

Abstract
Congenital long- or short-QT syndrome may lead to life-threatening ventricular tachycardia and sudden cardiac death. Apart from the rare disease-causing mutations, common genetic variants in CAPON, a neuronal nitric oxide synthase (NOS1) regulator, have recently been associated with QT interval variations in a human whole-genome association study. CAPON had been unsuspected of playing a role in cardiac repolarization; indeed, its physiological role in the heart (if any) is unknown. To define the biological effects of CAPON in the heart, we investigated endogenous CAPON protein expression and protein–protein interactions in the heart and performed electrophysiological studies in isolated ventricular myocytes with and without CAPON overexpression. We find that CAPON protein is expressed in the heart and interacts with NOS1 to accelerate cardiac repolarization by inhibition of L-type calcium channel. Our findings provide a rationale for the association of CAPON gene variants with extremes of the QT interval in human populations.
Keywords: NOS1, QT interval, cardiac electrophysiology
Rare disease-causing mutations leading to congenital long- or short-QT syndrome are well recognized, but there is little insight into genetic sources of QT interval variation in normal populations. A whole-genome association approach has recently implicated common genetic variants in CAPON as contributing to QT interval differences in a community-based German population (1). This association has since been confirmed in other populations (2, 3). The genetic findings challenge our current understanding of QT physiology. CAPON, first identified in rat brain neurons (4), is a highly conserved protein (≈92% conceptual amino acid sequence identity between rat and human) with an N-terminal phosphotyrosine-binding (PTB) domain and a C-terminal PDZ-binding [postsynaptic density-95 (PSD95)/drosophila discs large/zona occludens-1] domain (4–6). In brain, CAPON competes with PSD95 for the binding of neuronal nitric oxide synthase (NOS1) through the interaction of its C terminus with the PDZ domain of NOS1 (4), thus uncoupling the NMDA–NOS1–NO-mediated signaling pathways. CAPON is also an adaptor protein of NOS1, capable of directing NOS1 to specific target proteins (5, 6). Nowhere, however, has CAPON been suspected of playing a role in cardiac physiology.
Both NOS1 and endothelial NOS (NOS3) are constitutively expressed in cardiomyocytes (7). NOS1 in the sarcolemma has been proposed to interact with Na+-K+ ATPase (8) and with the plasma membrane Ca2+/calmodulin-dependent Ca2+ ATPase (PMCA) through the interaction of PDZ domain of NOS1 and the C terminus of PMCA4b isoform (9). In the sarcoplasmic reticulum (SR), NOS1 is structurally associated with ryanodine receptor 2 (RyR2) (10) and cardiac SR Ca2+ ATPase (SERCA2) (11) to regulate intracellular calcium cycling and excitation–contraction coupling. Conditional transgenic overexpression of NOS1 in heart leads to additional association of NOS1 and the sarcolemmal L-type calcium channel and thus suppresses the L-type calcium currents (ICa,L) (12). All of these lines of evidence highlight the notion that NOS1 plays a key role in regulating cardiac physiology.
Here, we hypothesized that CAPON is expressed in the heart and interacts with NOS1 to exert its biological effects. Therefore, we first sought to identify endogenous CAPON protein in ventricular myocytes and to characterize its physiological relevance by overexpression of CAPON using in vivo somatic gene transfer. We then probed the interaction between CAPON and the NOS–NO signaling pathways to dissect the potential mechanisms underlying the biological effects of CAPON in the heart.
Results
Identification of Endogenous CAPON Protein in the Heart.
Because CAPON expression has not been documented in the heart, we first sought to probe the endogenous CAPON protein in guinea pig ventricular myocardium by Western blotting. The protein bands from guinea pig heart were compared with those from lung and brain tissues and from HEK293 cells with heterologous overexpression of CAPON by in vitro gene transfer with a bicistronic adenoviral vector (AdCAPON-GFP). The ventricular myocardium exhibited a band migrating near the 60-kDa marker, as expected for endogenous full-length CAPON, comparable with the comigration bands from HEK293 cells with heterologously overexpressed CAPON and from brain, which has abundant endogenous CAPON (Fig. 1A). The higher bands (≈70 kDa) could reflect either posttranslational modification of CAPON or nonspecific cross-reactivity. Notably, the ventricular myocardium also displayed a band ≈30 kDa in size detected by antibody against the C terminus of CAPON, but not by antibody against an N-terminal epitope, which is consistent with the short-form of CAPON (13). Immunofluorescent staining of freshly isolated ventricular myocytes revealed a cytoplasmic distribution pattern of the soluble CAPON protein with focal accentuation over the perinuclear region and the sarcolemmal membrane (Fig. 1B). Additionally, enhancement of CAPON immunofluorescence can be appreciated in CAPON-overexpressing myocytes. Therefore, expression of the endogenous CAPON protein in ventricular myocytes and overexpression of CAPON by AdCAPON-GFP-mediated gene transfer were both confirmed biochemically.
Fig. 1.

Identification of endogenous CAPON protein in the heart. (A) Tissue homogenates from normal guinea pig ventricular myocardium (VM), brain, and lung, and HEK293 cells with (T) and without (NT) in vitro transduction with AdCAPON-GFP were subjected to SDS/PAGE Western blotting. Both full-length (CAPON-L) and short-form of CAPON (CAPON-S) are expressed in VM. The higher bands (arrowhead) could be caused by either posttranslational modification of CAPON or nonspecific cross-reactivity. (B) Immunostaining of CAPON in AdCAPON-GFP-transduced (3) and nontransduced (2) ventricular myocytes. Negative controls (secondary antibody only) are depicted in 1.
Adenovirus Alone Does Not Affect Electrophysiology.
Both action potential duration (APD) and peak ICa,L density were equivalent between AdGFP-transduced and nontransduced myocytes (Fig. 2 A and B). See supporting information (SI) Results for details. After ascertaining that adenoviral transduction alone does not alter cellular electrophysiology, we examined the effects of CAPON overexpression by comparing the electrophysiological parameters between AdCAPON-GFP-transduced and nontransduced myocytes.
Fig. 2.

Electrophysiological effects of CAPON overexpression in guinea pig ventricular myocytes. (A and B) Adenovirus alone does not affect electrophysiology. For details, see SI Results. (C–H) CAPON overexpression abbreviates the action potential duration and reduces ICa,L. (C) APD was markedly shortened in a representative AdCAPON-GFP-transduced myocyte compared with a representative control myocyte. (D) Significant abbreviation of APD can be seen spanning from APD10 to APD90 at 1-Hz stimulation in CAPON-overexpressing myocytes (CAPON, n = 9; control, n = 13). (E) The actual APD50 and APD90 measured from individual myocyte comprising each group reveal a consistent reduction of APD in CAPON-overexpressing myocytes. (F) Representative ICa,L recordings elicited by depolarizing voltage steps (500 ms) from −40 to +60 mV in 10-mV increments after a prepulse from −80 mV to −40 mV show reduction of ICa,L in a AdCAPON-GFP-transduced myocyte compared with a control myocyte. (G) The peak ICa,L density in CAPON-overexpressing myocytes was smaller than in control myocytes. (H) Averaged peak current–voltage relationships demonstrate attenuation of ICa,L at multiple depolarizing pulses in CAPON-overexpressing myocytes (CAPON, n = 8; control, n = 12). The number inside each bar graph indicates the number of cells studied.
CAPON Overexpression Accelerates Action Potential.
To determine whether CAPON modulates cardiac repolarization, we compared the APD in freshly isolated control and CAPON-overexpressing ventricular myocytes at 1, 2, and 0.05 Hz. At 1-Hz stimulation, the mean APD10, APD50, APD75, and APD90 were significantly shortened to 54.0 ± 7.2, 198.8 ± 8.1, 221.8 ± 7.9, and 233.9 ± 7.7 ms in CAPON-overexpressing cardiomyocytes (n = 9) compared with 96.6 ± 11.5, 291.0 ± 16.4, 316.1 ± 17.2, and 327.5 ± 17.9 ms in control myocytes (n = 13, P < 0.05, respectively) (Fig. 2 C–E). The abbreviation of APD with CAPON overexpression was maintained at 2 Hz and 0.05 Hz (data not shown), and the magnitude of APD90 reduction by CAPON overexpression was 28.6% at 1-Hz stimulation. We next explored the ionic basis for the abbreviation of APD.
L-Type Calcium Current.
In CAPON-overexpressing myocytes, the peak ICa,L density was significantly reduced (−7.2 ± 0.5 pA/pF, at +20 mV, n = 8) compared with that of control myocytes (−11.0 ± 1.0 pA/pF, at +10 mV, n = 12; P < 0.05) (Fig. 2 F and G). The reduction of the peak ICa,L density was 35% (Fig. 2G). Averaged peak current density–voltage relationships revealed significant suppression of peak ICa,L density from −30 mV to +40 mV (P < 0.05, respectively) by CAPON overexpression (Fig. 2H). By a decrease in net inward current, the CAPON-induced inhibition of peak ICa,L density would contribute to the abbreviation of APD.
Sodium Current.
Sodium current (INa) was not affected by CAPON overexpression (Fig. 3 A and B). For details see SI Results.
Fig. 3.

Electrophysiological effects of CAPON overexpression in guinea pig ventricular myocytes. (A and B) CAPON overexpression does not affect sodium current. For more details, see SI Results. (C–E) CAPON overexpression enhances IKr. (C) Delayed rectifier K+ tail current was measured in response to a depolarizing pulse to +40 mV for 5 s followed by repolarization to −40 mV. IKr was recorded after steady-state suppression of IKs by chromanol 293B. Representative recordings show a larger IKr tail current in a CAPON-overexpressing myocyte compared with a control myocyte. (D) Neither peak IK nor IKs tail current densities were significantly different between CAPON-overexpressing and control myocytes. However, peak IKr tail current density was modestly enhanced in CAPON-overexpressing myocytes. (E) Instantaneous IK1 current density elicited by ramp protocol from −100 to + 70 mV (5 s) was not different between CAPON-overexpressing (n = 10) and control myocytes (n = 9).
Outward Rectifier Potassium Current.
In guinea pig ventricular myocytes, there are two types of delayed rectifier potassium currents, the rapidly activating component (IKr) and the slowly activating component (IKs) (14), responsible for terminating the action potential plateau. To probe the potential contribution of these currents to CAPON overexpression-mediated APD changes, we first recorded the total IK tail currents and then used a specific IKs blocker, chromanol 293B (15), to separate IKr and IKs in CAPON-overexpressing and control myocytes. Although neither the total IK nor the IKs tail current density was changed between nontransduced control and CAPON-overexpressing myocytes, we found the IKr peak tail current density to be modestly enhanced in CAPON-overexpressing myocytes (0.92 ± 0.07 pA/pF, n = 10) compared with control myocytes (0.61 ± 0.08 pA/pF, n = 7; P < 0.05) (Fig. 3 C and D). These results indicate that in addition to the reduction of ICa,L, enhancement of IKr also contributes to the abbreviation of APD in CAPON-overexpressing myocytes.
Inward Rectifier Potassium Current.
Inward rectifier potassium current (IK1) was not changed by CAPON overexpression (Fig. 3E). See SI Results for details.
Protein–Protein Interaction of CAPON and NOS1 in the Heart.
After identifying that both full-length and short-form CAPON proteins are expressed in the heart and overexpression of CAPON accelerates APD by suppression of ICa,L and augmentation of IKr, the important next step is to answer whether the electrophysiological changes induced by CAPON overexpression were caused by modifications of the NOS–NO pathways. First, we determined whether CAPON interacts with NOS in the heart. To assess protein–protein interactions of CAPON and NOS, normal guinea pig ventricular tissue homogenates were immunoprecipitated with anti-CAPON- or anti-NOS1-bound protein G complex, separated by SDS/PAGE, and probed by CAPON, NOS1, and NOS3 antibodies, respectively. The crude tissue homogenates served as the positive controls while probing NOS1, NOS3, and CAPON (Fig. 4). We found that NOS1 specifically coimmunoprecipitated with the bound CAPON antibodies (Fig. 4 A and B), whereas NOS3 was not detected in the anti-CAPON immunoprecipitates (Fig. 4 C and D). To include a negative control, the ventricular myocardial homogenates were also incubated overnight with the protein G complex without prebound CAPON antibody; as a result, neither NOS1 nor CAPON could be detected in the eluted precipitates (Fig. 4 A and B). These findings indicate that CAPON–NOS1, but not CAPON–NOS3, exists as a physiological complex in ventricular myocytes.
Fig. 4.

CAPON interacts with NOS1, but not NOS3, in ventricular myocytes. Normal guinea pig ventricular tissue homogenates were immunoprecipitated overnight with anti-CAPON-bound protein G complex, separated by SDS/PAGE, and probed by anti-NOS1 (A), anti-CAPON (B and D), and anti-NOS3 (C), respectively. To include a negative control, the ventricular tissue homogenates were also incubated overnight with anti-CAPON-free protein G complex (beads). As a result, neither NOS1 nor CAPON could be detected in the eluted precipitates. WB, Western blotting.
CAPON Overexpression Stabilizes NOS1.
Because CAPON interacts with NOS1 in the heart, we next wanted to know how CAPON overexpression affects NOS1 protein level and activity. Freshly isolated ventricular myocytes were transduced in vitro with AdCAPON-GFP, or AdGFP, or not transduced with either virus. The gene transfer efficiency was confirmed by ≈100% GFP-positive cells in AdGFP-transduced myocytes and ≈50% GFP positive cells in AdCAPON-GFP myocytes, compared with only background autofluorescence (i.e., 0% GFP positivity) in nontransduced myocytes (Fig. 5A). The in vitro AdCAPON-GFP transduction caused a 1.9-fold increase of the CAPON protein level (Fig. 5B). At 0 h of culture, the NOS1 level was not different between AdCAPON-GFP-transduced and nontransduced myocytes (5.47 ± 2.51% vs. 6.08 ± 2.27%, n = 3 animals; p = NS) (Fig. 5C). After 40.3 ± 2.3 h of cell culture, NOS1 was down-regulated in nontransduced and AdGFP-transduced myocytes but was up-regulated in AdCAPON-GFP-transduced myocytes. Therefore, the mean NOS1 level from nontransduced and AdGFP-transduced myocytes was lower than that of AdCAPON-GFP-transduced myocytes (2.78 ± 1.75% vs. 18.54 ± 5.99%; P < 0.05) (Fig. 5D). Of note, NOS3 protein levels were not changed in AdCAPON-GFP-transduced myocytes (data not shown). Noncultured ventricular tissue homogenates served as positive control for the NOS1/NOS3 expression assays. These findings suggest that CAPON overexpression may stabilize NOS1 in ventricular myocytes.
Fig. 5.

CAPON overexpression stabilizes NOS1 in ventricular myocytes. (A) Freshly isolated ventricular myocytes were transduced in vitro with AdCAPON-GFP, or AdGFP, or not transduced with either virus. BF, bright-field microscopic images. (B) The in vitro AdCAPON-GFP transduction caused a 1.9-fold increase of the CAPON level. (C) At 0 h of culture, the NOS1 level was not different between AdCAPON-GFP-transduced and nontransduced myocytes (n = 3 animals). (D) After 40.3 ± 2.3 h of cell culture, NOS1 became down-regulated in both ventricular myocytes transduced with AdGFP and in nontransduced myocytes, whereas NOS1 was well preserved in the AdCAPON-GFP-transduced ventricular myocytes. The mean NOS1 level from nontransduced and AdGFP-transduced myocytes was lower than that of AdCAPON-GFP-transduced myocytes (*, P < 0.05 vs. control, n = 3 animals). Noncultured ventricular tissue homogenates (VM) served as positive control for NOS1 detection. The signal intensities of CAPON and NOS1 are normalized against the GAPDH signals.
Enhanced Intracellular NO Production in CAPON-Overexpressing Myocytes.
Because NO generation reflects NOS activity, we further imaged intracellular NO production by using a rhodamine-based chromophore, DAR-4M AM, in living ventricular myocytes isolated 3–5 days after in vivo gene transfer of CAPON. To test the specificity of DAR-4M AM for NO imaging, we measured fluorescence intensity in ventricular myocytes loaded with DAR-4M AM only or DAR-4M AM + sodium nitroprusside (SNP) or DAR-4M AM + NG-nitro-l-arginine methyl ester (l-NAME). Fluorescence intensified with the addition of SNP and waned with l-NAME compared with that in myocytes incubated with DAR-4M AM only (Fig. 6A). Next, we compared NO production between CAPON-overexpressing myocytes and control myocytes incubated with DAR-4M AM only or DAR-4M AM + 2 mM l-arginine (Fig. 6 B–D). The baseline fluorescent intensity was equivalent in CAPON-overexpressing and control myocytes (921.6 ± 32.5 a.u., n = 30 vs. 888.6 ± 17.3 a.u., n = 90; P = NS), whereas with the addition of l-arginine, the fluorescence intensity increased disproportionately in CAPON-overexpressing myocytes (1,420.9 ± 28.1 a.u., n = 43) compared with controls (1,329.0 ± 19.1 a.u., n = 166; P < 0.05) (Fig. 6D). These results indicate that CAPON overexpression may enhance intracellular NO production, particularly in the presence of additional NOS substrates.
Fig. 6.

Enhanced intracellular NO production in CAPON-overexpressing myocytes. (A) The specificity of DAR-4M AM was verified by a differential fluorescent intensity with the addition of SNP or l-NAME. (B) The NO fluorescence was enhanced after l-arginine stimulation. In GFP (+), CAPON-overexpressing myocytes, the NO fluorescence was stronger than in control myocytes. (C) Representative high-powered images illustrate a modest enhancement of the NO fluorescent marker in a CAPON-overexpressing myocyte. (D) The baseline fluorescent intensity was equivalent in CAPON-overexpressing (n = 30) and control myocytes (n = 90), whereas with the addition of l-arginine, the fluorescence intensity increased disproportionately in CAPON-overexpressing myocytes (CAPON-l-arg) (n = 43) compared with controls (Control-l-arg) (n = 166).
l-NAME Reverses CAPON Overexpression-Mediated APD Abbreviation and ICa,L Reduction.
After ascertaining that CAPON overexpression resulted in up-regulation of NOS1–NO activity, we examined the effects of l-NAME on ICa,L and APD in ventricular myocytes isolated 3–5 days after in vivo gene transfer of CAPON. Pretreatment with 1 mM l-NAME reversed the APD90 abbreviation (1 Hz, 348.3 ± 47.8 ms, n = 7 vs. 378.5 ± 50.1 ms, n = 5; P = NS) (Fig. 7 A and B) and the ICa,L density reduction (−6.2 ± 0.6 pA/pF, n = 14 vs. −6.6 ± 0.4 pA/pF, n = 14; P = NS) (Fig. 7 C–E) in CAPON-overexpressing ventricular myocytes. Interestingly, APD90 tends to become longer (378.5 ± 50.1 ms, n = 5 vs. 333.7 ± 20.2 ms, n = 7; P = NS) (Fig. 7 A and B), and the peak ICa,L density tends to increase, too (−6.6 ± 0.4 pA/pF, n = 14 vs. −6.3 ± 0.3 pA/pF, n = 14; P = NS) (Fig. 7 C–E) in control myocytes after pretreatment with l-NAME. However, these changes were not statistically significant. In contrast, significant lengthening of APD90 (1 Hz, 348.3 ± 47.8 ms, n = 7 vs. 221.6 ± 19.9 ms, n = 6; P < 0.05) (Fig. 7 A and B) and the increase of the peak ICa,L density (−6.2 ± 0.6 pA/pF, n = 14 vs. −4.8 ± 0.3 pA/pF, n = 14; P < 0.05) (Fig. 7 C–E) were only seen in CAPON-overexpressing myocytes after pretreatment with l-NAME.
Fig. 7.

l-NAME reverses CAPON overexpression-mediated APD abbreviation and ICa,L reduction. (A) Representative action potential recordings show reversal of APD abbreviation with l-NAME in a CAPON-overexpressing myocyte. (B) Pretreatment with l-NAME significantly increased APD90 and reversed APD abbreviation in CAPON-overexpressing ventricular myocytes, whereas the APD90 was not significantly changed with l-NAME in control myocytes. (C) Representative ICa,L recordings show reversal of ICa,L suppression with l-NAME in a CAPON-overexpressing myocyte. (D) Averaged peak current–voltage relationships reveal significant increase of peak ICa,L densities with l-NAME in CAPON-overexpressing myocytes but not in control myocytes. The number of cells studied in each group is indicated in bar graphs in E. (E) Pretreatment with l-NAME significantly increased peak ICa,L density and rescued ICa,L suppression in CAPON-overexpressing myocytes, whereas the peak ICa,L density was not significantly changed with l-NAME in control myocytes.
Discussion
We demonstrate that CAPON, the brain-enriched NOS1 adaptor/regulator protein, is expressed in the heart and interacts with NOS1. Overexpression of CAPON results in up-regulation of the NOS1–NO signaling pathways, which further leads to abbreviation of APD through the inhibition of ICa,L and enhancement of IKr.
Expression of Endogenous CAPON Protein in the Heart.
Since the first identification of CAPON protein in the rat brain (4), Xu et al. (13) further found that CAPON protein has two isoforms that are translated from a full-length and a C-terminal splicing variant (13). The full-length transcript encompassing 10 exons contains both PTB- and PDZ-binding domains, whereas the C-terminal transcript containing the last 2 exons encodes only the PDZ-binding domain (13). We found that both full-length and short CAPON isoforms are expressed in guinea pig ventricular myocytes using the same CAPON antibody (13) predicted to react with the C terminus of CAPON. The full-length isoform comigrated at the same level as a brain-enriched protein band and a band from HEK293 cells expressing CAPON. Immunostaining reveals a cytoplasmic distribution of CAPON with focal accentuation around the perinuclear region and the cell membrane.
CAPON Overexpression Up-Regulates NOS1–NO Pathways.
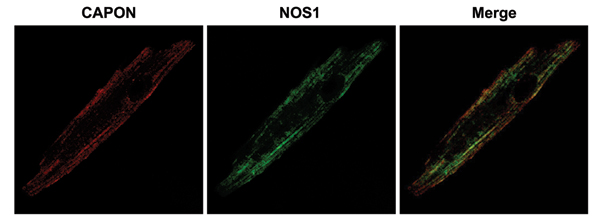
Given that CAPON acts as a regulator and adaptor of NOS1 in brain, we speculated that the CAPON overexpression-induced electrophysiological changes may be mediated by modifications of NOS–NO signaling pathways. To investigate this hypothesis, we began by probing the interactions of CAPON and NOS1 in the heart. We found that CAPON interacts with NOS1, but not NOS3, to form a physiological complex in guinea pig ventricular myocytes. This interaction was further supported by an intracellular colocalization of CAPON and NOS1 by immunostaining and confocal microscopy (SI Fig. 8). Interestingly, only the full-length CAPON isoform is responsible for the interaction with NOS1, because only a single full-length CAPON band (≈60 kDa) was detected in the eluted immunoprecipitates. These findings are in line with the previous observations that the full-length CAPON of a similar molecular mass was accountable for the interactions with NOS1 in neurons (4).
To explore how CAPON overexpression affects NOS1, we compared NOS1 protein level in cultured guinea pig ventricular nontransduced or AdGFP- or AdCAPON-GFP-transduced myocytes. Interestingly, the NOS1 protein becomes down-regulated both in ventricular myocytes transduced with AdGFP and in nontransduced myocytes, whereas NOS1 levels were well preserved in AdCAPON-GFP-transduced ventricular myocytes after 40 h of cell culture. However, NOS3 protein level was not obviously affected by AdCAPON-GFP transduction compared with control myocytes. This finding provides direct evidence to support the notion that CAPON has a functional role in stabilizing NOS1 in ventricular cardiomyocytes.
Because intracellular NO production directly reflects NOS activity, we further compared the intracellular NO generation between AdCAPON-GFP-transduced ventricular myocytes and nontransduced myocytes by using DAR-4M AM. The baseline intracellular NO production was equivalent between CAPON-overexpressing ventricular myocytes and control myocytes; however, with the addition of l-arginine, CAPON-overexpressing myocytes generated more NO than did the controls. Therefore, overexpression of CAPON in ventricular myocytes not only stabilizes NOS1 but may also enhance NOS1 activity. The absolute increase in NO production was only modest in CAPON-overexpressing compared with control myocytes (7%), yet it has to be kept in mind that these small but significant increases in global NO production may translate into biologically relevant local NO concentrations. Given the very short elimination half-life of NO, the biological effects of NOS1–NO signaling depend largely on the subcellular localization of NOS1, where CAPON might play a role to direct NOS1 to the specific target proteins. In addition, CAPON may compete with other PDZ-binding proteins for the PDZ domain. Therefore, it is possible that CAPON may exert its biological effects by mechanisms other than modulation of the NOS1–NO pathway.
CAPON Overexpression Shortens APD by Suppression of ICa,L and Enhancement of IKr.
The electrophysiological studies revealed that CAPON overexpression resulted in abbreviation of APD mediated by a diminished ICa,L and an enhanced IKr. Because guinea pig ventricular myocytes lack endogenous Ito (16), ICa,L and IK represent the two principal currents controlling the length of the action potential plateau and APD. Accordingly, a 35% decrease of ICa,L combined with a 50% increase in IKr is sufficient to explain the 30% shortening of APD90 observed with CAPON overexpression.
Reversal of CAPON Overexpression-Induced Electrophysiological Changes by l-NAME.
Because we found that overexpression of CAPON stabilizes NOS1 and enhances NOS1-derived NO production, pharmacological intervention with a NOS blocker should be able to counteract CAPON overexpression-mediated electrophysiological changes. As expected, pretreatment with l-NAME significantly increased APD90 and ICa,L, thereby reversing APD abbreviation and ICa,L suppression in CAPON-overexpressing ventricular myocytes. The magnitudes of increments in APD90 (57% vs. 13%) and ICa,L (29% vs. 5%) in CAPON-overexpressing ventricular myocytes obviously surpass those of the control myocytes when both are exposed to l-NAME, excluding the possibility that a significant background effect of l-NAME is responsible for the changes. The differential electrophysiological responsiveness to l-NAME between CAPON-overexpressing myocytes and control myocytes further supports the notion that CAPON overexpression activates the NOS1–NO pathway to modulate cellular electrophysiology.
In guinea pig ventricular myocytes, NO can shorten APD by suppression of ICa,L and augmentation of IKs (17, 18). The inhibition of ICa,L by NO is caused by cGMP-dependent or cGMP-independent (direct S-nitrosylation/redox) pathways (17, 19, 20). Similarly, NO can modulate the delayed rectifier K+ currents by cGMP-dependent or -independent pathway (17, 18, 21). Interestingly, although CAPON overexpression activates NOS1-derived NO in our work, the electrophysiological changes mediated by this NO activation are comparable with those in previous reports (17–19, 21). Recently, evidence emerged that NOS1 may play a critical role in the regulation of calcium handling and myocyte contraction in the heart (9, 10, 12, 22). Burkard et al. (12) observed that in a conditional NOS1-overexpressing transgenic mouse model, the peak ICa,L density was significantly reduced by 39% in NOS1-overexpressing ventricular myocytes, which was attributed to the translocation of NOS1 to sarcolemma where NOS1 interacted with L-type calcium channel and caused inhibition of ICa,L. Conceivably, the observation that activated NOS1 is translocated to the sarcolemma where it interacts with ion channels may help explain our finding of ICa,L inhibition and IKr augmentation.
In conclusion, CAPON protein is expressed in the heart and interacts with NOS1 to exert its biological effects. Overexpression of CAPON may stabilize NOS1 and activate NOS1-derived NO signaling cascades to shorten action potential duration via inhibition of l-type calcium channels and activation of delayed rectifier potassium channels. Our findings provide a rationale for the association of CAPON gene variants with extremes of the QT interval in human populations. Specifically, we propose that CAPON gene variants, which are in noncoding regions (1–3), act by influencing the basal expression level of CAPON in the heart, thereby affecting cardiac repolarization and the QT interval by the mechanisms identified here.
Study Limitations.
In addition to the study of sarcolemmal ion channels, we did not examine the role of CAPON on calcium transients and excitation–contraction coupling in myocardium. This investigation might be of particular importance, because NOS1–NO pathways have recently been related to play a role in regulating cardiac contractility (9, 10, 12). Moreover, because CAPON may compete with other PDZ-binding proteins for PDZ binding, it is likely that CAPON, in addition to modulating the NOS1–NO pathway, might also have other biological effects, particularly regarding stabilization of cell membrane proteins, including ion channels. Therefore, the changes in electrophysiology might be attributed not only to NO-dependent mechanisms (as laid out in the present work), but also to NO-independent mechanisms that have not been addressed herein. Finally, our experimental design did not allow us to examine the ECG phenotype in CAPON-overexpressing animals because only a small area of the left ventricular myocardium was injected, and the transduction efficiency was in the range of 1–10%. Therefore, our approach only allowed us to study changes in cellular electrophysiology, but not the global ECG phenotype. Future studies using transgenic models with CAPON overexpression and knockdown are needed to address this question.
Methods
This work conformed to the standards of the National Institutes of Health regarding care and use of laboratory animals and was performed in accordance with the guidelines of Animal Care and Use Committee of The Johns Hopkins University.
Cloning of CAPON cDNA, Plasmid Construction, and Adenovirus Preparation.
The full-length CAPON cDNA was cloned from guinea pig ventricular myocardium and subcloned into a bicistronic adenoviral vector (AdCAPON-GFP) coexpressing the reporter GFP. For details see SI Methods.
Western Blot Analysis.
To probe endogenous CAPON protein expression, tissue homogenates from normal guinea pig ventricular myocardium, brain, and lung and HEK293 cells with and without in vitro transduction with AdCAPON-GFP were subjected to SDS/PAGE Western blotting. See SI Methods for further details.
Coimmunoprecipitation of CAPON and NOS.
Normal guinea pig ventricular tissue homogenates were immunoprecipitated overnight with anti-CAPON-bound protein G complex, separated by SDS/PAGE, and probed by anti-NOS1, anti-CAPON, and anti-NOS3, respectively. Further details are described in SI Methods.
Cell Culture and in Vitro Gene Transfer.
For details, see SI Methods.
Immunofluorescent Staining and Confocal Microscopic Imaging.
For details, see SI Methods.
In Vivo Gene Transfer and Myocyte Isolation.
In vivo gene transfer was performed by open-chest intramyocardial injection of the adenoviral vectors into the left ventricular apex. The ventricular myocytes were isolated 72 h after gene transfer for further studies. For details, see SI Methods.
Electrophysiological and Pharmacological Studies.
Recording techniques and conditions are described in SI Methods.
Intracellular NO Imaging with DAR-4M AM.
Intracellular NO production was imaged by DAR-4M AM in living ventricular myocytes isolated 3–5 days after in vivo transduction with AdCAPON-GFP. For further details, see SI Methods.
Statistical Analysis.
All data are expressed as mean ± SEM. Comparison between groups was performed by using the unpaired Student's t test, and P values <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments.
This work was supported in part by a grant from the Donald W. Reynolds Cardiovascular Clinical Research Center and by a Transatlantic Network of Excellence grant from the Le Ducq Foundation. K.-C.C. was supported by a research grant from China Medical University Hospital, Taichung, Taiwan; A.S.B. was supported by German Research Foundation (DFG) Grant BA 3341/1-1; and Y.Z. was supported by a Research Fellowship from the Heart and Stroke Foundation of Canada.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0709118105/DC1.
References
- 1.Arking D-E, et al. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38:644–651. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- 2.Post W, et al. Associations between genetic variants in the NOS1AP (CAPON) gene and cardiac repolarization in the old order Amish. Hum Hered. 2007;64:214–219. doi: 10.1159/000103630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aarnoudse A-J, et al. Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam study. Circulation. 2007;116:10–16. doi: 10.1161/CIRCULATIONAHA.106.676783. [DOI] [PubMed] [Google Scholar]
- 4.Jaffrey S-R, Snowman A-M, Eliasson M-J, Cohen N-A, Snyder S-H. CAPON: A protein associated with neuronal nitric oxide synthase that regulates its interactions with PSD95. Neuron. 1998;20:115–124. doi: 10.1016/s0896-6273(00)80439-0. [DOI] [PubMed] [Google Scholar]
- 5.Fang M, et al. Dexras1: A G protein specifically coupled to neuronal nitric oxide synthase via CAPON. Neuron. 2000;28:183–193. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- 6.Jaffrey S-R, Benfenati F, Snowman A-M, Czernik A-J, Snyder S-H. Neuronal nitric-oxide synthase localization mediated by a ternary complex with synapsin and CAPON. Proc Natl Acad Sci USA. 2002;99:3199–3204. doi: 10.1073/pnas.261705799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hare J-M, Stamler J-S. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest. 2005;115:509–517. doi: 10.1172/JCI200524459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu K-Y, et al. Nitric oxide protects cardiac sarcolemmal membrane enzyme function and ion active transport against ischemia-induced inactivation. J Biol Chem. 2003;278:41798–41803. doi: 10.1074/jbc.M306865200. [DOI] [PubMed] [Google Scholar]
- 9.Oceandy D, et al. Neuronal nitric oxide synthase signaling in the heart is regulated by the sarcolemmal calcium pump 4b. Circulation. 2007;115:483–492. doi: 10.1161/CIRCULATIONAHA.106.643791. [DOI] [PubMed] [Google Scholar]
- 10.Barouch L-A, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–340. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- 11.Xu K-Y, Huso D-L, Dawson T-M, Bredt D-S, Becker L-C. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proc Natl Acad Sci USA. 1999;96:657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burkard N, et al. Conditional neuronal nitric oxide synthase overexpression impairs myocardial contractility. Circ Res. 2007;100:e32–e44. doi: 10.1161/01.RES.0000259042.04576.6a. [DOI] [PubMed] [Google Scholar]
- 13.Xu B, et al. Increased expression in dorsolateral prefrontal cortex of CAPON in schizophrenia and bipolar disorder. PLoS Med. 2005;2:999–1007. doi: 10.1371/journal.pmed.0020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanguinetti M-C, Jurkiewicz N-K. Two components of cardiac delayed rectifier K+ current: Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosch R-F, et al. Effects of the chromanol 293B, a selective blocker of the slow, component of the delayed rectifier K+ current, on repolarization in human and guinea pig ventricular myocytes. Cardiovasc Res. 1998;38:441–450. doi: 10.1016/s0008-6363(98)00021-2. [DOI] [PubMed] [Google Scholar]
- 16.Inoue M, Imanaga I. Masking of A-type K+ channel in guinea pig cardiac cells by extracellular Ca2+. Am J Physiol. 1993;264:C1434–C1438. doi: 10.1152/ajpcell.1993.264.6.C1434. [DOI] [PubMed] [Google Scholar]
- 17.Bai C-X, Takahashi K, Masumiya H, Sawanobori T, Furukawa T. Nitric oxide-dependent modulation of the delayed rectifier K+ current and the L-type Ca2+ current by ginsenoside Re, an ingredient of Panax ginseng, in guinea pig cardiomyocytes. Br J Pharmacol. 2004;142:567–575. doi: 10.1038/sj.bjp.0705814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai C-X, et al. Role of nitric oxide in Ca2+ sensitivity of the slowly activating delayed rectifier K+ current in cardiac myocytes. Circ Res. 2005;96:64–72. doi: 10.1161/01.RES.0000151846.19788.E0. [DOI] [PubMed] [Google Scholar]
- 19.Campbell D-L, Stamler J-S, Strauss H-C. Redox modulation of L-type calcium channels in ferret ventricular myocytes: Dual mechanism regulation by nitric oxide and S-nitrosothiols. J Gen Physiol. 1996;108:277–293. doi: 10.1085/jgp.108.4.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furukawa T, et al. Ginsenoside Re, a main phytosterol of Panax ginseng, activates cardiac potassium channels via a nongenomic pathway of sex hormones. Mol Pharmacol. 2006;70:1916–1924. doi: 10.1124/mol.106.028134. [DOI] [PubMed] [Google Scholar]
- 21.Han N-L, Ye J-S, Yu A, Sheu F-S. Differential mechanisms underlying the modulation of delayed-rectifier K+ channel in mouse neocortical neurons by nitric oxide. J Neurophysiol. 2006;95:2167–2178. doi: 10.1152/jn.01185.2004. [DOI] [PubMed] [Google Scholar]
- 22.Sears C-E, et al. Cardiac neuronal nitric oxide synthase isoform regulates myocardial contraction and calcium handling. Circ Res. 2003;92:e52–e59. doi: 10.1161/01.RES.0000064585.95749.6D. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}