C5 [19], CYP2C8 [20] and CYP2C9 [21]. Using the Clustal-X [34] and T-coffee alignment packages [35], the CYP26A1 sequence was aligned with the sequences of the above mentioned mammalian CYP450 proteins. The alignment data are given in Figure 4. As assigned by Williams et al. [19], helices A-L and β-sheets were identified and marked to facilitate the following discussion. The three residues R173, F186, and C358 found in the present study are marked in bold. The observed sequence homology between CYP26A1 and the mammalian CYP proteins was 39-40%. A comparison of CYP26A1 with these four structures suggests this approach is useful for structure-function relationship predictions [36]. In all four X-ray crystal structures of mammalian CYP proteins, the corresponding amino acid for F186 in CYP26A1 is an identical phenylalanine residue conserved at the end of the E-helix (Fig 4). This residue in the X-ray structures is involved in the formation of a hydrophobic core with the conserved phenylalanine residue at 299 in CYP26A1 (Fig. 5). This hydrophobic interaction may play a key role in the correct positioning of the I-helix, since the F299 is in the I-helix (Fig. 5). There is a high sequence identity in the I-helix residues of the CYP proteins shown in Figure 4. In fact, Hasemann et al. [37] proposed that the conserved Thr (T315 in CYP26A1) and neighboring acidic residue (E314 in CYP26A1), along with the water molecules in the grove of the I-helix, are believed to comprise the essential elements of a proton transfer network [37]. For example, the proton in CYP450BM3 is proposed to be transferred to the heme from the side chain of the conserved Thr, which is protonated by a water molecule [38]. The F186L change in the CYP26A1 protein may possibly lead to a weakening of the hydrophobic interaction that is required to keep the I-helix in the correct position of the water channel [36].
Fig. 4. Sequence alignments of CYP3A4, CYP2C5, CYP2C8, CYP2C9 and CYP26A1.

CYP26A1 coding variants are indicated in bold. Alignments were done using Clustal-X and T-Coffee alignment programs. Helices A-L are identified and marked along with beta-sheets and short helices as assigned by Williams et al [29]. Residues affected by polymorphism have been highlighted.
Fig. 5. A ribbon diagram showing a putative template structure of CYP26A1 based on the known crystal structure of CYP2C5.
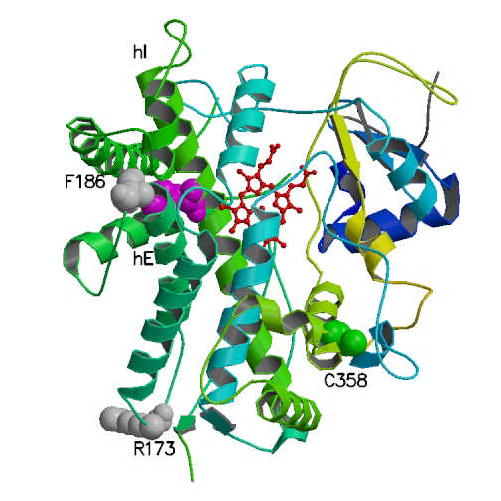
The heme moiety is shown in red. The F299 residue in I-helix is shown between heme and F186. The residues for which coding mutations were identified are indicated using space-filling.
R173 is observed in a loop region between helices D and E in Figure 5. However, the corresponding position in CYP2C5, CYP2C8 and CYP2C9 is a Ser, while it is a Thr in CYP3A4. This is consistent with our observation that the R173S change would not have a significant influence on the function of CYP26A1. Sequence alignment indicated that the C358R residue is in the highly conserved segment of the K-helix (YXXOVXXE in which O represents the position of Cys, and it is a variable residue in the other CYP crystal structures (Ala and Met). However, this residue is in a position that can interact with a small helical segment of residues composed of the conserved FXPXXF (residues 414-419 in CYP26A1 and the small helix in Figure 5 next to C358). This small helix is the only structured segment in the region between residues 404-440. Interestingly, it is inferred from the crystal structures that weakening of the helix-helix interaction could introduce large fluctuations in the spatial arrangements of this region. We also noticed that in the X-ray crystal structures, the C-terminal segment of these residues is in direct contact with the heme moiety. Thus, one can speculate that the C358R could cause a conformational change responsible for the decreased catalytic activity seen in this study despite its distal location from the active site.
Previous studies have reported metabolism of at-RA by the full length CYP26A1 containing 497 amino acids (RefSeq NM_000783) [26]. A potential second shorter transcript (RefSeq NM_057157) lacking 69 amino acids has been predicted. However, preliminary transfection experiments in the present study indicate that this variant has little or no activity. Therefore this putative variant was not used for mutagenesis studies.
In the humans genome, there are three members of the CYP26 subfamily. CYP26A1 and CYP26B1,metabolize at-RA>>9-cisRA [8]. CYP2C6C1 metabolizes both at- and 9-cis-RA [25]. Among these three genes, CYP26A1 has been the most extensively studied in humans and mice. CYP26A1 mRNA expression in humans has been reported by microarray analysis in various tissues, such as intestinal, liver, and endothelial tissues [39-42], [http://genome.ucsc.edu/cgi-bin/hgNear]. Cyp26a1 is expressed early in the mouse embryo in a stage and region-specific manner: in the neural plate, neural crest cells for cranial ganglia, hindgut and tailbud mesoderm [7], Cyp26a1 knock-out mice exhibit embryonic lethality, spina bifida, severe caudal agenesis, abnormalities of the kidneys and hindgut, and hindbrain abnormalities [12, 29], These data suggest that expression of CYP26B1 and CYP26C1 is not sufficient to compensate for the absence of CYP26A1, and that a significant amount of RA metabolism during embryonic development is dependent upon the expression of CYP26A1.
The contribution of other cytochrome P450s to RA metabolism would differ depending on the tissue and the stage of development. Among the other cytochrome P450 enzymes which metabolize at-RA in the liver, recombinant CYP3A7 was identified as showing greater formation of 4-OH-RA from at-RA than CYP3A5>CYP2C8>CYP3A4 [18]. However, contribution depends on relative concentration. CYP3A7 is highly expressed in fetal liver [43]. Studies with adult human liver microsomes have estimated that CYP2C8 and CYP3A4 account for 50% of the 4-hydroxylation of all-trans-RA in liver, while CYP2C9 accounts for ~10% [44]. There are no studies comparing turnover numbers of CYP26 enzymes with any of the classic drug metabolizing CYP enzymes, but levels of CYP26 in human liver are probably much lower than those of CYP2C8, CYP3A4, and CYP2C9. However, CYP26A1 activity can be induced by RA [6, 13, 39, 42] resulting in variations in at-RA metabolism in humans. When at-RA is used therapeutically in the treatment of various cancers, plasma clearance is increased contributing to tolerance [45]. Since at-RA has been used for the treatment of acute promyelotic leukemia [40, 41] and uterine cervical cancer [3, 39, 46], nonlethal mutations might counteract tolerance. Therefore, CYP26A1 could make a contribution to hepatic metabolism of this compound in individuals treated with retinoids.
Polymorphisms in human CYP26A1 would be predicted to affect embryonic development and null polymorphisms might affect viability of the fetus. CYP26A1 maps to chromosome 10q23-24 [47]. Several disease loci including hand-split foot and infantile onset spinocerebellar atrophy have been mapped to this region. In humans, a syndrome known as caudal regression syndrome is characterized by congenital sacral agenesis, and involvement of the nervous system, urinary, and gastrointestinal system [31-33]. De Marco and coworkers [33] have suggested that the most severe forms of caudal regression syndrome could resemble the phenotype in the Cyp26a1-null mouse.
Genetic polymorphisms in CYP26A1 would be expected to result in serious outcomes. Importantly, our present study reports coding polymorphisms in the human CYP26A1 gene as well as changes in introns. Coding polymorphisms could produce partially defective alleles. Based on data with recombinant enzymes, individuals with the CYP26A1 defective alleles F186L and C358R could be defective in the metabolism of at-RA. The identification of genetic variants of human CYP26A1 which affect at-RA metabolism will particularly useful in clinical studies of various developmental disorders. at-RA has been used for the treatment of acute promyelotic leukemia [40, 41] and uterine cervical cancer [3, 39, 46], nonlethal mutations might affect blood levels of retinoids.
Acknowledgments
The authors wish to thank the NIH Fellows Editorial Board for editorial comments concerning the manuscript.
This research was supported in part by the intramural division of the NIH, National Institute of Environmental Health Sciences. Work at LLNL was performed under the auspices of the US DOE by LLNL; contract No. W-7405-ENG-48 and supported by interagency agreement Y1-ES-8054-05 from NIEHS.
References
- 1.Perlmann T. Retinoid metabolism: a balancing act. Nat Genet. 2002;31(1):7–8. doi: 10.1038/ng877. [DOI] [PubMed] [Google Scholar]
- 2.Clagett-Dame M, DeLuca HF. The role of vitamin A in mammalian reproduction and embryonic development. Annu Rev Nutr. 2002;22:347–81. doi: 10.1146/annurev.nutr.22.010402.102745E. [DOI] [PubMed] [Google Scholar]
- 3.Abu J, Batuwangala M, Herbert K, Symonds P. Retinoic acid and retinoid receptors: potential chemopreventive and therapeutic role in cervical cancer. Lancet Oncol. 2005;6(9):712–20. doi: 10.1016/S1470-2045(05)70319-3. [DOI] [PubMed] [Google Scholar]
- 4.Horton C, Maden M. Endogenous distribution of retinoids during normal development and teratogenesis in the mouse embryo. Dev Dyn. 1995;202(3):312–23. doi: 10.1002/aja.1002020310. [DOI] [PubMed] [Google Scholar]
- 5.Niederreither K, Abu-Abed S, Schuhbaur B, Petkovich M, Chambon P, Dolle P. Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat Genet. 2002;31(1):84–8. doi: 10.1038/ng876. [DOI] [PubMed] [Google Scholar]
- 6.White JA, Guo YD, Baetz K, Beckett-Jones B, Bonasoro J, Hsu KE, Dilworth FJ, Jones G, Petkovich M. Identification of the retinoic acid-inducible all-trans-retinoic acid 4-hydroxylase. J Biol Chem. 1996;271(47):29922–7. doi: 10.1074/jbc.271.47.29922. [DOI] [PubMed] [Google Scholar]
- 7.Fujii H, Sato T, Kaneko S, Gotoh O, Fujii-Kuriyama Y, Osawa K, Kato S, Hamada H. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. Embo J. 1997;16(14):4163–73. doi: 10.1093/emboj/16.14.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White JA, Ramshaw H, Taimi M, Stangle W, Zhang A, Everingham S, Creighton S, Tam SP, Jones G, Petkovich M. Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc Natl Acad Sci U S A. 2000;97(12):6403–8. doi: 10.1073/pnas.120161397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chambon P. A decade of molecular biology of retinoic acid receptors. Faseb J. 1996;10(9):940–54. [PubMed] [Google Scholar]
- 10.Fiorella PD, Giguere V, Napoli JL. Expression of cellular retinoic acid-binding protein (type II) in Escherichia coli. Characterization and comparison to cellular retinoic acid-binding protein (type I) J Biol Chem. 1993;268(29):21545–52. [PubMed] [Google Scholar]
- 11.Pijnappel WW, Hendriks HF, Folkers GE, van den Brink CE, Dekker EJ, Edelenbosch C, van der Saag PT, Durston AJ. The retinoid ligand 4-oxo-retinoic acid is a highly active modulator of positional specification. Nature. 1993;366(6453):340–4. doi: 10.1038/366340a0. [DOI] [PubMed] [Google Scholar]
- 12.Abu-Abed S, Dolle P, Metzger D, Beckett B, Chambon P, Petkovich M. The retinoic acid-metabolizing enzyme, CYP26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001;15(2):226–40. doi: 10.1101/gad.855001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SY, Adachi H, Koo JS, Jetten AM. Induction of the cytochrome P450 gene CYP26 during mucous cell differentiation of normal human tracheobronchial epithelial cells. Mol Pharmacol. 2000;58(3):483–90. doi: 10.1124/mol.58.3.483. [DOI] [PubMed] [Google Scholar]
- 14.Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998;8(12):1229–31. doi: 10.1101/gr.8.12.1229. [DOI] [PubMed] [Google Scholar]
- 15.Barnes HJ, Arlotto MP, Waterman MR. Expression and enzymatic activity of recombinant cytochrome P450 17 alpha-hydroxylase in Escherichia coli. Proc Natl Acad Sci U S A. 1991;88(13):5597–601. doi: 10.1073/pnas.88.13.5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SJ, Bell DA, Coulter SJ, Ghanayem B, Goldstein JA. Recombinant CYP3A4*17 is defective in metabolizing the hypertensive drug nifedipine, and the CYP3A4*17 allele may occur on the same chromosome as CYP3A5*3, representing a new putative defective CYP3A haplotype. J Pharmacol Exp Ther. 2005;313(1):302–9. doi: 10.1124/jpet.104.078758. [DOI] [PubMed] [Google Scholar]
- 17.Lee SJ, Usmani KA, Chanas B, Ghanayem B, Xi T, Hodgson E, Mohrenweiser HW, Goldstein JA. Genetic findings and functional studies of human CYP3A5 single nucleotide polymorphisms in different ethnic groups. Pharmacogenetics. 2003;13(8):461–72. doi: 10.1097/00008571-200308000-00004. [DOI] [PubMed] [Google Scholar]
- 18.Marill J, Cresteil T, Lanotte M, Chabot GG. Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol Pharmacol. 2000;58(6):1341–8. doi: 10.1124/mol.58.6.1341. [DOI] [PubMed] [Google Scholar]
- 19.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5(1):121–31. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 20.Schoch GA, Yano JK, Wester MR, Griffin KJ, Stout CD, Johnson EF. Structure of human microsomal cytochrome P450 2C8. Evidence for a peripheral fatty acid binding site. J Biol Chem. 2004;279(10):9497–503. doi: 10.1074/jbc.M312516200. [DOI] [PubMed] [Google Scholar]
- 21.Williams PA, Cosme J, Angove HC, Matak Vinkovic D, Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424(6947):464–8. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 22.Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF. The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J Biol Chem. 2004;279(37):38091–4. doi: 10.1074/jbc.C400293200. [DOI] [PubMed] [Google Scholar]
- 23.Antonarakis SE. Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Hum Mutat. 1998;11(1):1–3. doi: 10.1002/(SICI)1098-1004(1998)11:1<1::AID-HUMU1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 24.Loudig O, Babichuk C, White J, Abu-Abed S, Mueller C, Petkovich M. Cytochrome P450RAI(CYP26) promoter: a distinct composite retinoic acid response element underlies the complex regulation of retinoic acid metabolism. Mol Endocrinol. 2000;14(9):1483–97. doi: 10.1210/mend.14.9.0518. [DOI] [PubMed] [Google Scholar]
- 25.Taimi M, Helvig C, Wisniewski J, Ramshaw H, White J, Amad M, Korczak B, Petkovich M. A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J Biol Chem. 2004;279(1):77–85. doi: 10.1074/jbc.M308337200. [DOI] [PubMed] [Google Scholar]
- 26.White JA, Beckett-Jones B, Guo YD, Dilworth FJ, Bonasoro J, Jones G, Petkovich M. cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J Biol Chem. 1997;272(30):18538–41. doi: 10.1074/jbc.272.30.18538. [DOI] [PubMed] [Google Scholar]
- 27.Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286(5439):487–91. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 28.Evans WE, Johnson JA. Pharmacogenomics: the inherited basis for interindividual differences in drug response. Annu Rev Genomics Hum Genet. 2001;2:9–39. doi: 10.1146/annurev.genom.2.1.9. [DOI] [PubMed] [Google Scholar]
- 29.Sakai Y, Meno C, Fujii H, Nishino J, Shiratori H, Saijoh Y, Rossant J, Hamada H. The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001;15(2):213–25. doi: 10.1101/gad.851501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kessel M, Gruss P. Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell. 1991;67(1):89–104. doi: 10.1016/0092-8674(91)90574-i. [DOI] [PubMed] [Google Scholar]
- 31.Cama A, Palmieri A, Capra V, Piatelli GL, Ravegnani M, Fondelli P. Multidisciplinary management of caudal regression syndrome (26 cases) Eur J Pediatr Surg. 1996;6(Suppl 1):44–5. [PubMed] [Google Scholar]
- 32.Renshaw TS. Sacral agenesis. J Bone Joint Surg Am. 1978;60(3):373–83. [PubMed] [Google Scholar]
- 33.De Marco P, Merello E, Mascelli S, Raso A, Santamaria A, Ottaviano C, Calevo MG, Cama A, Capra V. Mutational screening of the CYP26A1 gene in patients with caudal regression syndrome. Birth Defects Res A Clin Mol Teratol. 2006;76(2):86–95. doi: 10.1002/bdra.20225. [DOI] [PubMed] [Google Scholar]
- 34.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Notredame C, Higgins DG, Heringa T. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302(1):205–17. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 36.Gorokhov A, Negishi M, Johnson EF, Pedersen LC, Perera L, Darden TA, Pedersen LG. Explicit water near the catalytic I helix Thr in the predicted solution structure of CYP2A4. Biophys J. 2003;84(1):57–68. doi: 10.1016/S0006-3495(03)74832-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hasemann CA, Kurumbail RG, Boddupalli SS, Peterson JA, Deisenhofer J. Structure and function of cytochromes P450: a comparative analysis of three crystal structures. Structure. 1995;3(1):41–62. doi: 10.1016/s0969-2126(01)00134-4. [DOI] [PubMed] [Google Scholar]
- 38.Warman AJ, Roitel O, Neeli R, Girvan HM, Seward HE, Murray SA, McLean KJ, Joyce MG, Toogood H, Holt RA, Leys D, Scrutton NS, Munro AW. Flavocytochrome P450 BM3: an update on structure and mechanism of a biotechnologically important enzyme. Biochem Soc Trans. 2005;33(Pt 4):747–53. doi: 10.1042/BST0330747. [DOI] [PubMed] [Google Scholar]
- 39.Villani MG, Appierto V, Cavadini E, Valsecchi M, Sonnino S, Curley RW, Formelli F. Identification of the fenretinide metabolite 4-oxo-fenretinide present in human plasma and formed in human ovarian carcinoma cells through induction of cytochrome P450 26A1. Clin Cancer Res. 2004;10(18 Pt 1):6265–75. doi: 10.1158/1078-0432.CCR-04-0655. [DOI] [PubMed] [Google Scholar]
- 40.Idres N, Marill J, Chabot GG. Regulation of CYP26A1 expression by selective RAR and RXR agonists in human NB4 promyelocytic leukemia cells. Biochem Pharmacol. 2005;69(11):1595–601. doi: 10.1016/j.bcp.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 41.Ozpolat B, Mehta K, Tari AM, Lopez-Berestein G. all-trans-Retinoic acid-induced expression and regulation of retinoic acid 4-hydroxylase (CYP26) in human promyelocytic leukemia. Am J Hematol. 2002;70(1):39–47. doi: 10.1002/ajh.10099. [DOI] [PubMed] [Google Scholar]
- 42.Ozpolat B, Mehta K, Lopez-Berestein G. Regulation of a highly specific retinoic acid-4-hydroxylase (CYP26A1) enzyme and all-trans-retinoic acid metabolism in human intestinal, liver, endothelial, and acute promyelocytic leukemia cells. Leuk Lymphoma. 2005;46(10):1497–506. doi: 10.1080/10428190500174737. [DOI] [PubMed] [Google Scholar]
- 43.Lacroix D, Sonnier M, Moncion A, Cheron G, Cresteil T. Expression of CYP3A in the human liver--evidence that the shift between CYP3A7 and CYP3A4 occurs immediately after birth. Eur J Biochem. 1997;247(2):625–34. doi: 10.1111/j.1432-1033.1997.00625.x. [DOI] [PubMed] [Google Scholar]
- 44.McSorley LC, Daly AK. Identification of human cytochrome P450 isoforms that contribute to all-trans-retinoic acid 4-hydroxylation. Biochem Pharmacol. 2000;60(4):517–26. doi: 10.1016/s0006-2952(00)00356-7. [DOI] [PubMed] [Google Scholar]
- 45.Marill J, Idres N, Capron CC, Nguyen E, Chabot GG. Retinoic acid metabolism and mechanism of action: a review. Curr Drug Metab. 2003;4(1):1–10. doi: 10.2174/1389200033336900. [DOI] [PubMed] [Google Scholar]
- 46.Osanai M, Petkovich M. Expression of the retinoic acid-metabolizing enzyme CYP26A1 limits programmed cell death. Mol Pharmacol. 2005;67(5):1808–17. doi: 10.1124/mol.104.005769. [DOI] [PubMed] [Google Scholar]
- 47.White JA, Beckett B, Scherer SW, Herbrick JA, Petkovich M. P450RAI (CYP26A1) maps to human chromosome 10q23-q24 and mouse chromosome 19C2-3. Genomics. 1998;48(2):270–2. doi: 10.1006/geno.1997.5157. [DOI] [PubMed] [Google Scholar]