

Abstract
Bacillus subtilis strain ATCC6633 has been identified as a producer of mycosubtilin, a potent antifungal peptide antibiotic. Mycosubtilin, which belongs to the iturin family of lipopeptide antibiotics, is characterized by a β-amino fatty acid moiety linked to the circular heptapeptide Asn-Tyr-Asn-Gln-Pro-Ser-Asn, with the second, third, and sixth position present in the D-configuration. The gene cluster from B. subtilis ATCC6633 specifying the biosynthesis of mycosubtilin was identified. The putative operon spans 38 kb and consists of four ORFs, designated fenF, mycA, mycB, and mycC, with strong homologies to the family of peptide synthetases. Biochemical characterization showed that MycB specifically adenylates tyrosine, as expected for mycosubtilin synthetase, and insertional mutagenesis of the operon resulted in a mycosubtilin-negative phenotype. The mycosubtilin synthetase reveals features unique for peptide synthetases as well as for fatty acid synthases: (i) The mycosubtilin synthase subunit A (MycA) combines functional domains derived from peptide synthetases, amino transferases, and fatty acid synthases. MycA represents the first example of a natural hybrid between these enzyme families. (ii) The organization of the synthetase subunits deviates from that commonly found in peptide synthetases. On the basis of the described characteristics of the mycosubtilin synthetase, we present a model for the biosynthesis of iturin lipopeptide antibiotics. Comparison of the sequences flanking the mycosubtilin operon of B. subtilis ATCC6633, with the complete genome sequence of B. subtilis strain 168 indicates that the fengycin and mycosubtilin lipopeptide synthetase operons are exchanged between the two B. subtilis strains.
In the struggle for nutrients, microorganisms have a wide arsenal of chemical compounds at their disposal to inhibit competing organisms. Many of these compounds have a peptide origin and can be synthesized ribosomally as well as nonribosomally.
The Gram-positive bacterium Bacillus subtilis produces a variety of nonribosomally synthesized circular oligopeptides that are modified by a fatty acid. The lipopeptides surfactin and fengycin contain a β-hydroxy fatty acid, whereas members of the iturin lipopeptide family, such as mycosubtilin, bacillomycin, and iturin (Fig. 1), carry a β-amino fatty acid modification (1–3). The peptide moiety of the iturin lipopeptides contains a tyrosine in the D-configuration at the second amino acid position and two additional d-amino acids at positions three and six (3). The members of the iturin family exhibit strong antifungal and hemolytic activities and a limited antibacterial activity (3). Although multienzyme complexes responsible for the biosynthesis of surfactin and fengycin have been identified and characterized in detail, the enzymes responsible for the biosynthesis of the iturin lipopeptides are still unknown (4–8). However, on the basis of their structural similarity to surfactin and fengycin, a comparable mechanism of synthesis has been proposed.
Figure 1.

Schematic structure of mycosubtilin, iturin A, and bacillomycin D. In the carbon chain structure of the fatty acid, n is generally 14 (myristate).
Surfactin and fengycin are synthesized by large multienzyme complexes. Such peptide synthetases can be found in prokaryotes as well as eukaryotes (4, 7, 9–12). Genetic and biochemical analyses of peptide synthetases have revealed a modular structure of these multifunctional proteins (4–15). A module is defined as the unit that catalyzes the incorporation of a specific amino acid into the peptide product. The arrangement of the modules of a peptide synthetase is usually colinear with the amino acid sequence of the peptide. The modules can be further subdivided into different domains that are characterized by a set of short conserved sequence motifs (14). The core of each module is an amino acid adenylation domain that recognizes and activates a specific amino acid (16). The thiolation domain, located C terminally of the adenylation domain, contains an invariant serine residue essential for the binding of a 4′-phosphopantetheine cofactor (17). An N-terminal condensation domain is required for the coupling of two consecutively bound amino acids (18). In addition, modules can be supplemented with domains that catalyze the modification of the activated amino acid, such as N-methylation, and epimerization from the L-configuration to the D-configuration (18, 19). The conserved modular organization of peptide synthetases provides the means for the creation of peptide synthetases by genetic engineering strategies. In this way, the exchange of different modules in the surfactin synthetase has been successfully used to produce new lipopeptide compounds (20). The prospect of creating numerous bioactive peptides by the engineering of existing peptide synthetases has stimulated the search for additional peptide synthetases.
Following this aim, we set out to identify the biosynthetic genes for iturin lipopeptides. In this report, we describe the molecular analysis of the mycosubtilin synthetase operon. The unique characteristics of mycosubtilin synthetase are discussed.
Materials and Methods
General Methods and Materials.
Molecular cloning and PCR procedures were carried out by using standard techniques (21–23). Media for growth of B. subtilis and Escherichia coli have been previously described (23–25). B. subtilis chromosomal DNA was purified as previously described (26).
Isolation of Lipopeptides.
Isolation of lipopeptides produced by B. subtilis ATCC6633 was essentially performed as described by Ebata et al. (27). Cells were grown in ammonium citrate/sucrose medium at 37°C (25). After 4 days of cultivation, the supernatant was collected by centrifugation and adjusted to pH 2.0 by using concentrated HCl and stirred overnight. The precipitate was collected by centrifugation and extracted twice, first with 95% ethanol and subsequently with 70% ethanol. The concentrated ethanolic extracts were mixed with 2 volumes of water and, after centrifugation, the precipitate was recrystallized three times and fractionated by using LH-20 gel permeation chromatography (column size 2 × 25 cm) and eluent system C: chloroform/methanol/ethanol/water 70:30:35:15 (vol/vol/vol/vol).
Mass Spectrometry of the Lipopeptide Products.
Lipopeptides were analyzed by using fast atom bombardment (FAB) mass spectrometry, and matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry. For FAB mass spectrometry, a vacuum generator ZAB-3HF spectrometer (BEB configuration) was used (Vg-instruments, Manchester). Samples were dissolved in dimethyl sulfoxide/glycerol, and positive ions were detected. MALDI-TOF mass spectra were recorded on a Bruker (Bruker Daltonik, Bremen) Reflex MALDI-TOF instrument with a 337-nm nitrogen laser for desorption and ionization. A saturated solution of α-cyano-4-hydroxycinnamic acid in 70% acetonitrile/0.1% trifluoroacetic acid (vol/vol) was used as matrix. Ions were accelerated with a voltage of 20 kV. The positive-ion and reflector mode was applied.
TLC.
For the analysis of the amino acid composition, the lipopeptide fractions were separated by TLC by using silica gel 60-plates (Merck). Three eluent systems (A, B, and C) were used: system A, consisting of N-butanol-acetic acid-water 4:1:1 (vol/vol/vol); system B, consisting of chloroform/methanol/water 65:25:4 (vol/vol/vol); and system C, consisting of chloroform/methanol/ethanol/water 70:30:35:15 (vol/vol/vol/vol). The various spots were visualized with ninhydrin by using the 4,4′-tetramethyldiamino-diphenylmethane assay and by charring after spraying with concentrated H2SO4 (28).
Amino Acid Analysis.
For preparative isolation of the lipopeptide fractions, the corresponding spots were scratched out from the thin-layer chromatograms, and the silica gel material was extracted with methanol. The purified lipopeptides were hydrolyzed by using 6 M HCl in the presence of 0.2% phenol and 0.1% thioglycolic acid in sealed evacuated tubes. Amino acid analysis of the hydrolysates was performed by using an automated amino acid analyzer (Durrum D-500, Biotronic, Munich). Determination of the absolute configuration of the amino acid components was performed by derivatizing the amino acids with 1-fluoro-2,4-dinitro-5-l-alanine amide (Marfey’s reagent) and separation of the obtained diastereomers with reversed phase HPLC (LKB) using Hypersil ODS RP18 (Knauer, Berlin; column size 250 × 6 mm) (29).
Incorporation of Radioactive Precursor Amino Acids.
Radioactive precursor amino acids (1 μCi; Amersham) were added to growing cultures 24 hr after incubation. The cells were harvested 3–4 days after inoculation. HCl-precipitates of the culture supernatants were extracted with 5 ml of 95% and 70% ethanol, respectively. Separation and quantification of the labeled lipopeptides were performed by TLC (eluent system A–C) and radioscanning, using a Berthold (Nashua, NH) Analyzer LB2832.
Identification of a Peptide Synthetase Operon.
A genomic library of B. subtilis ATCC6633 was constructed by partial digestion of chromosomal DNA with Sau3A and ligation into the BamHI site of the vector λ-DashII (Stratagene). The DNA was packaged in vitro by using GigapackII (Stratagene) according to the manufacturer’s protocols, and the resulting phages were used to infect E. coli P2392 (Stratagene). Plaques were transferred to nylon membranes (Qiagen, Chatsworth, CA). A mixture of oligonucleotide probes, directed against DNA regions encoding the highly conserved sequence motifs KAGGAYVP and GTTGKPKG of peptide synthetases, was used to screen the genomic DNA library, as has been previously described for screening in other microorganisms (30). Positive λ-DashII clones were restricted with EcoRI, blotted on a nylon membrane and hybridized at medium stringency with the oligonucleotide probes. This Southern hybridization was used to discriminate insert size and possible surfactin operon fragments. A selected λ clone was restricted with EcoRI, subcloned into pBluescript KS(+) (Stratagene), and its sequence was determined.
Sequence Determination of the Synthetase Operon.
To obtain the complete sequence of the peptide synthetase-encoding operon, inverse PCR was applied by using the Expand PCR system (Boehringer Mannheim) (22). The PCR fragments were fragmented by a DNaseI treatment, subcloned into pUC18, and sequenced (23, 31).
Purification of the Second Amino Acid-Activating Module.
A His6-tag fusion with the second module of the mycosubtilin synthetase was obtained by PCR cloning by using primers mycB1up: CGC GGA TCC ATG TCG GTG TTT AAA AAT CAA GTA ACG; and mycB1down: GGC GTC GAC TTA GGA CGC CAG CAG TTC TTC TAT TGA G. These primers contained restriction sites for BamHI and SalI, respectively (underlined), which were used to clone this module C-terminal to a His6-tag in the expression plasmid pQE30 (Qiagen). The resulting plasmid was transformed into E. coli DH5α containing plasmid pREPGroESL (32). For overexpression, an overnight culture, grown in Luria–Bertani medium at 28°C, was diluted 100-fold, and at A590 = 0.5, the culture was induced with 0.5 mM (final concentration) isopropyl-β-d-thiogalactopyranoside. The cells were harvested after 3 hr and disrupted by using a French press. The expressed second module was purified by using Ni+-affinity chromatography. The protein was eluted from the Ni+-column with 100 mM imidazole in a 20 mM Tris buffer (pH 6.5). Purification was followed by SDS/PAGE.
ATP-PPi Exchange Assay.
To determine the substrate specificity of the purified second module of the mycosubtilin synthetase, an ATP-PPi exchange assay was performed in the presence of different amino acids, as previously described (16).
Mutational Analysis.
To obtain a stable integrational mutant of the mycosubtilin synthetase, a two-plasmid system, developed for gene disruption in Lactococcus lactis, was used (33). This system uses a temperature-sensitive replication-initiation protein (RepA), delivered in trans on plasmid pVE6007 (33). In the SmaI restriction site of the plasmid pORI28 (EmR), lacking RepA, an internal fragment of the second ORF of the peptide synthetase operon was ligated. The PCR primers used to isolate this fragment were, myc33: CCG GAA TTC GAC CAC TTT CTG TCT CTG G; and myc32: AAT TTG ATG ATA TCT ATT CTC. Both plasmids were cotransformed into B. subtilis ATCC6633 by using protoplast transformation at the permissive temperature of 30°C (34). Site-specific integration was achieved by selection for chloramphenicol resistant colonies at the restrictive temperature of 37°C. Integration was verified by PCR control.
Results
Lipopeptides Produced by B. subtilis ATCC6633.
The lipopeptide fraction obtained from the culture supernatant of B. subtilis ATCC6633 was analyzed by FAB mass spectrometry and MALDI-TOF mass spectrometry. With FAB mass spectrometry, two clusters of ions were detected: (i) at m/z = 1,008.5, 1,022.5, and 1,036.5, and (ii) at m/z = 1,071, 1,085, 1,093, and 1,107. By MALDI-TOF mass spectrometry measurements, two series of ions were found: (i) at m/z = 1,045.7, 1,061.1, and 1,075.4, and (ii) at m/z = 1,109.9 and 1,123.9 (Fig. 2a). These ions indicate isoforms of surfactin (i) and mycosubtilin (ii), respectively. The FAB mass spectrometry data are consistent with [M + H]+-ions, whereas the m/z-values determined by MALDI-TOF mass spectrometry represent alkali adducts of these species.
Figure 2.
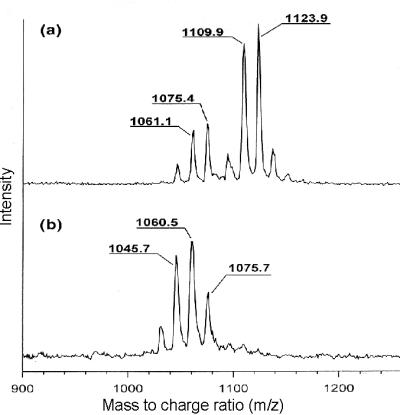
(a) MALDI-TOF mass spectrum of the lipopeptide fraction of B. subtilis ATCC6633. The first cluster (Left) corresponds to surfactin, and the second cluster (Right) corresponds to mycosubtilin. (b) MALDI-TOF mass spectrum of the lipopeptide fraction of B. subtilis ATCC6633 containing an integrational mutation in mycA, the gene encoding one of the mycosubtilin subunits. Only the cluster corresponding with surfactin is present.
The mass spectrometric results were corroborated by biochemical analysis. Table 1 presents the data obtained from the analysis of the amino acid composition of both lipopeptide fractions by using a Durrum Analyzer and reversed-phase HPLC, as described in Materials and Methods. The amino acid composition of the separated lipopeptide fractions corresponds to that of surfactin and mycosubtilin. In addition, when B. subtilis ATCC6633 was grown in medium containing 14C-labeled amino acids, 14C-labeled l-Asp, l-Glu, l-Leu, and l-Val were incorporated in the surfactin fraction, whereas 14C-labeled l-Asp, l-Glu, l-Ser, l-Pro, and l-Tyr were incorporated in the mycosubtilin fraction. These results substantiate that, under the growth conditions used, B. subtilis ATCC6633 produces two lipopeptides, surfactin and mycosubtilin.
Table 1.
Results of the biochemical analyses of the iipopeptide fraction of B. subtllis strain ATCC6633, determined as described in Material and Methods
| Surfactin | Myocosubtilin | |
|---|---|---|
| TLC | RfA = 0.90 | RfA = 0.23 |
| RfB = 0.65 | RfB = 0.27 | |
| RfC = 0.80 | RfC = 0.51 | |
| Durrum analyzer | Asx (1.0) Glx (1.0) | Asx (3.2) Glx (1.0) |
| Val (1.1) | Ser (0.8) Pro (1.0) | |
| Leu (3.2) | Tyr (0.8) | |
| Reverse-phase HPLC | Asx (1L) Glx (1L) | Asx: (1D, 2L) Glx (1L) |
| Val (1L) | Ser (0.8) Pro (1.0) | |
| HPLC | Leu (2L, 2D) | Tyr (1D) |
In the first row, the retention factors (Rf) for the three eluent systems used (A, B, and C) are given. The second row gives the results of the amino acid composition of the two lipopeptides determined with the Durrum analyzer. The thrid row gives the results of the absolute configuration determined with reverse-phase HPLC after derivatization with Marfey’s reagents.
Identification and Sequence Determination of a Peptide Synthetase.
The structural similarity of mycosubtilin with (lipo)peptide antibiotics suggests that mycosubtilin is synthesized by a member of the peptide synthetase family. Southern hybridization analyses, by using oligonucleotide probes directed against the coding regions of the highly conserved KAGGAYVP and GTTGKPKG amino acid motifs of peptide synthetases, was used to search in a genomic DNA library of B. subtilis ATCC6633 for the presence of peptide synthetases. The inserts of positive λ clones were analyzed, and a 22-kb insert with a restriction profile different from that of the known surfactin operon was selected. Alignments of the nucleotide sequence of the insert with other peptide synthetase genes revealed a typical modular structure and indicated that the isolated DNA fragment encoded part of an unknown peptide synthetase. Consecutive steps of inverse PCR were then used to complete the sequence of the peptide synthetase operon.
Specific Adenylation of Tyrosine by the Second Module.
Analysis of the peptide synthetase indicated the presence of seven consecutive modules. According to the colinearity rule, the second module of the mycosubtilin synthetase is responsible for the incorporation of tyrosine. To determine whether this assumption was correct, we attempted to overexpress and purify the tyrosine-activating module. For this purpose, we constructed an N-terminal (His)6-tag fusion with the first 775-aa residues of MycB, comprising the second amino acid-activating module. Overexpression of the 86-kDa fusion protein in E. coli resulted in the formation of inclusion bodies. Only by coexpression of the E. coli chaperones GroEL/ES did we obtain soluble fusion protein. The amino acid specificity of the purified module was determined in an ATP/PPi exchange assay. Of the 20 amino acids tested, only adenylation of l-tyrosine was detected. Because this module contains an epimerization domain, we conclude that the second module is responsible for the incorporation of d-tyrosine, which points to a mycosubtilin-producing peptide synthetase.
Insertional Mutagenesis Results in a Mycosubtilin-Negative Phenotype.
To unambiguously prove that we have cloned the mycosubtilin operon, the operon was genetically disrupted, and the effect on mycosubtilin production was examined. Because the competence development of B. subtilis ATCC6633 was insufficient to follow the protocol for natural transformation, we resorted to protoplast transformation by using a two-plasmid system initially developed for gene disruption in L. lactis (33). A 2-kb internal fragment of the second ORF of the operon was cloned into the integration plasmid pORI28 (EmR), which lacks repA, encoding the replication initiation protein. Protoplasts of B. subtilis ATCC6633 were transformed subsequently with plasmid pVE6007 (CmR), containing the gene for a temperature-sensitive RepA protein, and pORI28, containing 2 kb of ORF2. At the permissive temperature (30°C), RepA is stable, and both plasmids replicate. However, at the restrictive temperature (37°C), RepA is inactive, and pORI28 can be stably maintained only when integrated in the genome of the cell. Because integration occurs preferentially via homologous recombination, selection for chloramphenicol-resistant colonies at 37°C results in B. subtilis ATCC6633 mutants with pORI28 inserted into the second ORF of the peptide synthetase. PCR control by using primers flanking the expected integration site of pORI28 indicated that the plasmid had integrated at the expected locus (data not shown). Analysis by using MALDI-TOF mass spectrometry revealed that such mutants failed to produce mycosubtilin, because only mass peaks of surfactin were visible (Fig. 2b). Therefore, we conclude that we have identified the mycosubtilin synthetase.
Analysis of the Mycosubtilin Synthetase Gene Cluster.
Sequence analysis revealed the presence of a cluster of four ORFs, designated fenF, mycA, mycB, and mycC, respectively (Fig. 3). The cluster spans about 38 kb and is flanked by two putative transcriptional terminators located 685 bp upstream of the first ORF (fenF) and 71 bp downstream of the last ORF (mycC). The intergenic regions between the four ORFs, each containing a putative ribosomal binding site, varied between 35 and 100 bp.
Figure 3.

Schematic representation of the entire mycosubtilin operon comprising the ORFs fenF, mycA, mycB, and mycC. The deduced domain organizations of the different proteins specified by the operon are indicated (see Results for details).
The first ORF of the mycosubtilin operon, fenF, encodes a protein of 45.2 kDa, with 50% similarity to malonyl-CoA transacylases. The protein is 94% identical to FenF, encoded by a gene located upstream of the fengycin synthetase operon of B. subtilis F29–3 (35). MycA, mycB, and mycC encode proteins with estimated molecular masses of 449.3 kDa, 612.3 kDa, and 297.9 kDa, respectively. They show strong similarity with members of the peptide synthetase family and display the ordered assembly of conserved condensation, adenylation, and thiolation domains characteristic for such multienzymes [for reviews, see Marahiel et al. (14) and von Döhren et al. (36)] . The most conserved amino acid motifs, characteristic for the different domains commonly found in peptide synthetases, are indicated in Fig. 4. As shown in Fig. 3, a total of seven amino acid-activating modules can be distinguished: one in mycosubtilin synthetase subunit A (MycA), four in MycB, and two in MycC. Modules two, three, and six contain an epimerization domain, indicating that the activated amino acids are converted into the D-configuration. The number of modules and the location of epimerization domains correspond to the number of amino acids and the position of d-amino acids in the peptide moiety of mycosubtilin. The last domain of this multienzyme system is a thioesterase domain, which is presumably required for release and possibly for cyclization of the synthesized lipopeptide molecule (14).
Figure 4.

Comparison of the highly conserved peptide synthetase motifs of the various domains of the mycosubtilin synthetase subunits. The numbers represent the number of amino acids between the motifs in the mycosubtilin synthetase subunits.
MycA exhibits a remarkable complexity. The C-terminally located amino acid module is preceded by several domains with homology to proteins involved in the synthesis of fatty acids and polyketides. The conserved motifs, characteristic for these domains, were also identified in these domains and are aligned with a number of domains and proteins from other organisms, with a comparable enzymatic activity (Fig. 5). As indicated in Fig. 3, four different domains could be distinguished. The first domain (AL) shows 49% similarity to long-chain fatty acid CoA-ligases and contains a putative ATP binding box. The second and fourth domains (ACP) show 45% similarity to acyl carrier proteins, and both contain a putative 4′-phosphopantetheine binding box. The third domain (KS) is 53% similar to β-ketoacyl synthetases. These fatty acid synthase domains are followed by a domain (AMT) with 54% similarity to glutamate-1-semialdehyde aminotransferases, which contains a typical pyridoxal phosphate-binding box. The fatty acid synthase and amino transferase domains are connected to the first module of this peptide synthetase via an extra condensation and thiolation domain.
Figure 5.

Motif sequence alignment of MycA and fatty acid synthetases and amino transferases of various organisms. AL, acyl-CoA ligase; LCFA, long-chain fatty acid-CoA ligase; PksJ, polyketide synthetase J; ACP, acyl carrier protein; PksL-m5, polyketide synthetase L module 5; KS, β-keto acyl synthetase; PksL-m2, polyketide synthetase L module 2; 0156-m5, oleandomycine synthetase module 5; FAS, fatty acid synthetase; AMT, aminotransferase; GSA, glutamate semialdehyde aminotransferase.
Discussion
B. subtilis strain ATCC6633 produces a potent antifungal lipopeptide, mycosubtilin. In this report, we describe the identification of a peptide synthetase operon specifying the synthesis of this antibiotic. This conclusion is based on the following results: (i) The number of modules of the synthetase and the position of epimerization domains correspond to the amino acid sequence of mycosubtilin. (ii) The predicted amino acid specificity of the second module was confirmed in vitro. (iii) Genetic disruption of the operon abolished mycosubtilin production. The mycosubtilin operon is the first operon encoding a member of the iturin lipopeptide family that has been identified and sequenced.
The mycosubtilin operon containing 38 kb of DNA consists of four ORFs: fenF, mycA, mycB, and mycC. Although the overall structure of the synthetase resembles that of other peptide synthetases, mycosubtilin synthetase exhibits a number of important deviations. Most striking is the large N-terminal multifunctional part of MycA, with domains that show homology to fatty acid and polyketide synthetases. It is likely that these domains play a role in the incorporation of the β-amino fatty acid moiety into the mycosubtilin molecule. In bacteria and higher plants, fatty acids are synthesized by separate polypeptides, so-called type II FAS systems (37). In contrast, vertebrates and fungi use type I FAS systems, in which these polypeptides are fused into large multifunctional enzymes. Although the multifunctional domain organization of MycA resembles that of type I FAS systems, the presence of an acyl CoA-ligase and amino transferase as part of such a multifunctional protein, to our knowledge, is new. On the basis of the function of similar domains in fatty acid, polyketide, and peptide synthetases, as well as in amino transferases, we propose the following model, schematically presented in Fig. 6, for the synthesis of mycosubtilin and other iturin lipopeptides. In the first step (Fig. 6, step I), the acyl CoA-ligase domain couples coenzymeA (CoA) to a long-chain fatty acid, presumably myristate, in an ATP-dependent reaction. The activated fatty acid is then transferred to the 4-phosphopantetheine cofactor of the first acyl carrier domain. In addition, a malonyl-CoA is attached to the 4-phosphopantetheine cofactor of the second acyl carrier domain, a reaction catalyzed by the malonyl-CoA transacylase, encoded by fenF. We assume that in these early steps of synthesis, asparagine is activated and coupled to the 4-phosphopantetheine cofactor of the first module. The second step is the condensation of the malonyl and acyl thioesters (Fig. 6, step II), catalyzed by the β-ketoacyl synthetase domain, resulting in a β-ketoacyl thioester. In contrast to normal fatty acid synthesis, this β-ketoacyl thioester is not converted into a fatty acid by subsequent reduction and dehydration reactions but is converted into a β-amino fatty acid by a transamination reaction, catalyzed by the domain homologous to amino transferases (Fig. 6, step III). Subsequently, the β-amino fatty acid is transferred to a thiolation domain, catalyzed by the preceding condensation domain. The β-amino fatty acid is then coupled to the activated asparagine thioester, catalyzed by the condensation domain preceding the first module of the peptide synthetase (Fig. 6, step IV). In subsequent condensation reactions, the mycosubtilin molecule is then synthesized, analogous to other nonribosomal peptide synthetase reactions (Fig. 6, step V).
Figure 6.

Model for the initiation of iturin lipopeptides synthesis. For details, see Discussion. The reaction intermediates are attached to MycA as carboxy thioesters via 4′-phosphopantheteine cofactors. The carbon chain structure of the fatty acid R is CH3(CH2)n, where n is generally 14 (myristate).
Research into the synthesis of surfactin indicated that lipopeptide synthesis is initiated by the coupling of a β-hydroxy fatty acid thioester to the first activated amino acid (5), although enzymes catalyzing this reaction have not yet been identified. The model presented in Fig. 6 suggests that a condensation domain in front of the first module is sufficient to catalyze this coupling. In all lipopeptide synthetases identified so far, comprising the surfactin, fengycin, lichenysin and mycosubtilin synthetases, the first module is preceded by a condensation domain (4, 7, 11). If the fatty acid is delivered as an activated thioester attached to an acyl carrier protein, a common intermediate in bacterial fatty acid synthesis, it is likely that these condensation domains catalyze the transfer of the fatty acid to the first amino acid to be incorporated into the peptide chain.
The mycosubtilin operon encodes a hybrid synthetase, integrating fatty acid synthesis and peptide synthesis. The unique combination of enzymatic domains in MycA provides several additional possibilities to engineer lipopeptides. Recently, modifications of the peptide moiety of lipopeptides have been achieved (20). A hybrid synthetase subunit such as MycA offers the potential to modify the fatty acid moiety of lipopeptides as well. In addition, MycA might be fused to other peptide synthetases, to turn them into lipopeptide synthetases.
In many peptide synthetases, the successive modules are distributed over several protein subunits. The positions of the epimerization domains generally determine the subunit size, because epimerization domains constitute the last domain in a synthetase subunit. As a result, the successive subunit starts with a condensation domain. For example, this organization applies to the fengycin and surfactin synthetases of B. subtilis (4, 7). The subunit organization of mycosubtilin synthetase deviates from this rule. As shown in Fig. 3, the subunit separation in the mycosubtilin synthetase is between condensation and adenylation domains, as a consequence of which the subunits MycB and MycC start with an adenylation domain and not with a condensation domain. In addition, the epimerization domains of mycosubtilin synthetase are positioned within the synthetase subunits. Apart from mycosubtilin synthetase, only the HC-toxin synthetase of Cochliobolus carbonum and the bacitracin synthetase of Bacillus licheniformis contain internal epimerization domains (10, 12).
The complete genome of B. subtilis 168 has been sequenced (38). Only the biosynthetic gene clusters for surfactin and fengycin appeared to be present in this strain. Analysis of the DNA sequences adjacent to the mycosubtilin operon revealed that this operon is bounded by the same genes as the fengycin operon in B. subtilis 168 (Fig. 7). Because we were not able to detect fengycin biosynthetic genes in B. subtilis ATCC6633 (data not shown), the fengycin and mycosubtilin operons appear to be exchanged in both B. subtilis strains. FenF, the first gene of the mycosubtilin operon, is not present in B. subtilis 168, but has been identified immediately downstream of the fengycin operon of B. subtilis F29–3 (35). Several B. subtilis strains are known that produce both fengycin and iturin lipopeptides. These preliminary data suggest that in such strains the fengycin and iturin operons may be tandemly arranged on the chromosome. Apparently a high degree of genetic flexibility exists in this region of the B. subtilis genome.
Figure 7.

Comparison of the location of the mycosubtilin operon in B. subtilis ATCC6633 (shaded) and the location of the fengycin operon in B. subtilis 168 (white). The genes pbp and yngL (black) are identical in the two B. subtilis strains. Transition points of intergenic DNA where base sequence identity discontinues is presented above the operons (not to scale).
Acknowledgments
This work was supported by the European Union (EU Grant CT950176).
Abbreviations
- MycA
B, C
- mycosubtilin synthetase subunits
FAB, fast atom bombardment
- MALDI-TOF
matrix-assisted laser desorption/ionization-time of flight
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
The sequence reported in this paper has been deposited in the GenBank database (accession no. AF184956).
References
- 1.Kakinuma A, Hori M, Isono M, Tamura G, Arima K. Agric Biol Chem. 1969;33:973–976. [Google Scholar]
- 2.Marget-Dana R, Peypoux F. Toxicology. 1994;87:151–174. doi: 10.1016/0300-483x(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 3.Vanittanakom N, Loeffler W, Jung G. J Antibiot. 1986;39:888–901. doi: 10.7164/antibiotics.39.888. [DOI] [PubMed] [Google Scholar]
- 4.Cosmina P, Rodriguez F, de Ferra F, Grandi G, Perego M, Venema G, van Sinderen D. Mol Microbiol. 1993;8:821–831. doi: 10.1111/j.1365-2958.1993.tb01629.x. [DOI] [PubMed] [Google Scholar]
- 5.Menkhaus M, Ullrich C, Kluge B, Vater J, Vollenbroich D, Kamp R M. J Biol Chem. 1993;268:7678–7684. [PubMed] [Google Scholar]
- 6.Steller S, Vollenbroich D, Leenders F, Stein T, Conrad B, Hofemeister J, Jaques P, Thonart P, Vater J. Chem Biol. 1999;6:31–41. doi: 10.1016/S1074-5521(99)80018-0. [DOI] [PubMed] [Google Scholar]
- 7.Tosato V, Albertini A M, Zotti M, Sonda S, Bruschi C V. Microbiology. 1997;143:3443–3450. doi: 10.1099/00221287-143-11-3443. [DOI] [PubMed] [Google Scholar]
- 8.Ullrich C, Kluge B, Palacz Z, Vater J. Biochemistry. 1991;30:6503–6508. doi: 10.1021/bi00240a022. [DOI] [PubMed] [Google Scholar]
- 9.Haese A, Schubert M, Herrmann M, Zocher R. Mol Microbiol. 1993;7:905–914. doi: 10.1111/j.1365-2958.1993.tb01181.x. [DOI] [PubMed] [Google Scholar]
- 10.Konz D, Klens A, Schörgendorfer K, Marahiel M A. Chem Biol. 1997;4:927–937. doi: 10.1016/s1074-5521(97)90301-x. [DOI] [PubMed] [Google Scholar]
- 11.Konz D, Doekel S, Marahiel M A. J Bacteriol. 1999;181:133–140. doi: 10.1128/jb.181.1.133-140.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott-Craig J S, Panaccionem D G, Pocard J A, Walton J D. J Biol Chem. 1992;267:26044–26049. [PubMed] [Google Scholar]
- 13.Stachelhaus T, Marahiel M A. J Biol Chem. 1995;270:6163–6169. doi: 10.1074/jbc.270.11.6163. [DOI] [PubMed] [Google Scholar]
- 14.Marahiel M A, Stachelhaus T, Mootz H D. Chem Rev. 1997;97:2651–2673. doi: 10.1021/cr960029e. [DOI] [PubMed] [Google Scholar]
- 15.Stein T, Vater J, Kruft V, Otto A, Wiimann-Liebold B, Franke P, Panico M, McDowell R, Morris H R. J Biol Chem. 1996;271:15428–15435. doi: 10.1074/jbc.271.26.15428. [DOI] [PubMed] [Google Scholar]
- 16.Turgay K, Krause M, Marahiel M A. Mol Microbiol. 1992;6:529–546. doi: 10.1111/j.1365-2958.1992.tb01498.x. [DOI] [PubMed] [Google Scholar]
- 17.Schlumbohm B, Stein T, Ullrich C, Vater J, Krause M, Marahiel M A, Kruft V, Wittmann-Liebold B. J Biol Chem. 1991;266:23135–23141. [PubMed] [Google Scholar]
- 18.De Crécy-Lagard V, Marlière P, Saurin W. C R Acad Sci Ser III. 1995;318:927–936. [PubMed] [Google Scholar]
- 19.Stein T, Kluge B, Vater J, Franke P, Otto A, Wittmann-Liebold B. Biochemistry. 1995;34:4633–4642. doi: 10.1021/bi00014a017. [DOI] [PubMed] [Google Scholar]
- 20.Stachelhaus T, Schneider A, Marahiel M A. Science. 1995;269:69–72. doi: 10.1126/science.7604280. [DOI] [PubMed] [Google Scholar]
- 21.Birnboim H C, Doly J. Nucleic Acids Res. 1979;7:1513–1523. doi: 10.1093/nar/7.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ochman H, Medhora M M, Garza D, Hart D L. PCR Protocols. San Diego: Academic; 1990. [Google Scholar]
- 23.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 24.Spizizen J. Proc Natl Acad Sci USA. 1958;44:1072–1078. doi: 10.1073/pnas.44.10.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walton R P, Woodruff H B. J Clin Invest. 1949;28:924–926. doi: 10.1172/JCI102180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venema G, Pritchard R H, Venema-Schröder T. J Bacteriol. 1965;89:1250–1255. doi: 10.1128/jb.89.5.1250-1255.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ebata M, Miyazaki K, Takahashi Y. J Antibiot. 1991;22:467–472. [PubMed] [Google Scholar]
- 28.Von Arx E, Faupel M, Brugger M. J Chromatogr. 1976;12:329–341. [Google Scholar]
- 29.Marfey P. Carlsburg Res Commun. 1984;49:591–596. [Google Scholar]
- 30.Bernhard F, Demel G, Soltani K, Döhren H V, Blinov V. DNA Sequence. 1996;6:319–330. doi: 10.3109/10425179609047570. [DOI] [PubMed] [Google Scholar]
- 31.Yanisch-Perron C, Vielra J, Messing J. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- 32.Cole P A. Structure (London) 1996;4:235–242. doi: 10.1016/s0969-2126(96)00028-7. [DOI] [PubMed] [Google Scholar]
- 33.Leenhouts K, Buist G, Bolhuis A, ter Berge A, Kiel J, Mierau I, Dabrowska M, Venema G, Kok J. Mol Gen Genet. 1996;253:217–224. doi: 10.1007/s004380050315. [DOI] [PubMed] [Google Scholar]
- 34.Chang S, Cohen S N. Mol Gen Genet. 1979;168:111–115. doi: 10.1007/BF00267940. [DOI] [PubMed] [Google Scholar]
- 35.Chen C L, Chang L K, Chang Y S, Liu S T, Tshen J S. Mol Gen Genet. 1995;248:121–125. doi: 10.1007/BF02190792. [DOI] [PubMed] [Google Scholar]
- 36.von Döhren H, Keller U, Vater J, Zocher R. Chem Rev. 1997;97:2675– 2705. doi: 10.1021/cr9600262. [DOI] [PubMed] [Google Scholar]
- 37.Hopwood D A, Sherman D H. Annu Rev Genet. 1990;24:37–66. doi: 10.1146/annurev.ge.24.120190.000345. [DOI] [PubMed] [Google Scholar]
- 38.Kunst F, Ogasawara N, Moszer I, Albertini A M, Alloni G, Azevedo V, Bertero M G, Bessiers P, Bolotin A, Borchert S, et al. Nature (London) 1997;390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]