

Abstract
Considering carcinogenesis as a microevolutionary process, best described in the context of metapopulation dynamics, provides the basis for theoretical and empirical studies that indicate it is possible to estimate the relative contribution of genetic instability and selection to the process of tumor formation. We show that mutational load distribution analysis (MLDA) of DNA found in pancreatic fluids yields biometrics that reflect the interplay of instability, selection, accident, and gene function that determines the eventual emergence of a tumor. An in silico simulation of carcinogenesis indicates that MLDA may be a suitable tool for early detection of pancreatic cancer. We also present evidence indicating that, when performed serially in individuals harboring a p16 germ-line mutation bestowing a high risk for pancreatic cancer, MLDA may be an effective tool for the longitudinal assessment of risk and early detection of pancreatic cancer.
Keywords: biomarkers, cancer, early detection, modeling, microevolution
Studies of advanced colorectal carcinomas in humans have suggested the ecological theory of metapopulation dynamics contributes to the understanding of tumor microheterogeneity, but ecological theory has seldom been applied to the understanding of the initial phases of tumor formation (1). For the common epithelial tumors of humans, the period of tumor development spans a decade or more (2); during this time, tissues are constantly under the assault of environmental mutagens, but damage is largely neutralized by DNA repair mechanisms (3). Studies of nonneoplastic tissues in asymptomatic individuals that are unlikely to develop tumors show the presence of mutations (4, 5) and, even when occurring in cancer genes, mutations appear to be cleansed from the cells constituting a tissue. This constant low level of mutation and cleansing produces a random fluctuation of mutations that can be registered with sensitive detection technologies.
Under physiological conditions, the structural and functional integrity of tissues is ensured by a compartmental organization (6) whose spatial constraints regulate the coexistence of physiological clonal patches maintained by stem cells. The progeny of the stem cells undergo differentiation and eventually engage the apoptotic program. The introduction of mutation and aneuploidy in tissue stem cells (7, 8) alters the ecology of the clonal cell populations that compose a tissue and create a collection of subpopulations (metapopulations) of the same cell type occupying separate patches of a subdivided habitat. The widely accepted ecological concept that disturbances (exogenous agents of mortality) have pronounced effects on diversity (9, 10) suggests that repeated insults that affect tissues (e.g., repeated chronic inflammation, repeated exposure to toxins) are likely to influence the metapopulation dynamics of the clonal patches composing them. In ecological thought, biodiversity was originally considered to be highest in undisturbed systems. An alternative proposal, the “intermediate disturbance hypothesis,” proposes that diversity is highest when disturbance occurs neither too rarely nor too frequently or at an intensity that is neither very large nor very small. This was originally proposed by Connell (11) and supported by a number of subsequent empirical studies (10, 12, 13).
Under these circumstances, the three conditions necessary for an evolutionary process to occur, variation (mutation, epigenetic alterations), competition (differential fitness), and replication, are met, and hence carcinogenesis can be regarded as a microevolutionary process acting on a metapopulation of cells. The combination of mutations and epigenetic changes occurring in a small subset of several hundred cancer genes leads to the emergence of the complex cellular behavior that characterizes malignant tumors (14). It is thought that the accumulation of mutations in tumor cells is greatly enhanced by genetic instability, a property distinguishing tumor from normal cells (15, 16). Despite genetic instability and the so-called “mutator phenotype” of tumor cells, common tumor types are defined by a limited set of recurring genetic alterations [compare the Cancer Genome Anatomy Project (CGAP)] that represent the most frequent final states of an evolutionary process about whose exact genealogy we remain largely ignorant. For each of the altered cancer genes known to be responsible for the emergence of a tumor, several alleles are found in a cohort of fully developed malignant tumors (17) and, although each tumor harbors a dominant mutated allele, recent studies suggest that microheterogeneity exists in fully developed tumors (18, 19). We reasoned that, under a metapopulation-based view of carcinogenesis, the frequency of a given allele will correspond to the number of cells bearing the particular allele. Thus, examined over time, the mutational spectrum (the relative frequencies of the different alleles for each altered cancer gene) will reflect the degree of instability and strength of selection as carcinogenesis unfolds. As the selective forces that shape the tumor genotype exert their influence, the distribution of the relative frequencies of mutated alleles will depart from randomness. We posit that the sequential analysis of the mutational spectra at loci involved in the pathogenesis of tumors arising in any given tissue [mutational load distribution analysis (MLDA)] reflects the dynamic changes resulting from genetic instability and the strength of selection responsible for the emergence of a tumor.
Here, we present empirical data obtained in human pancreatic juice showing that MLDA profiles can be obtained from human pancreatic juice, and that two metrics derived from the MLDA profile, namely the total mutational load (TML) and the highest allele (HDA), can be used for risk measurement and early detection of pancreatic cancer. Studies of MLDA profiles in members of families predisposed to pancreatic carcinoma show the potential of MLDA as a putative risk assessment tool. In silico modeling illustrates how MLDA can be used for early detection of cancer.
Results
MLDA of Pancreatic Juice.
We obtained empirical data in human subjects by analyzing the soluble DNA found in pancreatic or duodenal juice after stimulation with secretin. An oligonucleotide zip-code microarray with rolling circle-amplification signal enhancement enables the simultaneous interrogation of tissue fluids for a moderate number of alleles (20) and the detection of low-prevalence allelic variants. Alleles of both the Ki-ras and p53 genes are well suited for MLDA of pancreatic juice, because both are often found to be altered in a high proportion of pancreatic carcinomas (21). From the mutational spectrum of these two genes, we selected 22 somatic point mutations (Fig. 1) that were both prevalent enough to be informative and technically compatible for being simultaneously interrogated in an RCA enhanced zip-array format.
Fig. 1.

MLDA profiles in three distinct populations. Each row represents one subject; Top is composed of subjects with no known pancreatic pathology (n = 9), Middle groups patients with chronic pancreatitis at increased risk for pancreatic carcinoma (n = 12), and Bottom depicts the results obtained in patients with pancreatic carcinoma (n = 21). The number above the triplet sequences identifies the codon. Each column represents one allele, and the color in each box denotes the proportion of each allele constituting the population of molecules encoding Ki-ras p21 or the p53 protein. Although many alleles in cancer patients were >5%, the actual representation is cut off to depict the dynamic range of values between 0.000% and 5.000%. The enhanced SI Fig. 5 shows the results from the addition of the fractional values for an individual gene (Ki-Ras or p53) and the total mutational load resulting from the addition of all fractional values (Ki-Ras and p53). The actual fractional values for the each allele are provided http://genecube.med.yale.edu:8080/montebello.
We initially tested the ability of MLDA to discriminate among three distinct cohorts: subjects without known pancreatic pathology or risk factors for pancreatic cancer (n = 9), patients thought to be at increased risk for pancreatic cancer because of repeated bouts of pancreatitis (n = 12), and patients with clinically evident pancreatic carcinoma (n = 21). Fig. 1 shows the mutational profiles for each of the cases examined [see supporting information (SI) Fig. 5 for an enhanced Fig. 1 that includes the aggregate (allelic) and total mutational load values]. The total mutational load does not overlap among the three groups of cases (normal, 5.8–7.3; pancreatitis, 12.3–21.0; and cancer, 22.8–67.9) (Fig. 2), and we conjecture that it reflects the degree of genetic instability present in the population of pancreatic cells. The observed differences in aggregate and total mutational load values among the three groups are statistically significant with a P value <0.0001 (Kruskal–Wallis test) for Ki-ras, p53, and the sum of both loci. The predictive value of the TML metric was assessed with a supervised classification method based on multinomial regression (a generalization of logistic regression for more than two groups) by using the 42 cases studied. Perfect classification of the individuals into their diagnostic groups was possible with the model. Sensitivity was 100% (95% C.I., 84.4–100) for cancer and 100% (95% C.I., 75.7–100) for pancreatitis. Specificity was 100% (95% C.I., 70.1–100). Because the sample size was small, bootstrap techniques were used to estimate the expected misclassification rate under similar conditions. The 0.632+ estimate of the misclassification error was 2% (based on 1,000 replicates) (22). The inspection of the bootstrap replicates revealed that, of the 42 samples, three were often misclassified: sample 9B, with the lowest total mutational load in the pancreatitis category, was usually assigned a high probability to belong to the “normals”; sample 18B, with the highest total mutational load in the pancreatitis group, was usually misclassified as pancreatic cancer; and sample Y10, with the lowest total mutational load in the carcinoma group, was usually misclassified as pancreatitis.
Fig. 2.

Total mutational load. Individual cases (arrayed along the x axis) are presented by increasing total mutational load values (y axis). The total mutational load parameter derived from the MLDA profiles separates the three groups of subjects with a narrow band of overlap between pancreatitis and cancer. The increase in total mutational load can be interpreted as a reflection of progressive genetic instability.
To identify whether a subset of specific alleles was equally predictive, we explored stepwise methods for selection of alleles and methods based on classification trees (random forests analysis) (23). The misclassification error rates were 41% for the stepwise procedure based on multinomial regression and 19% for the random forest, showing that none of the alternative classification strategies improved the results. Inspection of the selected alleles in different bootstrap samples revealed great variability, suggesting that the information provided by each allele is similar, and that a subset of alleles more informative than the rest is unlikely to exist. For discrimination purposes, the addition of the contribution of each allele into the TML metric is the most informative. Qualitative inspection of the MLDA profiles (Fig. 1) reveals two scenarios underlying a high TML: it can be due to uniformly high values distributed throughout most of the alleles examined (case 7B) or to a very high predominant allele (case 4L), presumably produced by selection acting on background genetic instability.
The second metric derived from MLDA is the highest allele value in a profile [highest dominant allele (HDA)]. Among the subjects with no known pancreatic pathology, the highest allele was between 0.7 and 1.2; for cases with pancreatitis, the highest HDA value was between 1.1 and 2.7; and for cases with carcinoma, the values were between 2.9 and 37.3. Although the HDA metric was differentially distributed through the three distinct clinical groups, its discriminating capability was found to be lower than TML (the estimate of the misclassification error rate was 5%). It is noteworthy that six (29%) cancer samples show a pattern of high total mutational load without one dominant allele but several alleles with increased mutation frequency and relatively low highest value. Despite the slightly inferior performance of this second metric, the correlation of TML with HDA and with the standard deviation among the individual allele values indicates the validity of these additional metrics (compare SI Fig. 6).
The observation that MLDA-derived metrics are effective in identifying individuals belonging to three different risk groups for pancreatic cancer motivated us to analyze pancreatic juice from members of families predisposed to pancreatic cancer by a germ-line p16 mutation. Blinded examination of the mutational profiles and the TML values in the 16 samples examined showed two homogeneous groups: a “normal-like” and a “pancreatitis-like” pattern (see Fig. 3 and legend). After unblinding the series of samples, two individuals with normal p16 genotype showed profiles in the normal “no-risk” zone, and four individuals, harboring a p16 germ-line mutation and belonging to three independent families, turned out to have iterative studies that provided data on the time-dependent variation of MLDA-derived metrics. The random fluctuation of the values for specific alleles obtained at different times can be appreciated in the serial samples of individuals exhibiting a normal-like pattern as well as in some of the alleles in pancreatitis-like patterns. Of the six individuals with a p16 germ-line mutation, two had initial low-risk samples (normal-like pattern) and moved to the high-risk category (pancreatitis-like pattern), two were classified as “at risk” and remained in this class, and two had a single time-point study (see SI Fig. 7 for an enhanced Fig. 3). It is important to note that, for a given individual, the alleles that show the highest values vary from time point to time point. However, in two instances, the ascending allele remains identical, raising the possibility that the “persistence of dominance” may constitute an additional qualitative predictive factor for the development of cancer. These observations underscore the value of using a wide mutational spectrum for each locus interrogated by MLDA. Not only is it impossible to predict which of the alleles will be driven by selection to be ultimately and predominantly expressed in the invasive tumor state, but also the allele that is dominant within the risk boundaries may vary because of chance events. A disturbance or a deleterious mutation can eliminate an expanding oncodeme(s) and thus alter the subsequent MLDA pattern (see below and Fig. 4).
Fig. 3.

Longitudinal MLDA profiling of a population at risk. MLDA profiles from subjects belonging to different families with increased risk for pancreatic cancer due to inherited p16 mutation. Notation is: Family.Subject.Serial sample (SI Fig. 7 provides the enhanced version of the figure indicating the age at which the sample was obtained and the genotype. Two patterns can be recognized in the samples: normal- (e.g., family 5, sample N5) and pancreatitis-like patterns (e.g., family 4, sample N4). For subjects with sequential samples, the profiles change with time from normal- to pancreatitis-like, indicating an increase in risk. Note that in instances when the risk increases, the alleles with high values do not necessarily persist. The total load for Ki-ras and p53, the age at time of sampling, and the p16 genotype are provided for each subject on the right.
Fig. 4.

Simulated mutational load over time. Graphic representation of the MLDA values obtained at each time step in runs for the three classes of outcome. Time series of mutational load at each 25th step over the entire 5,000 iterations (200 measurements per run). Rows represent the mutational load at a single time point proceeding from bottom (t = 0) to top (t = 5,000). Columns represent the mutational load for 10 alleles of three genes as in Fig. 1. (Top) Low risk (undisturbed); (Middle) high risk (no tumor formation for duration of simulation); and (Bottom) tumor. The transition from “low risk” to groups Middle or Bottom is determined by setting the disturbance parameter. Specific synthetic histories are classified a posteriori into “high risk” or “tumor” by outcome. The differences in MLDA profile are clearly apparent. For the run ending in tumor, MLDA provides a time zone of early detection. Note the similarity of the risk and tumor profiles during the early time period preceding the “early detection band,” The simulation indicates that the progressive increase in risk identifies the individual runs marked by the emergence of a “tumor” and enables the prediction of the tumor class.
In Silico Modeling.
The aim of our simulation is to show that a relatively simple stochastic model can provide a plausible explanation for the dynamics and distribution of mutational load and provide insight into the relation of parameters reflecting metapopulation dynamics to the emergence of tumors and therefore to the measure of cancer risk. The model, based on a microevolutionary view of carcinogenesis, takes into account intermittent global disturbances applied to a spatially structured tissue containing metapopulations of cells. Without disturbance and for an arbitrary length of time representing the life span of the organism host, it is possible to parameterize the model in such a way that, despite the occurrence of mutations, no tumors emerge. Within a broad range of parameters, we observed that intermediate frequencies and intensities of disturbance would lead to higher probabilities of tumor formation than in states with more extreme or no disturbances, but with equivalent mutation rates, types of mutation, mutated phenotypes, and otherwise identical model parameters (SI Fig. 8). In the model, demes evolve on a grid with periodic boundary conditions. The fitness of a deme or clone is a function of mutations affecting three general biological functions: the proliferative rate, the death rate (either promoting deme survival or more commonly, by several orders of magnitude, deleterious to deme survival), and susceptibility to disturbances. Demes were initially randomly distributed throughout the grid at a fixed density. The parameters of a single run included initial cell density, baseline mutation rate, wild-type and mutated growth, death, and susceptibility probabilities, as well as disturbance frequency and intensity. Runs consisted of 5,000 Monte Carlo iterations.
The simulations show that the hypothetical transition, from a randomly varying mutational spectrum to a spectrum persistently dominated by a dominant allele(s), does take place during in silico carcinogenesis and distinguishes a population at risk from a population developing a tumor (SI Fig. 9). For any specific run, we can ascertain the in silico MLDA profile at each of the time steps for the entire time length of the simulation. Because the model is nondeterministic, we can select runs that do not terminate in tumor formation and compare the MLDA profiles for each step to those of runs that terminate in tumor formation. We find that the MLDA profile does cross the “cancer threshold with no return” in the instances in which disturbance acts as a factor causing the emergence of a tumor (Fig. 4). Simulations designed to explore the role of disturbance showed that the effect of disturbance frequency appears to lead to relatively gradual changes in the risks of developing tumor. The maximum risk of tumor formation occurs at intermediate disturbance intervals. However, the effect of disturbance intensity shows a fairly steep bifurcation between lower and higher intensities with a maximum risk at intermediate intensity (SI Fig. 8).
Discussion
MLDA of pancreatic juice yields two biometrics, TML and HAD, that distinguish normal individuals from those at risk and from patients suffering from pancreatic carcinoma with a high degree of specificity and sensitivity. Statistical analysis of the data suggests that MLDA is equivalent to a conventional biomarker in phase IIB of development although before a phase III is undertaken validation studies are required to test the reproducibility of the MLDA profiles and derived metrics in stored samples and thus ready to support the design of more extensive validation studies (24). Additional studies will also be required to determine whether the source of the DNA sampled by the MLDA assay (pancreatic vs. duodenal juice) influences the results of the test. The technological platform used for our initial studies is relatively cumbersome and thus not widely applicable. Technologies are emerging, however, that will enable sequencing of allele mixtures with quantitation at the 1% level.
The potential of MLDA is reinforced by in silico simulations that suggest both TML and HDA will enable the early detection of pancreatic cancer. The agent-based model also validates the notion that disturbance is a powerful factor that underlies the emergence of tumors independent of the rate of mutation. It is possible to think that stool could serve as a surrogate substrate for pancreatic juice, thus facilitating investigation of an asymptomatic population. Although the Rolling Circle Amplification enhanced zip-code array has been carefully validated (see Material and Methods), it is hoped that technological improvements will facilitate the quantitative analysis of the variation present in other cancer loci, making MLDA a more robust and reliable test.
We believe TML and HDA may be particularly effective for early detection and risk measurement, because they reflect the pathogenetic process of carcinogenesis seen through the prism of metapopulation dynamics. Future studies should explore whether other ways to measure diversity will be more relevant to the measurement of cancer risk in specific tissues (25). The interrogation of soluble DNA found in biological fluids derived from the tissue of interest, in this case pancreatic juice, is a crucial element of this approach, because it provides a sample of the entire cell population at risk and can be thought off as an index of “molecular dysplasia.” Furthermore, it provides the means to repeatedly sample and monitor events occurring in the tissues without physical disruption. Work in progress shows that fluids contain a broader spectrum of mutated alleles that partially overlaps with that found in paired tissue samples (data not shown). Because cells harboring mutations are more likely to die, either spontaneously or under the effect of disease (disturbance), we speculate that fluids are enriched for mutations with respect to tissue.
As opposed to conventional biomarkers that are based on a single molecule (protein or nucleic acid) specific for the tumor cell, MLDA exploits variability as the source of information. Whereas conventional markers will miss tumors failing to express the specific molecule, MLDA reports the emergence of any dominant tumor genotype within the alleles used as probes. Most useful for future longitudinal studies is the potential generation of a scale that enables the measurement of risk. The risk scale is based on the identification, in cross-sectional studies, of two boundaries separating normal individuals from individuals at risk and the latter from patients harboring a tumor.
Longitudinal records of the metrics derived from MLDA provide a real-time description of the dynamics of carcinogenesis and thus can be used as a relative measurement of cancer risk for an individual organ. A number of factors contribute to an individual's risk of developing cancer and, at least for some high-penetrance genes and environmental exposures, epidemiologists have devised an array of means to compute risk. However, for most common cancer types, the risk factors acting on the vast majority of the population are weak, and even for those with powerful risk factors, we have no way to monitor the outcome of the process leading to tumor formation. In other words, although we have imperfect ways to identify who is at risk, we lack tools to forecast when the tumor will emerge. The MLDA strategy should be well suited to implement early-detection protocols.
Our preliminary longitudinal data in subjects at risk for pancreatic cancer due to a germ-line p16 mutation suggest that MLDA-derived metrics do reflect the evolution of risk in time and can classify samples according to risk levels that are coherent and correlate well with the understanding of pancreatic cancer. The risk of individuals harboring a p16 germ-line mutation increases with age, and no increase in risk is suggested by the MLDA profiles of family members with wild-type p16 genotype.
MLDA biometrics measured in tissue or biological fluids can be applied to a wide spectrum of organ sites. Colorectal cancer (stools or colonic lavage), breast cancer (nipple aspirates or ductal lavage), epithelial malignancies of the lower urinary tract (urine), bronchopulmonary cancer, and other tumors should be detectable at an early stage.
Materials and Methods
Simulation/Model Description.
We have simulated the distribution of mutational load and its relation to tumor development in a population of cells located within a spatially structured tissue subject to periodic disturbances by using a stochastic lattice model (see SI Fig. 8 for a detailed description and SI Fig. 9 for sensitivity analyses). The lattice is a 100 × 100 square grid with periodic boundary conditions. Demes are represented as individual (stem-like) cells that have the capacity to mutate, reproduce by expanding into vacant neighboring tissue niches or die. Demes were initially randomly distributed throughout the grid at various overall densities, each occupying a single location in the grid. We simulate metapopulation barriers by limiting colonization into unoccupied neighboring sites only. A deme is characterized by its “genotype,” which is initially wild type but subsequently altered by accumulated mutations. During each time step, mutation, expansion, and death were randomly assigned to each cell according to transition probabilities based on the genotype of the deme. For the studies presented here, a single baseline mutation rate was used throughout. For the purpose of monitoring mutation load, the genotype of a deme was represented by 10 alleles, for 3 target genes or 10 possible values for each gene (one for each type of potential advantage; proliferative rate, death avoidance, and susceptibility to disturbance). The frequencies of each mutated allele in the population were explicitly monitored to track relative changes in mutational load profile in the tissue over time. The mutational load at a number of other loci (with deleterious mutations) was also monitored. The parameters of a single run included growth, death, and susceptibility probabilities (for both wild-type and mutated genotypes) and mutation rate, as well as disturbance frequency and intensity. Runs consisted of 5,000 Monte Carlo iterations. Global disturbance events were fatal for a cell as a randomized function of its overall death and susceptibility rates and the global disturbance intensity parameter. Average disturbance intervals ranged from every 2 to 1,000 iterations. Disturbance intensities ranging from 0.3 to 0.999 were evaluated. For the studies presented here, either no disturbances occurred (undisturbed state) or disturbance frequency was 25 iterations, with disturbance intensity of 0.9. Individual demes within patches were subject to random culling if they persisted without a sufficient threshold of mutations before the potential transformation to an oncodeme, defined for the current studies as the appearance of a third mutation. Tumor formation was considered to have occurred with the accumulation of three mutations in a continuously expanding clone (see SI Appendix and SI Text).
The software for this model is available through the model repository from the Harvard Integrative Cancer Biology Center for the Development of a Virtual Tumor (CViT) Program and can be found at http://genecube.med.yale.edu:8080/montebello.
Human Subjects.
All samples were collected under Institutional Review Board-approved protocols (Yale Human Investigation Committee Protocol 10926 and Evanston Northwestern Healthcare and University of Nebraska Medical Center, and informed consent was obtained from each patient). The “no-risk” or normal samples (n = 9) were collected from individuals undergoing Endoscopic retrograde cholangio pancreatography for biliary disease with no evidence of pancreatic pathology or malignancy. The pancreatitis group (n = 12) had impaired exocrine function and clinical and imaging findings of chronic pancreatitis. Twenty-one patients with histologically documented diagnosis of pancreatic cancer were used to establish the profile associated with malignancy. Eight members belonging to three families at risk for hereditary pancreatic cancer due to a p16 germ-line mutation are being followed at the Creighton University Hereditary Cancer Institute and Evanston Northwestern Healthcare. Six of the eight are asymptomatic carriers, and two are of wild-type genotype with no known pancreatic pathology.
Assessment of Mutational Load.
Pancreatic juice was obtained during endoscopic examination either by canulation of the pancreatic duct or by i.v. injection of secretin 1 unit/kg, 0.2 mgr/kg (Repligen) and collection of duodenal juice in 3- to 5-min intervals. Soluble DNA in the fluid was extracted, and 50 ng of genomic DNA was used to PCR-amplify Ki-ras exon 1 and p53 exons 5 and 7 in a final volume of 30 μl, as described elsewhere (20). Amplified DNA was used for a multiplex ligation detection reaction and its products hybridized, 36 replicates for each spot, onto a generic zip-code 3D-Link slide microarray according to Ladner et al. (20). Hybridization was detected by rolling-circle amplification decorated with complementary fluor-oligonucleotides. Slides were scanned at 635 nm on a GSI Lumonics 4000 Scanarray and analyzed with GenePix Pro 3.0 software (Axon Instruments) or Spot (CISRO Biotech Imaging Group). Truncated median values of the 36 replicates were used to make the calculations for each zone of the array. Normalization of a given subarray was performed by using the signal intensity of three sample-control replicates and the added intensity of all controls. Repeated assessment of MLDA starting with the genomic amplifications gave reproducible measurements with 0.52% standard deviation. The profiles obtained when three aliquots of the original fluid were analyzed did not vary significantly, and values were within 0.67 standard deviation. Spiking of fluids with human Ki-ras-mutated DNA was also used to verify the fidelity of the procedure. In 12 cases with a dominant allele, >20% the presence of the mutation was confirmed by sequencing the PCR product.
Total mutational load represents the sum of mutated allele frequencies for a given case. Aggregate mutational load refers to the sum of frequencies of mutated alleles for one gene, which, in this case, is either K-Ras or p-53.
Statistical Analysis.
Mutational load profiles of the distinct groups (normal, pancreatitis, and cancer) were compared by using the Kruskal–Wallis test. The predictive value of the TML metric was assessed with a supervised classification method based on multinomial regression, also named polytomous logistic regression. This is a generalization of logistic regression for a response variable with more than two groups. The model aims to predict the probability of multiple class assignment using TML as a quantitative predictor. Because the sample size was small, bootstrap techniques were used to estimate the expected misclassification rate under similar conditions. The 0.632+ estimate of the misclassification error was 2% (based on 1,000 replicates).
The model was internally validated by using bootstrap techniques. The expected misclassification rate was estimated by using the bias-corrected 0.632+ method (22).
The predictive ability of the total mutational load was assessed with a supervised classification method based on multinomial regression (a generalization of logistic regression for more than two groups). The model was internally validated by using bootstrap techniques. The expected misclassification rate was estimated by using the bias-corrected 0.632+ method (21).
Supplementary Material
Acknowledgments
We thank Zenta Walther, M.D., Ph.D., for comments on the manuscript; Katherine Henderson for help with the graphic representation of the data; and Xavier Solé i Joan Valls for help with the statistical analysis. This work was supported by the Early Detection Research Network (National Cancer Institute) and by a gift from Marcia Israel-Curley. The collection of pancreatic juice from families and sporadic cases was funded in part by the Jacqueline Sercussi Memorial Foundation for Cancer Research and by the National Institutes of Health.
Footnotes
Conflict of interest statement: J.C. serves on the Board of Directors of Aureon Laboratories. Aureon Laboratories have exclusive license from Yale University to use MLDA as a potential diagnostic cancer test.
This article contains supporting information online at www.pnas.org/cgi/content/full/0708250105/DC1.
References
- 1.Gonzalez-Garcia I, Sole RV, Costa J. Proc Natl Acad Sci USA. 2002;99:13085–13089. doi: 10.1073/pnas.202139299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatia S, Yasui Y, Robison LL, Birch JM, Bogue MK, Diller L, DeLaat C, Fossati-Bellani F, Morgan E, Oberlin O, et al. J Clin Oncol. 2003;21:4386–4394. doi: 10.1200/JCO.2003.11.059. [DOI] [PubMed] [Google Scholar]
- 3.Rouse JJ, Stephen P. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 4.Dolle MET, Giese H, Hopkins CL, Martus HJ, et al. Nat Genet. 1997;17:431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- 5.King CM, Gillespie ES, McKenna PG, Barnett YA. Mutat Res. 1994;316:79–90. doi: 10.1016/0921-8734(94)90010-8. [DOI] [PubMed] [Google Scholar]
- 6.Mintz B. Symp Soc Exp Biol. 1971;25:345–370. [PubMed] [Google Scholar]
- 7.Cairns J. Nature. 1975;255:197–200. doi: 10.1038/255197a0. [DOI] [PubMed] [Google Scholar]
- 8.Cairns J. Proc Natl Acad Sci USA. 2002;99:10567–10570. doi: 10.1073/pnas.162369899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rainey PB, Buckling A, Kassen R, Travisano M. TREE. 2000;15:243–247. doi: 10.1016/s0169-5347(00)01871-1. [DOI] [PubMed] [Google Scholar]
- 10.Buckling A, Kassen R, Bell G, Rainey PB. Nature. 2000;408:961–964. doi: 10.1038/35050080. [DOI] [PubMed] [Google Scholar]
- 11.Connell JH. Science. 1978;199:1302–1310. doi: 10.1126/science.199.4335.1302. [DOI] [PubMed] [Google Scholar]
- 12.Floder S, Sommer U. Limnol Oceanogr. 1999;44:1114–1119. [Google Scholar]
- 13.Li J, Loneragan WA, Duggin JA, Grant CD. Plant Ecol. 2004;172:11–26. [Google Scholar]
- 14.Hahn WC, Weinberg RA. Nat Rev Cancer. 2002;2:331–341. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 15.Bardelli A, Cahill DP, Lederer G, Speicher MR, Kinzler KW, Vogelstein B, Lengauer C. Proc Natl Acad Sci USA. 2001;98:5770–5775. doi: 10.1073/pnas.081082898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lengauer C, Kinzler KW. Nature. 1998;396:643–649. doi: 10.1038/25292. Rev. [DOI] [PubMed] [Google Scholar]
- 17.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. Hum Mutat. 2002;19:607–614. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 18.Losi L, Baisse B, Bouzourene H, Benhattar J. Carcinogenesis. 2005;26:916–922. doi: 10.1093/carcin/bgi044. [DOI] [PubMed] [Google Scholar]
- 19.Kabbarah O, Chin L. Cancer Cell. 2005;8:439–441. doi: 10.1016/j.ccr.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Ladner DP, Leamon JH, Hamann S, Tarafa G, Strugnell T, Dillon D, Lizardi P, Costa J. Lab Invest. 2001;81:1079–1086. doi: 10.1038/labinvest.3780320. [DOI] [PubMed] [Google Scholar]
- 21.Hruban RH, Goggins M, Parsons J, Kern SE. Clin Cancer Res. 2000;6:2969–2972. [PubMed] [Google Scholar]
- 22.Efron B. J Am Stat Assoc. 1997;92:548–560. [Google Scholar]
- 23.Breiman L. Machine Learn. 2001;45:5–32. [Google Scholar]
- 24.Pepe MS, Etzioni R, Feng S, Potter JD, Thomson ML, Thornquist M, Winget M, Yasui Y. J Natl Cancer Inst. 2001;93:1054–1061. doi: 10.1093/jnci/93.14.1054. [DOI] [PubMed] [Google Scholar]
- 25.Magurran AE. Measuring Biological Diversity. Boston: Blackwell; 2004. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
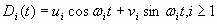{kind=link}
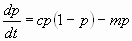{kind=link}