

Abstract
Genetically encoded sensors of glutamate concentration are based on FRET between cyan and yellow fluorescent proteins bracketing a bacterial glutamate-binding protein. Such sensors have yet to find quantitative applications in neurons, because of poor response amplitude in physiological buffers or when expressed on the neuronal cell surface. We have improved our glutamate-sensing fluorescent reporter (GluSnFR) by systematic optimization of linker sequences and glutamate affinities. Using SuperGluSnFR, which exhibits a 6.2-fold increase in response magnitude over the original GluSnFR, we demonstrate quantitative optical measurements of the time course of synaptic glutamate release, spillover, and reuptake in cultured hippocampal neurons with centisecond temporal and spine-sized spatial resolution. During burst firing, functionally significant spillover persists for hundreds of milliseconds. These glutamate levels appear sufficient to prime NMDA receptors, potentially affecting dendritic spike initiation and computation. Stimulation frequency-dependent modulation of spillover suggests a mechanism for nonsynaptic neuronal communication.
Keywords: fluorescence resonance energy transfer, hippocampal neurons, synaptic release
Glutamate is the primary excitatory neurotransmitter in the brain, and precise measurement of its spatiotemporal pattern of synaptic release and propagation would provide insight into diverse brain processes, including synaptic crosstalk, cerebral ischemia, and mechanisms of learning and memory. In hippocampal slices, synaptic glutamate spillover to the dendrite and neighboring synapses induces homeostatic regulation of glutamate release through extrasynaptic mGluR activation (1), limits synaptic independence (2), lengthens EPSC durations (3), and permits heterosynaptic LTP/LTD (4). Spillover is a primary means of chemical neurotransmission between mitral cells in the rat olfactory bulb (5) and between climbing fibers and molecular layer interneurons in cerebral cortex (6). Estimates of glutamate concentration and dynamics in synaptic, extrasynaptic, and extracellular compartments have been made by NMDAR antagonist displacement (7), glutamate uptake inhibitor application (2), whole “sniffer” cells (8), outside-out “sniffer” patch electrodes (9, 10), patch recording of astrocyte synaptically evoked transporter currents (STCs) (11), enzymatically coupled electrochemical probes (12), enzymatically coupled metabolite imaging (13), and other methods. Although each method provided a new perspective on glutamate action, all were hampered by a lack of resolution in the spatial or temporal domains because of single-site measurement, reliance on partially coupled or confounded currents, desensitizing receptors, or indirect and slow secondary cascades.
Recently, the glutamate reporters glutamate-sensing fluorescent reporter (GluSnFR)‖ (14) and fluorescent indicator protein for glutamate (FLIPE) (15) were constructed by linear genetic fusions of the glutamate periplasmic binding protein GltI (also known as ybeJ) with enhanced cyan fluorescent protein (ECFP) and a yellow fluorescent protein, Citrine (16) or Venus (17). These reporters provide a sensitive optical readout of glutamate concentration in vitro by FRET-dependent changes in the CFP/YFP emission ratio. When expressed on the surface of hippocampal neurons, synaptic glutamate release was detectable‖ (14, 15). However, quantitative measurements of rapid glutamate transients have been hampered by the low signal-to-noise ratio of these sensors when used in physiological buffers and by suboptimal glutamate affinity.
Intramolecular FRET reporter responses have been dramatically improved by restricting the rotational freedom of the chromophores, adjusting their orientation by linker variation and fluorescent protein substitution (18–20). Therefore, to maximize GluSnFR response magnitude for neuronal measurements, we performed a comprehensive, mammalian cell based screen of linker truncations of linearly fused constructs, and circularly permuted fluorescent protein substitution. To optimize sensor affinity, we rationally mutated GltI residues known to coordinate ligand binding. We have identified a greatly improved variant of GluSnFR, SuperGluSnFR, which exhibits 44% change in emission ratio upon glutamate binding with a dissociation constant (Kd) of 2.5 μM when expressed on the extracellular surface of neurons.
We have used SuperGluSnFR to directly measure the time course of glutamate propagation after synaptic release. We demonstrate that submicromolar glutamate persists along the dendritic surface for hundreds of milliseconds after coordinated synaptic release. Spillover concentration is strongly modulated by stimulation number and frequency. Active uptake and buffering by neuronal and glial glutamate transporters appears insufficient to prevent extrasynaptic NMDA receptor activation after bursts of synaptic release. Furthermore, uptake transporter capacity may regulate the dependence of extrasynaptic glutamate signaling on action potential frequency and provide an avenue for nonsynaptic neuron and astrocyte communication.
Results
The initial construct design of GluSnFR bracketed the mature length GltI domain with truncated ECFP (AA1–228) and full-length Citrine (Fig. 1a). Later experiments anchored GluSnFRs to the extracellular surface of mammalian cells by fusion to a truncated PDGF receptor (Fig. 1b). Minimal linkers were used to maximize interdomain rigidity. This first GluSnFR was designated GluSnFR0N0C, where the subscript indicates the amino acids truncated from the N and C termini of mature GltI before fusion. When dilute purified GluSnFR0N0C in 50 mM Tris buffer was excited at 420 nm, its emission spectrum showed two peaks corresponding to CFP and YFP emissions, whose ratio was modestly sensitive to glutamate (Fig. 1c). Trypsin digestion extinguished the 526-nm YFP peak and raised the 476-nm CFP peak by 15.2%, indicating a FRET efficiency of 13.2% [supporting information (SI) Fig. 6a]. Application of increasing glutamate increased the emission ratio from ≈0.66 to ≈0.77, with a maximum ratio change (ΔRmax) of 18% and apparent Kd of ≈150 nM (Fig. 1d). Replacing Tris buffer with HBSS reduced ΔRmax to 7% (Fig. 1 c and d). Addition of NaCl to Tris buffer caused a concentration-dependent reduction in ΔRmax to 0% in 50 mM Tris plus 1 M NaCl (Fig. 1e). Substitution of 100 mM Na-gluconate or KCl for 100 mM NaCl reduced ΔRmax equally, indicating a general effect of buffer ionic strength rather than substrate interactions with a specific ion (data not shown). Because neurons require buffers with ionic strength of ≈0.15, observed ΔRmax in mammalian cells with surface-displayed reporters had been limited to ≈10% (15).
Fig. 1.

In vitro characterization of prototype GluSnFR constructs. (a) Genetic construction of GluSnFR for protein purification (Upper) or mammalian cell surface display (Lower). His-6 is a hexahistidine protein purification tag. Ig-κ is a murine Ig κ-chain V-J2-C signal peptide. (b) Schematic of surface displayed GluSnFR in ligand-free state. (c) In vitro emission spectra changes of soluble GluSnFR0N0C to 1 mM glutamate in 50 mM Tris buffer (black) or HBSS (red). (d) Titration curves of soluble GluSnFR0N0C in 50 mM Tris (black triangle) or HBSS (red square). (e) Maximum ratio change of GluSnFR0N0C (circle) decreases with increasing ionic strength of buffer.
Glutamate-dependent ratio changes were partially reversible by glutamate deamination by glutamate-pyruvate transaminase (SI Fig. 6b). Titrations with aspartate and glutamine gave Kd values of ≈700 nM and ≈30 μM, respectively, consistent with fluorescently labeled GltI (21). Application of 1 mM serine, arginine, and sucrose had no effect (data not shown).
The high glutamate affinity (150 nM Kd) of GluSnFR0N0C might cause partial saturation at background glutamate levels in neuronal systems, limiting dynamic range. Furthermore, native PBPs have kon rates of ≈108 M−1 s−1 (22), implying a koff of ≈15 s−1 for this GluSnFR that would filter the response decay to brief impulses of glutamate. We reduced the glutamate affinity by site-directed mutagenesis of key glutamate binding residues (21), which produced a range of glutamate Kd (T93A, E26A, and E26D were 300 nM; S73T was 2.5 μM; R25K was 20 μM; and E26R was 700 μM). Mutations are numbered from start of the mature GltI product. S73T was preferred for neuronal measurements.
To maximize response magnitude, we screened a linker-truncation library of GluSnFRs on the surface of mammalian cells in HBSS. GluSnFRs with N- and C-terminal truncations of GltI(S73T) (Fig. 2a) fused to a truncated PDGF receptor (Fig. 1 a and b) expressed spatially uniformly on the extracellular surface of mammalian cells (Fig. 2b). Constructs were scored by efficiency of membrane targeting and ratio change between zero and saturating [glutamate] (Fig. 2c). The best mutant, GluSnFR8N5C(S73T), had a dramatically lower glutamate-free CFP/YFP ratio and a 44% average ΔRmax (n = 18), a 6.2-fold improvement over the 7.1% average of GluSnFR0N0C (Fig. 2 d and e). We refer to this construct as SuperGluSnFR. In vitro tests of soluble SuperGluSnFR demonstrated ΔRmax of 46% and 34% in 50 mM Tris and Ringer's solution, respectively (Fig. 2f). The slightly lower ΔRmax in Ringer's in solution vs. on cell surface may be due to the increased conformational freedom of free solution. Glutamate titration curves of SuperGluSnFR expressed on HEK293 or HeLa cells demonstrated a 2.5 μM apparent Kd and a 1.0 Hill coefficient (Fig. 2g).
Fig. 2.

Optimization of GluSnFR response on mammalian cell surface. (a) Amino acid sequence of GltI N-terminal (Upper) and C-terminal (Lower) truncations between ECFP and Citrine. Truncation sites are indicated with thin vertical lines. The truncations for the original GluSnFR0N0C and best responding construct GluSnFR8N5C are noted in bold lines. (b) FRET channel showing clean extracellular membrane expression on transfected HEK293 cells. (c) Map of the truncation combination to response magnitude. Color indicates the percentage of maximal ratio change relative to GluSnFR8N5C. Black indicates improper construct folding. (d) (Left) HEK293 cells transfected with the nonoptimized GluSnFR0N0C. (Right) The best responder GluSnFR8N5C. Color bar values encompass the same range of relative CFP/YFP change for both constructs. (e) Ratio change between glutamate free and 100 μM glutamate solutions of HBSS for GluSnFR0N0C (n = 9 fields) and GluSnFR8N5C (n = 18). (f) In vitro emission spectra changes of soluble GluSnFR8N5C to 1 mM glutamate in 50 mM Tris buffer (black) or HBSS (red). (g) Glutamate titration curves of GluSnFR8N5C HEK/HeLa cells in Ringer's (squares and black dashed line; n = 20) and neurons in Ringer's plus 100 μM TBOA (diamonds and red line; n = 5). (Scale bars: 10 μm.)
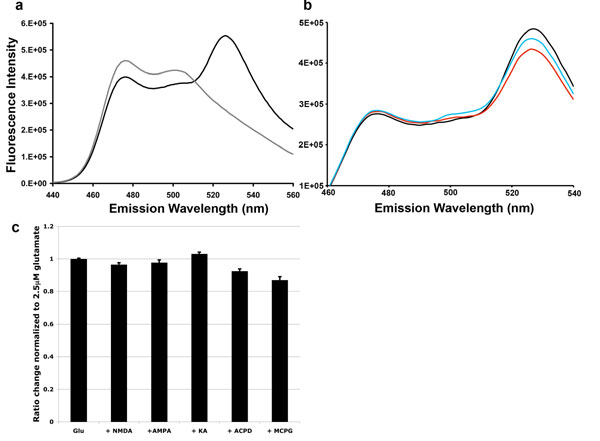
To test the selectivity of SuperGluSnFR for glutamate, a panel of glutamate receptor agonists and antagonists consisting of 2,3-dihydroxy-6-nitro-7-sulfamoylbenzo(F)quinoxaline (NBQX), 2-amino-5-phosphonovaleric acid (APV), α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA), N-methyl-d-aspartic acid (NMDA), kainic acid (KA), (+/−)-trans-1-amino-1,3-cyclopentane dicarboxylic acid (ACPD), and (RS)-α-methyl-4-carboxyphenylglycine (MCPG) was sequentially applied to SuperGluSnFR transfected HEK293 cells. No tested compound directly induced a ratio change or reduced the ΔRmax. However, 300 μM NMDA, 25 μM ACPD, and 250 μM MCPG each caused a small reduction in ratio change from 2.5 μM glutamate (SI Fig. 6c). Addition of 100 μM DL-threo-β-benzyloxyaspartate (TBOA) or 1 mM γ-aminobutyric acid had no effect (data not shown).
We used SuperGluSnFR to detect glutamate on the surface of cultured dissociated hippocampal neurons. After transfection, protein was localized on the extracellular surface of the neuron with even distribution across dendritic spines and shaft and significant intracellular fluorescence confined to the soma or occasional tiny inclusions in the dendritic shaft (Fig. 3a). The glutamate affinity of SuperGluSnFR in neurons was determined by bath changes of Ringer's solution (2 mM Ca2+, 1.3 mM Mg2+, 25 μM NBQX, and 50 μM APV) with increasing glutamate from 0 to 100 μM and was identical to the affinity in HEK293 cells (Fig. 2g). Background glutamate levels in the bath were negligible, but inclusion of 100 μM TBOA in the titration buffer was required to prevent local depletion of glutamate by astrocyte-mediated uptake (SI Fig. 7 a–d).
Fig. 3.

Spatial resolution of glutamate measurement after synaptic release. (a) (Far Left) FRET channel of SuperGluSnFR expressed on the dendritic surface. (Magnification: ×150.) (Scale bar: 10 μm.) Spatially resolved percent ratio change before (Left), during (Middle), and after (Right) a 10-AP, 30-Hz field stimulus in Ringer's (Upper) and with 100 μM TBOA added (Lower). Images are intensity-modulated. Temporal averages of 10 frames are indicated by bars above the traces in 4b, spatially filtered with a 5-pixel wide (466 nm eq.) two-dimensional wiener filter to reduce noise. (b) Averaged ratio change across the total surface of the expressing neuron in a in Ringer's (black) or with TBOA (red). (c) Spatially resolved ratio change to average of 30 single AP stimuli. (Far Left) FRET as in a (Upper Left to Lower Right) sequential frames of response (33 ms per frame). Stimulation of one AP occurs with slight jitter between frames two and three. (d) Time course of average dendritic response.
We tested the ability of SuperGluSnFR to resolve electrically evoked glutamate transients with high spatial resolution. A brief train of 10 field stimulations was delivered at 30 Hz while a small segment of SuperGluSnFR expressing dendrite was imaged at high (×150) magnification. Each pulse in the train was designed to evoke a single action potential (AP) across all neurons (23). During burst stimulation, a rapid transient increase in CFP/YFP ratio was observed across all areas of the dendrite (Fig. 3a). Addition of 100 μM TBOA increased the peak response and dramatically prolonged the recovery toward baseline glutamate levels (Fig. 3b), indicating a glutamate-specific response. Lowering extracellular calcium to 0.1 mM and raising magnesium to 5 mM reversibly abolished responses to field stimulation, indicating the glutamate source was synaptic release (data not shown).
The average area of an active zone in hippocampal culture has been estimated at 0.027 μm2 (24) contributing to <2% of the total dendritic surface area. Furthermore, the high speed of intersynaptic glutamate diffusion (D = 0.76 μm2/ms) has been predicted to produce a relatively smooth glutamate distribution for timescales of >5 ms (25). Even if a significant proportion of the functional synapses are made directly on the dendritic shaft, the GluSnFR signal, particularly when integrated >33-ms intervals, should primarily report glutamate levels arising from synaptic spillover and pooling.
Because spike number may affect the spatial distribution of spillover, we reduced the stimulus to a single AP. The signal to noise ratio was insufficient to resolve individual responses without significant spatial averaging. Therefore, we averaged 30 single AP stimulations. This response also had very broad spread across dendrites, indicating that spillover affects spines and extrasynaptic areas to a similar extent (Fig. 3c). Although the trial-averaged spread was homogeneous, because of the stochastic nature of synaptic release (≈0.3 vesicles/synapse at 2 mM Ca2+, 1.3 mM Mg2+) (26), there may have been a greater heterogeneity on individual trials that we were unable to resolve because of signal/noise or camera speed.
We assessed the temporal resolution of SuperGluSnFR by high-speed imaging (770 fps) of glutamate transients evoked by a single AP. Twenty-seven CFP/YFP traces of single AP stimulation on a single neuron's dendrite were averaged and corrected by a fit bleach curve of a stimulation free trace (Fig. 4a). Individual responses were clearly resolvable (Fig. 4a gray). The trial-averaged ratio was converted to estimated glutamate concentration (Fig. 4b), using titration curves (Fig. 2g). The 20–80% rise time was 6.6 ms, peak glutamate concentration was 720 nM and the time to half decay was 40 ms from peak (Fig. 4b). Low camera resolution and binning prevented a spatial analysis of the signal.
Fig. 4.

Temporal resolution of SuperGluSnFR response. (a) Normalized CFP/YFP emission ratio of a SuperGluSnFR expressing dendritic arbor during single action potential field stimulus. Individual traces are in gray; average traces are in black (n = 27). (b) Corresponding glutamate concentration measurements after calibration.
For SuperGluSnFR to accurately report glutamate spillover decay time course, the rate of ligand unbinding, koff, must exceed the rate of decay of ligand at the neuronal surface. We were uncertain how increasing the sensor Kd to 2.5 μM would affect the kinetics, so we estimated them, using a numerical model of glutamate release and uptake. Total release amount and GluSnFR binding constants were allowed to vary while holding other parameters fixed. Glutamate release was assumed to be instantaneous and homogeneous with minimal buffering from GluSnFR. Because FRET ratio is a sublinear function of glutamate concentration (Fig. 2g), spatial averaging may underestimate the concentration of the initial portion (<5 ms) of the heterogeneously distributed synaptic release transient. Asynchronous release, or buffering by GluSnFR itself would also lower the apparent kinetic rates. Thus, the best-fit parameters of kon = 3.0 × 107 M−1·s−1 and koff = 75 s−1 (SI Fig. 8a) serve only as a lower bound. Even at this lower bound, SuperGluSnFR kinetics are sufficiently rapid to capture the essential waveform of spillover glutamate decay beyond the first 10 ms after synaptic release.
To determine how active glutamate reuptake regulates spillover, we imaged glutamate during a set of six field stimulation conditions. Trains of 1 AP, 10 AP–15 Hz and 10 AP–30 Hz were delivered in Ringer's with or without 100 μM TBOA (Fig. 5a). TBOA increased the peak glutamate concentration during stimulation and greatly slowed the decay back to baseline. In the single-AP case, TBOA increased the average peak [glutamate] at 67 ms from 270 nM to 440 nM and increased the time to half decay from 90 ms to 140 ms (Fig. 5b). Peak glutamate levels, particularly for the single AP cases, are likely underestimates because of the camera's broad temporal integration window. In the 10-AP cases, glutamate levels reached an apparent steady state after 4AP of stimulation with active uptake, while they continued to rise throughout the entire TBOA stimulation. TBOA raised peak [glutamate] from 540 to 1,200 nM and 830 to 1,320 nM, whereas half decay increased from 160 to 650 ms and 140 to 390 ms for 15 and 30 Hz respectively. Doubling stimulation frequency from 15 Hz to 30 Hz gave a 60% enhancement of peak [glutamate] with active uptake, whereas only a 10% enhancement with uptake blocked. Thus, active reuptake may serve multiple purposes, to recycle glutamate and to permit AP frequency dependent signaling by modulation of spillover concentration.
Fig. 5.

Time course of glutamate release and uptake. (a) Average responses to AP (black squares), 10 AP–15 Hz (red circles) and 10 AP–30 Hz (blue triangles) field stimulation in Ringer's solution (solid line) or with 100 μM TBOA added (open shapes, dotted line) to block active glutamate uptake. (b) The decay phase of a scaled to the maximum height to highlight the relative glutamate clearance rate. Colors are as in a. (c) Comparison of surface glutamate in model to observed levels. Average SuperGluSnFR responses to field stimulation in Ringer's solution or in 100 μM TBOA (black). Model of instantaneous glutamate levels in plane of neurons (gray). Model glutamate filtered by the camera integration (red). (d) Model of glutamate double-bound NR2A (black), NR2B (blue), and NR2D (red) NMDA receptors in response to 1 AP, 10 AP–15 Hz, and 10 AP–30 Hz stimulation in Ringer's.
To explore the effect of our measured glutamate transients on NMDA receptor activation, we made a simple numerical model of glutamate dynamics for 2 s after onset of field stimulation (Fig. 5c, SI Fig. 8 b–e, and SI Scheme 1). After a single action potential, only 1.4% of NR2A, 2.4% of NR2B, and 3.7% of NR2D receptors were doubly bound by glutamate (Fig. 5d). However, the prolonged spillover from a burst of 10AP at either 15 or 30 Hz caused a sustained activation of 20 or 22% NR2A, 36 or 34% NR2B, and 50 or 45% of NR2D receptors, respectively.
Discussion
Sensor Optimization.
FRET sensor optimization is an area of active research, with numerous recent reports on improved fluorescent proteins, FRET pairs, substrate and linker mutations and screening techniques. Optimization of GluSnFR demonstrates the delicate sensitivity of FRET reporters to their constituents. The fitness landscape of GluSnFR linkers was surprisingly peaked. A single construct, GluSnFR8N5C, of the 176 tested was far superior to all others (Fig. 2c). Although there was general improvement in sensor response as linkers were truncated, the process was nonmonotonic, limiting the predictability of this design strategy. We attempted systemic substitution of fluorescent proteins (FPs) and circularly permuted FPs to improve response, including the fluorescent protein variants ECFP-A206K, Cerulean (27), CyPet, YPet (28), Venus (17) and cpVenus (M145, 157, 173, 195). Unexpectedly, substitutions of improved components to linker-optimized GluSnFRs reduced either the quality of the reporter's surface expression or response magnitude. This is likely due to linker sequences being already highly tuned for the specific chromophore orientations and subtle electrostatic interactions of the GltI domain and the FP pair.
We inserted ECFP into the putative transmembrane loops of GltI, similar to FLI81PE reported by Deuschle et al. (18). When expressed as a purified protein, FLI81PE has a twofold ratio change to glutamate, but this and all tested ECFP insertion mutants fail to express properly on the surface of mammalian cells. Incorporation of superfolding GFP mutations (29) into the inserted ECFP improved folding and trafficking of many insertion mutants. However, our best-case ΔRmax when expressed on cell surface was only 4% (data not shown). Further improvements of sensor response may be possible by FP substitution or insertion but will require a rescreening of many linker combinations for that pair.
Screening of single circularly permuted fluorescent protein insertions into GltI may produce a single wavelength glutamate sensor analogous to camgaroos (19). A single-FP GluSnFR could be of practical use in more challenging preparations, such as 2-photon in vivo imaging, when using multiple optical sensors simultaneously or when quantitative calibration is not a priority.
SuperGluSnFR represents a major improvement over other optical indicators of glutamate. No previous membrane-tethered PBP-based FRET reporter has achieved more than a 10% FRET ratio change for glutamate or any other substrate. Another recently reported glutamate sensor, EOS (30), requires purification of recombinant protein followed by thiol-mediated dye labeling and cell surface immobilization through biotinylation. The apparent glutamate off-rate of EOS is on the order of hundreds of milliseconds. In contrast, SuperGluSnFR is genetically expressed, allowing cell-specific or subcellular targeting, has been quantitatively calibrated, and has adequate response size, sensitivity and kinetic rates to resolve single action potentials. Future versions of SuperGluSnFR may be genetically targeted to the active zone by fusion to specific synaptic proteins or targeting motifs, raising the possibility of direct comparison of synaptic vs. extrasynaptic glutamate dynamics. For synaptic targeting, a GluSnFR variant with lower glutamate affinity, such as R25K or E26, would be desired to prevent sensor saturation in the synaptic cleft.
Functional Significance.
Measurements of glial synaptic transporter currents (STCs) indicate that the bulk of synaptically released glutamate is rapidly buffered and internalized by glutamate transporters. Estimates of spillover glutamate time course, using STCs, primarily mediated by EAAT2 in the hippocampus, have provided an evolving range of estimates of clearance time in the low tens of milliseconds (31). Recently, deconvolution analysis, using partially blocked STC time courses, has suggested that the true clearance rate of glutamate is faster than the STC decay rate, with an exponential τdecay of 5.8 ms in P12–14 and 0.75 ms in adult hippocampal slices (32). Despite this rapid rate of apparent glutamate clearance, spillover in the Schaffer collateral pathway of hippocampal slice is thought to prolong NMDAR-mediated EPSCs by tens to hundreds of milliseconds in a stimulus intensity-dependent manner (3). Possible mechanisms include increased activation of extrasynaptic NR2B and NR2D subunit-containing receptors (33) or cooperative action of glutamate at neighboring synapses (2). Furthermore, extended activation of NMDARs can trigger dendritic calcium spikes in the basal dendrites of cortical and CA1 pyramidal neurons (34, 35). Priming of the NMDARs by residual glutamate has been suggested as the activation mechanism, but direct measurements of these glutamate transients have not been made.
SuperGluSnFR imaging may reconcile the discrepancy between STC estimates of millisecond glutamate clearance rates and prolonged NMDAR activation. After bursts of electrical stimulation, our imaging shows long, slowly decaying glutamate transients persist across both dendritic spines and shaft. These are of submicromolar concentration, but are sufficient to dramatically enhance NMDAR activation for hundreds of milliseconds in a stimulus strength-dependent manner (Fig. 5d). These glutamate transients may be missed in STC recordings, because STCs are confounded by a small slowly decaying potassium conductance, which is difficult to perfectly compensate (31). Given the estimated EAAT2 glutamate Kd of 18 μM (36), transporter currents at submicromolar extracellular glutamate concentrations are probably small enough to be obscured by errors associated with the potassium conductance. Although SuperGluSnFR measurements are insensitive to the first few milliseconds of spillover, because of sensor kinetics and heterogeneous initial glutamate distribution, their calibration, sensitivity, and spatial resolution provide a valuable complement to the fine temporal resolution of STCs.
The ease of imaging SuperGluSnFR in a reduced culture preparation forced some tradeoffs in potential physiological relevance. Spillover decay time course may vary from culture to brain slice because of differences in the density of synapses, transporters and buffering agents, temperature, neuropil geometry, and access to bath. We drove synaptic release with spatially broad field stimulations to heterogeneous circuits, which caused simultaneous release from many synaptic neighbors likely leading to pooling of extrasynaptic glutamate. In vivo, neuronal activity is generally sparser, and glutamate uptake may be more efficient, so spillover and NMDAR priming may be more temporally and spatially constrained. Nonetheless, our imaging results in culture are congruent with numerous observations of the functional consequences of spillover in slice (2, 3, 34, 35). Direct observation of glutamate propagation with GluSnFRs in slice models (37) or via 2-photon imaging and/or microendoscopy (38) in vivo would more directly address the extent and significance of glutamate spillover.
Steady-state spillover concentration during burst stimulation is strongly modulated by stimulation frequency (Fig. 5a). This suggests that nonsynaptically connected neurons or astrocytes may be able to estimate the firing rate of neighboring neurons by the degree of activation of high-affinity extrasynaptic glutamate receptors. This could allow induction of a measured amount of heterosynaptic LTD (4), homeostatic regulation (1), or vasoregulation (39). These effects would be sensitive to modulation of glutamate uptake by changes in astrocyte membrane potential, internal glutamate concentration, or other means.
Linker-optimized GluSnFRs may be adapted for a number of other neuronal applications beyond measuring synaptic spillover. The selectivity of GluSnFRs for true glutamate over standard glutamatergic agonists and antagonists should facilitate dissection of feedback loops where GluRs modulate release or uptake. SuperGluSnFR is currently being used in studies of glutamate release from astrocytes. GluSnFRs could be used as a calibration tool to assess the propagation of glutamate in uncaging or iontophoresis experiments, as a specific marker of presynaptic modulation in studies of LTP or LTD, or in mapping the functional connectivity of the brain. More clinically relevant uses may include GluSnFR imaging in the screening of drug candidates for glutamate release, transporter or receptor modulation, or in models of glutamate excitotoxicity in cerebral ischemia.
Materials and Methods
Sensor Construction and in Vitro Characterization.
Genomic DNA from Escherichia coli was isolated by chemical lysis and cartridge purification. GltI was amplified from the genomic DNA by PCR and subcloned into the bacterial expression vector pRSETB (Invitrogen). To make GluSnFR0N0C-pRSETB, GltI was ligated into the AKAR2-pRSETB PKA reporter (40) at the SphI and SacI sites between ECFP and Citrine. This construct was expressed in bacteria, extracted by chemical lysis (B-PER II; Pierce), purified by 6-His Ni-NTA gel column filtration (Qiagen), and dialyzed. Spectroscopy was performed as described in ref. 41. Ligand-induced ratio changes were assayed by progressive glutamate addition to an albumin-coated quartz cuvette containing 10 nM GluSnFR in 50 mM Tris or HBSS (pH 7.40). For mammalian cell surface expression, an ECFP-GltI fragment and Citrine fragment were amplified from GluSnFR0N0C-pRSETB by PCR with new BglII and PstI restriction sites flanking the N and C termini of the combined fragment. These were triple ligated into a pDisplay vector (Invitrogen) between the BglII and PstI sites to make GluSnFR0N0C-pDisplay.
Library Construction and Screening.
Affinity mutations were made in GltI-pRSETB, using QuikChange (Qiagen); transferred to GluSnFR0N0C-pRSETB by digestion and ligation at the SphI and SacI sites; and assayed in vitro as above. GltI(S73T) was incorporated into GluSnFR0N0C-pDisplay by digestion and ligation at SphI and SacI sites. Glutamate affinity was assayed by bath changes of HBSS with increasing [glutamate] on transfected HEK293 and HeLa cells.
Preliminary large truncations of the N and C termini of GltI indicated that deletions beyond the first putative α-helix element of the N and C termini (data not shown) caused misfolding. Therefore, the library was limited to 176 combinations of deletions of 0–15 aa of the N terminus and 0–10 of the C terminus of GltI. Truncation combinations were amplified with Phusion polymerase (NEB) and purified with 96-well PCR cleanup cartridges (Qiagen). Truncations were digested with SphI and SacI, ligated into the GSFR0N0C-pDisplay vector, replacing the full-length GltI domain, and plated on selective media. Two colonies of each transformation were cultured and miniprepped. Proper insert length was checked for all by analytical restriction digests.
HEK293 or HeLa cells were seeded on 96-well culture plates, grown for one day, and transfected with one of the 176 × 2 minipreps. Two days after transfection, glutamate responses we measured by thorough bath exchanges with 0 and 100 μM [glutamate] HBSS. Repeated optical measurements were made on selected fields in each well with a 20× air objective and a motorized stage. A random library sample and all large responders were sequenced. 0N0C, 8N0C and 8N5C truncations underwent confirmatory imaging in transfected HEK293 cells on 12-mm coverslips in a Warner imaging chamber with a 40× oil-immersion objective.
Additional Methods.
See SI Text.
Supplementary Material
Acknowledgments.
We thank P. Steinbach for imaging assistance and M. Lin, M. Häusser, C. Stevens, J. Isaacson, and J. Diamond for helpful discussions. This work was supported by National Institutes of Health Grant NS027177 and the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database [accession no. EU422995 (SuperGluSnFR)].
Hires SA, Zhu Y, Stevens CF, Tsien RY, Thirty-Fourth Annual Meeting of the Society for Neuroscience, October 23–27, 2004, San Diego, CA.
This article contains supporting information online at www.pnas.org/cgi/content/full/0712008105/DC1.
References
- 1.Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- 2.Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci. 2002;5:325–331. doi: 10.1038/nn825. [DOI] [PubMed] [Google Scholar]
- 3.Lozovaya NA, Kopanitsa MV, Boychuk YA, Krishtal OA. Enhancement of glutamate release uncovers spillover-mediated transmission by N-methyl-d-aspartate receptors in the rat hippocampus. Neuroscience. 1999;91:1321–1330. doi: 10.1016/s0306-4522(98)00638-1. [DOI] [PubMed] [Google Scholar]
- 4.Vogt KE, Nicoll RA. Glutamate and gamma-aminobutyric acid mediate a heterosynaptic depression at mossy fiber synapses in the hippocampus. Proc Natl Acad Sci USA. 1999;96:1118–1122. doi: 10.1073/pnas.96.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Isaacson JS. Glutamate spillover mediates excitatory transmission in the rat olfactory bulb. Neuron. 1999;23:377–384. doi: 10.1016/s0896-6273(00)80787-4. [DOI] [PubMed] [Google Scholar]
- 6.Szapiro G, Barbour B. Multiple climbing fibers signal to molecular layer interneurons exclusively via glutamate spillover. Nat Neurosci. 2007;10:735–742. doi: 10.1038/nn1907. [DOI] [PubMed] [Google Scholar]
- 7.Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- 8.Pasti L, Zonta M, Pozzan T, Vicini S, Carmignoto G. Cytosolic calcium oscillations in astrocytes may regulate exocytotic release of glutamate. J Neurosci. 2001;21:477–484. doi: 10.1523/JNEUROSCI.21-02-00477.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Copenhagen DR, Jahr CE. Release of endogenous excitatory amino acids from turtle photoreceptors. Nature. 1989;341:536–539. doi: 10.1038/341536a0. [DOI] [PubMed] [Google Scholar]
- 10.Allen TG. The “sniffer-patch” technique for detection of neurotransmitter release. Trends Neurosci. 1997;20:192–197. doi: 10.1016/s0166-2236(96)01039-9. [DOI] [PubMed] [Google Scholar]
- 11.Diamond JS, Bergles DE, Jahr CE. Glutamate release monitored with astrocyte transporter currents during LTP. Neuron. 1998;21:425–433. doi: 10.1016/s0896-6273(00)80551-6. [DOI] [PubMed] [Google Scholar]
- 12.Pomerleau F, Day BK, Huettl P, Burmeister JJ, Gerhardt GA. Real time in vivo measures of l-glutamate in the rat central nervous system using ceramic-based multisite microelectrode arrays. Ann N Y Acad Sci. 2003;1003:454–457. doi: 10.1196/annals.1300.051. [DOI] [PubMed] [Google Scholar]
- 13.Innocenti B, Parpura V, Haydon PG. Imaging extracellular waves of glutamate during calcium signaling in cultured astrocytes. J Neurosci. 2000;20:1800–1808. doi: 10.1523/JNEUROSCI.20-05-01800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsien RY. Building and breeding molecules to spy on cells and tumors. FEBS Lett. 2005:927–932. doi: 10.1016/j.febslet.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 15.Okumoto S, et al. Detection of glutamate release from neurons by genetically encoded surface-displayed FRET nanosensors. Proc Natl Acad Sci USA. 2005;102:8740–8745. doi: 10.1073/pnas.0503274102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griesbeck O, Baird GS, Campbell RE, Zacharias DA, Tsien RY. Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J Biol Chem. 2001;276:29188–29194. doi: 10.1074/jbc.M102815200. [DOI] [PubMed] [Google Scholar]
- 17.Nagai T, et al. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002;20:87–90. doi: 10.1038/nbt0102-87. [DOI] [PubMed] [Google Scholar]
- 18.Deuschle K, et al. Construction and optimization of a family of genetically encoded metabolite sensors by semirational protein engineering. Protein Sci. 2005;14:2304–2314. doi: 10.1110/ps.051508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baird GS, Zacharias DA, Tsien RY. Circular permutation and receptor insertion within green fluorescent proteins. Proc Natl Acad Sci USA. 1999;96:11241–11246. doi: 10.1073/pnas.96.20.11241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagai T, Yamada S, Tominaga T, Ichikawa M, Miyawaki A. Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci USA. 2004;101:10554–10559. doi: 10.1073/pnas.0400417101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Lorimier RM, et al. Construction of a fluorescent biosensor family. Protein Sci. 2002;11:2655–2675. doi: 10.1110/ps.021860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller DMr, Olson JS, Pflugrath JW, Quiocho FA. Rates of ligand binding to periplasmic proteins involved in bacterial transport and chemotaxis. J Biol Chem. 1983;258:13665–13672. [PubMed] [Google Scholar]
- 23.Murthy VN, Sejnowski TJ, Stevens CF. Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 24.Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barbour B, Hausser M. Intersynaptic diffusion of neurotransmitter. Trends Neurosci. 1997;20:377–384. doi: 10.1016/s0166-2236(96)20050-5. [DOI] [PubMed] [Google Scholar]
- 26.Murthy VN, Schikorski T, Stevens CF, Zhu Y. Inactivity produces increases in neurotransmitter release and synapse size. Neuron. 2001;32:673–682. doi: 10.1016/s0896-6273(01)00500-1. [DOI] [PubMed] [Google Scholar]
- 27.Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 28.Nguyen AW, Daugherty PS. Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat Biotechnol. 2005;23:355–360. doi: 10.1038/nbt1066. [DOI] [PubMed] [Google Scholar]
- 29.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 30.Namiki S, Sakamoto H, Iinuma S, Iino M, Hirose K. Optical glutamate sensor for spatiotemporal analysis of synaptic transmission. Eur J Neurosci. 2007;25:2249–2259. doi: 10.1111/j.1460-9568.2007.05511.x. [DOI] [PubMed] [Google Scholar]
- 31.Diamond JS, Jahr CE. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 2000;83:2835–2843. doi: 10.1152/jn.2000.83.5.2835. [DOI] [PubMed] [Google Scholar]
- 32.Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25:2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lozovaya NA, et al. Extrasynaptic NR2B and NR2D subunits of NMDA receptors shape “superslow” afterburst EPSC in rat hippocampus. J Physiol. 2004;558:451–463. doi: 10.1113/jphysiol.2004.063792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schiller J, Major G, Koester HJ, Schiller Y. NMDA spikes in basal dendrites of cortical pyramidal neurons. Nature. 2000;404:285–289. doi: 10.1038/35005094. [DOI] [PubMed] [Google Scholar]
- 35.Ariav G, Polsky A, Schiller J. Submillisecond precision of the input-output transformation function mediated by fast sodium dendritic spikes in basal dendrites of CA1 pyramidal neurons. J Neurosci. 2003;23:7750–7758. doi: 10.1523/JNEUROSCI.23-21-07750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arriza JL, et al. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14:5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Behrens CJ, van den Boom LP, de Hoz L, Friedman A, Heinemann U. Induction of sharp wave-ripple complexes in vitro and reorganization of hippocampal networks. Nat Neurosci. 2005;8:1560–1567. doi: 10.1038/nn1571. [DOI] [PubMed] [Google Scholar]
- 38.Flusberg BA, Jung JC, Cocker ED, Anderson EP, Schnitzer MJ. In vivo brain imaging using a portable 3.9 gram two-photon fluorescence microendoscope. Opt Lett. 2005;30:2272–2274. doi: 10.1364/ol.30.002272. [DOI] [PubMed] [Google Scholar]
- 39.Takano T, et al. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci. 2006;9:260–267. doi: 10.1038/nn1623. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Ma Y, Taylor SS, Tsien RY. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci USA. 2001;98:14997–15002. doi: 10.1073/pnas.211566798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heim R, Tsien RY. Engineering green fluorescent protein for improved brightness, longer wavelengths and fluorescence energy transfer. Curr Biol. 1996;6:178–182. doi: 10.1016/s0960-9822(02)00450-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}