

Abstract
The vacuolar ATPases (V-ATPases) are multisubunit complexes containing two domains. The V1 domain (subunits A–H) is peripheral and carries out ATP hydrolysis. The V0 domain (subunits a, c, c′ c″, d and e) is membrane integral and carries out proton transport. In yeast, there are three proteolipid subunits: subunit c (Vma3p), subunit c′(Vma11p) and subunit c″(Vma16p). The proteolipid subunits form a six-membered ring containing single copies of subunits c′ and c″ and four copies of subunit c. To determine the possible arrangements of proteolipid subunits in V0 that give rise to a functional V-ATPase complex, a series of gene fusions were constructed in order to constrain the arrangement of pairs of subunits in the ring. Fusions containing c′′ employed a truncated version of this protein lacking the first putative transmembrane helix (which we have previously shown to be functional), in order to insure that the N and C-termini of all subunits were located on the luminal side of the membrane. Fusion constructs were expressed in strains disrupted in c′, c′′ or both but containing a wild copy of c to insure the presence of the required number of copies of subunit c. The c-c′(ΔTM1), c′(ΔTM1)-c′ and c′-c constructs all complemented the vma− phenotype and gave rise to complexes possessing greater than 25% of wild type levels of activity. By contrast, neither the c-c′, the c′-c′(ΔTM1) nor the c′(ΔTM1)-c constructs complemented the vma− phenotype. These results suggest that functionally assembled V-ATPase complexes contain the proteolipid subunits arranged in a unique order in the ring.
The vacuolar (H+)-ATPases (V-ATPases1) are a family of ATP-dependent proton pumps that acidify intracellular compartments like endosomes, lysosomes and synaptic vesicles (1–6). Intracellular V-ATPases are important in processes such as membrane traffic, protein degradation, coupled transport and the entry of many viruses and toxins, including influenza virus and anthrax toxin (1,7). They also pump protons across the plasma membrane in certain cells, including renal intercalated cells, epididymal cells, osteoclasts and tumor cells (4,8–10). Plasma membrane V-ATPases in these cells are important in acid-base balance in the kidney, sperm maturation, bone resorption and tumor metastasis, respectively.
The V-ATPases are multisubunit complexes containing two domains (1–6). The peripheral V1 domain is composed of eight subunits (A–H) and functions to hydrolyze ATP. The integral V0 domain is composed of six subunits and is responsible for proton transport. V0 from both yeast and mammalian sources contain a set of common subunits (a,c,c″,d and e) (1,11). In addition, the yeast V0 domain contains an additional proteolipid subunit (c′) whereas the V0 domain from at least some mammalian sources contains a glycoprotein subunit termed Ac45 (12).
The proteolipid subunits of the V-ATPase (subunits c, c′ and c″) are homologous to each other and to the c subunit of the F-ATPase (13,14). The c subunit of the F-ATPase is an 8-kDa protein that contains two transmembrane helices (TMs). The second TM contains a buried acidic amino acid that is critical for proton transport (15). The F-ATPase c subunits are arranged in a ring. By contrast with the F-ATPase, which contains only a single type of proteolipid subunit, the V-ATPases contain either two or three different proteolipid subunits. In yeast, all three proteolipid subunits have been shown to be essential for activity (14).
Subunits c and c′ have molecular weights around 17 kDa and contain four transmembrane helices. Subunit c″ has a molecular weight of 23 kDa and contains a fifth transmembrane helix at the N-terminus (14). Each proteolipid subunit contains a buried glutamic acid residue critical for proton transport. This critical residue resides in TM4 of subunits c and c′ but TM3 of subunit c″(14). Subunit c″ contains an additional buried glutamic acid residue in TM5, but this residue is not essential for activity. The essential glutamic acid residues in each subunit are thought to undergo reversible protonation and deprotonation during proton transport through the V0 domain.
Although evidence from quantitative amino acid analysis suggests that the proteolipid subunits of the V-ATPase form a six membered ring (16), the arrangement of the proteolipids in the ring is not known. The purpose of this study was to determine which of the possible arrangements of the three proteolipid subunits give rise to a functional V-ATPase complex. This was addressed by construction of a series of gene fusions in which pairs of the proteolipid subunits were constrained to adopt a particular orientation. The results suggest that the V-ATPase proteolipid subunits in the functionally assembled complex adopt a unique orientation.
EXPERIMENTAL PROCEDURES
Materials
Zymolyase 100T was obtained from Seikagaku America, Inc. Protease inhibitors (leupeptin, pepstatin, aprotinin), the monoclonal antibody 3F10 (directed against the influenza hemagglutinin (HA) antigen) conjugated with horseradish peroxidase, mouse monoclonal antibody 8B1–F3 against the yeast V-ATPase A subunit and the mouse monoclonal antibody 10D7 against the 100-kDa subunit a were from Invitrogen- Molecular Probes. Bacterial and yeast culture media were purchased from Difco. Restriction endonucleases, T4 DNA ligase, and other molecular biology reagents were from GIBCO, Promega, and New England Biolabs. Concanamycin A, ATP, phenylmethylsulfonylfluoride and most other chemicals were purchased from Sigma.
Plasmid construction and epitope tagging
We genetically fused the different proteolipid subunits to each other in six different combinations (Fig 1a). The plasmid pYW3–16 encoding the fusion of c and c′ (ΔTM1) was constructed by ligation of plasmid pTN3 containing VMA3 with a PCR product derived from pTN16 encoding a VMA16 lacking amino acids 1–48 but containing a single HA tag at the C-terminus. The linker region connecting c and c′ (ΔTM1) was 14 amino acids in length and included the C-terminal 6 amino acids of c and amino acids 49–54 of c′′ (Fig. 1b). pYW16-3, pYW16-11, pYW11–16 and pYW11-3 were all constructed using the same strategy as pYW3-16 and contained linker regions of 14 amino acids (for the first three) or 10 amino acids (for pYW11-3) (Fig. 1b). pYW16-3, pYW11-16 and pYW11-3 contained single HA tags at the C-terminus while pYW16-11 contained 3 tandem HA tags at the C-terminus. Initial attempts to construct a fusion protein containing c followed by c′ using the same strategy failed due to cleavage of the fusion protein in the linker region connecting the two subunits. To solve this problem, the linker region was replaced with the 14 amino acid linker from pYW3-16 (Fig. 1b), which then gave a stable fusion construct. pYW3-11 again contained a single HA tag at the C-terminus. For construction of 11-3pRS316, an XhoI and SpeI fragment of pYW11-3 was recloned into pRS316. For construction of 3-16pRS316, an EcoRI and SalI frangment of pYW3-16 was recloned into pRS316. The oligonucleotides used for amplification of fusion genes are as follows: pYW3-11 forward, GTCGGATCCTTCTTGTTAAGGACATC CGCTCCCTTTTTCGGGTTCGC, and pYW3-11 reverse, GAAACTAGTTTAGGCGTAGTCGGGCA CGTCGTAGGG; pYW3-16 forward, GTCGGATCCTTCTTGTTAAGGACATC C, and the same reverse primer as pYW3-11. pYW11-3 forward, CGGATTCATGACTGAATTGTGTCCTG, and pYW11-3 reverse, CGGGAATCCTTAGGCGTAGTCGGGCA CGTCGTAGGG; pYW16-3 forward, GGGACGCGTACTGAATTGTGTCCTGT C, and the same reverse primer as pYW11-3. pYW16-11 forward, ACGCGTGTCGACATGTCAACGCAACT C, and pYW16-11 reverse, CGGGAATTCTTAGGCGTAGTCGGGCA CGTCGTAGGGGTAACAGACAACATCT TGAGT.
Figure 1. Proteolipid subunit fusion proteins.

A, Topological models of the six proteolipid fusion proteins created in this study. Transmembrane helices are numbered from the N-terminus and are shown as: subunit c (grey gradient); subunit c′ (white); and subunit c′′ (solid grey). B, The linker sequences used to fuse two proteolipids. The loop regions are shown in bold, and the black lines underneath the sequences denote the amino acids derived from each proteolipid subunit.
Strains and culture conditions
The yeast strains and plasmids used in this study are listed in Tables I and II. HA-tagged forms of each proteolipid subunit (and the c″( ΔTM1) construct) were expressed in the strain in which the corresponding gene was disrupted. All fusion proteins were also HA tagged. The c-c′ and c′-c fusions were expressed in a strain disrupted in subunit c′ but containing the endogenous copies of subunits c and c″. Similarly, the c-c″ and c″-c fusions were expressed in a strain disrupted in c″ but containing the endogenous copies of subunits c and c′. The c′-c″ and c″-c′ fusions were expressed in a strain disrupted in c′ and c″ but containing the endogenous copy of subunit c. By expressing all fusions in strains containing the wild-type c gene, we could insure that the requisite number of copies of subunit c to form a functional ring would be present. All yeast strains were cultured in synthetic dropout minimal media supplemented with the appropriate amino acids or YEPD media buffered to pH 5.5 using 50 mM succinate/phosphate. To test for a vma− phenotype, saturated cultures were diluted to an absorbance at 600 nm of 0.20. Serial dilutions of the cultures were prepared in YEPD media buffered to pH 7.5 and growth was monitored on YEPD plates buffered to the same pH.
Table I.
Yeast strains used in this study.
| Strain | Genetype | Source |
|---|---|---|
| TN101 | MATa, ura3–52, lys2–801, ade2–101, trp1-Δ63, his3-Δ200, leu2-Δ1, vma3:: TRP1 | Ref. 26 |
| TN102 | MATa, ura3–52, lys2–801, ade2–101, trp1-Δ63, his3-Δ200, leu2-Δ1, vma11:: TRP1 | Ref. 26 |
| TN103 | MATa, ura3–52, lys2–801, ade2–101, trp1-Δ63, his3-Δ200, leu2-Δ1, vma16:: TRP1 | Ref. 26 |
| YW100 | MATa, ura3–52, lys2–801, ade2–101, trp1-Δ63, his3-Δ200, leu2-Δ1, vma11::Kanr, vma16::TRP1 | This study |
| LGY125 | SF838-1D alpha ade6 leu2 ura3 pep4-3 his4 vma3::Kanr vma11::Hygr | Dr. Tom Stevens |
| LGY139 | SF838-1D alpha ade6 leu2 ura3 pep4-3 his4 vma3::Kanr vma16::Natr | Dr. Tom Stevens |
Table II.
Plasmids used in this study.
| Plasmid | Gene | Protein expressed | Strain expressed in | Source |
|---|---|---|---|---|
| pRS413 | None | None | TN103 | Ref. 26 |
| pTN3 | VMA3::1×HA | c | TN101 | Ref. 26 |
| pTN11 | VMA11::1×HA | c′ | TN102 | Ref. 26 |
| pTN16 | VMA16::1×HA | c′′ | TN103 | Ref. 26 |
| pTN16-TM1 | VMA16(Δ2–41)::1×HA | c′′(ΔTM1) | TN103 | Ref. 26 |
| pYW3-11 | VMA3::VMA11::1×HA | c-c′ | TN102 | This study |
| pYW11-3 | VMA11::VMA3::1×HA | c′-c | TN102 | This study |
| pYW3-16 | VMA3::VMA16(Δ1-48)::1×HA | c-c′′(ΔTM1) | TN103 | This study |
| pYW16-3 | VMA16(Δ2–41)::VMA3::1×HA | c′′(ΔTM1)-c | TN103 | This study |
| pYW11-16 | VMA11::VMA16(Δ1-41)::1×HA | c′-c′′(ΔTM1) | YW100 | This study |
| pYW16-11 | VMA16(Δ2–41)::VMA11(Δ2–10)::3×HA | c′′(ΔTM1)-c′ | YW100 | This study |
| 11-3pRS316 | VMA11::VMA3::1×HA | c′-c | LGY125 | This study |
| 3-16pRS316 | VMA3::VMA16(Δ1–48)::1×HA | c-c′′(ΔTM1) | LGY139 | This study |
Transformation and selection
Yeast cells were transformed using the lithium acetate method (17). The transformants were selected on ura minus plates (for 11-3pRS316 and 3–16pRS316) or histidine minus plates for other strains, and growth phenotypes of the mutants were assessed on YEPD plates buffered with 50 mM KH2PO4 or 50 mM succinic acid to either pH 7.5 or pH 5.5.
Protein preparation, SDS-PAGE, and immunoblot analysis
Whole cell lysates were prepared using a modification of the previously described protocol (18): 5 ml yeast cultures were grown overnight at 30 °C to an absorbance at 600 nm of 1.0. Cells were collected by centrifugation and washed with 1 ml cold extraction buffer (150 mM ammonium sulfate, 10% glycerol, 1 mM EDTA, 200 mM Tris (pH 8.0), 2mM DTT and protease inhibitors). Cells were resuspended in 200 μl extraction buffer and vortexed with glass beads for 5×1 min pulses. Unbroken cells were removed by sedimentation for 3 min at 4000 rpm and samples of the supernatant were collected. Vacuolar membrane vesicles were isolated as previously described (19). Whole cell lysates and vacuolar membranes were separated by SDS-PAGE on 4–15% gradient acrylamide gels, as previously described (20). Expression of the fusion proteins was analyzed by Western blotting using the horseradish peroxidase-conjugated monoclonal antibody 3F10 against HA, whereas subunit a and subunit A were detected using the monoclonal antibodies 10D7 and 8B1–F3, respectively, followed by a horseradish peroxidase-conjugated secondary antibody, as previously described (21,22). Blots were developed using a chemiluminescent detection method obtained from Kirkegaard & Perry Laboratories.
ATPase activity and ATP dependent proton transport
ATPase activity was measured using a coupled spectrophotometric assay (23). The reactions were carried out at 30 °C and vacuolar membrane vesicles were incubated with DMSO or 1μM concanamycin A (in DMSO) for 5 min prior to measurement of ATPase activity. ATP dependent proton transport was measured by fluorescence quenching with the fluorescence probe 9-amino-6-chloro-2- methoxyacridine in transport buffer (50 mM NaCl, 30 mM KCl, 20 mM HEPES, 0.2 mM EGTA, 10% glycerol (pH 7.0)) as described (23) in the presence or absence of 1 μM concanamycin A.
RESULTS
Construction of proteolipid fusion proteins
Previous results from studies of the bovine clathrin-coated vesicle and yeast V-ATPases suggest that the proteolipid ring of the yeast V-ATPase consists of four copies of subunit c and one copy of subunits c′ and c′′ (16,24). The arrangement of subunits in the proteolipid ring, however, is not known. In order to determine which arrangements of the subunits in the proteolipid ring are able to give rise to a functional V-ATPase complex, a series of six gene fusions were constructed between pairs of proteolipid subunit genes (Fig. 1a). These fusion constructs were then expressed in the appropriate deletion strains (Tables I and II). By expressing proteolipid subunits as fusion constructs, the resultant oligomer is restricted in the possible arrangements the proteolipid subunits can adopt. Assuming that each proteolipid subunit inserts in the ring with the same internal arrangement of transmembrane helices (see Discussion), there are five possible arrangements of the three proteolipid subunits in a six-membered ring (Fig. 2). In two of these, subunits c′ and c″ are adjacent to each other, either clockwise or counterclockwise relative to each other (models A and B). In two other arrangements, subunits c′ and c″ are separated by a single copy of subunit c (c′ and c″ are again either clockwise or counterclockwise relative to each other – models C and D). In the final arrangement, subunit c′ and c″ are on opposite sides of the six-membered ring separated by two copies of subunit c (model E). Each of these arrangements make specific predictions about which proteolipid fusions should give rise to a functional V-ATPase (Table III).
Figure 2. Possible arrangements of the three proteolipid subunits (c, c′ and c″) in a six-membered proteolipid ring of the yeast V-ATPase.
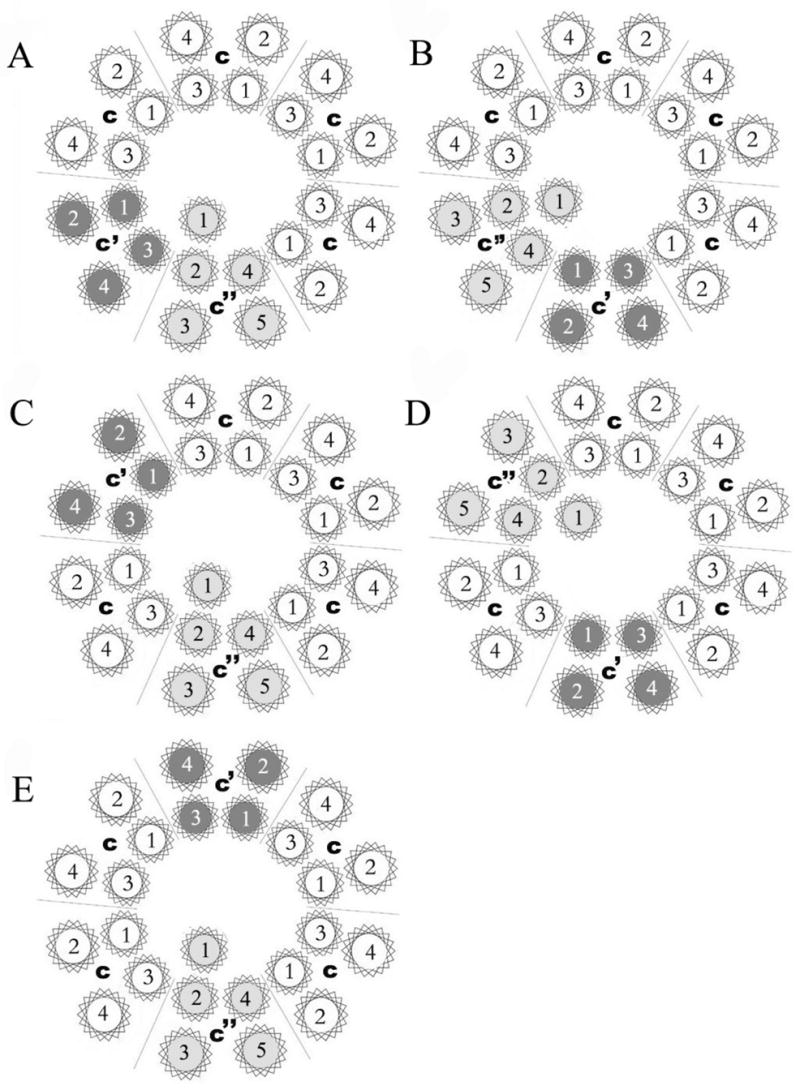
Subunit c is shown in white, subunit c′ is shown in dark gray and subunit c′′ is shown in light gray. The proteolipid ring is viewed from the lumenal side of the membrane, with the arrangement of helices within each subunit modeled after that observed in the c ring of the Na+ V-ATPase from Enterococcus hirae (27). A five TM model of subunit c″ is assumed in this diagram.
Table III.
| Model A | Model B | Model C | Model D | Model E | Result | |
|---|---|---|---|---|---|---|
| c-c′ | + | − | + | + | + | − |
| c-c′′(ΔTM1) | − | + | + | + | + | + |
| c′-c | − | + | + | + | + | + |
| c′-c′′(ΔTM1) | + | − | − | − | − | − |
| c′′(ΔTM1)-c | + | − | + | + | + | − |
| c′′(ΔTM1)-c′ | − | + | − | − | − | + |
“+” indicates the proteolipid ring shown in the corresponding model in Fig. 2 can be formed from the fusion construct indicated whereas “−” indicates the ring cannot be formed.
Topological studies of the yeast proteolipids suggest that subunit c″ has 5 transmembrane helices with the N-terminus on the cytoplasmic side of the membrane whereas subunits c and c′ both have 4 transmembrane helices with both the N and C-terminus on the luminal side of the membrane (25). It was therefore necessary to delete TM1 of subunit c″ in order to insure that construction of fusion proteins containing subunit c″ at the C-terminus did not change the way in which the transmembrane segments of subunit c″ were inserted (Fig. 1a). We and others had previously shown that strains expressing subunit c″ from which TM1 had been deleted still gave rise to a functional V-ATPase complex (25,26). Thus, c″ (ΔTM1) was used in place of c″ in construction of fusion proteins in this study.
Care was taken to define the transmembrane segments as well as the minimal length of the loop connecting the two proteins. We defined the transmembrane helix/connecting loop boundaries of the yeast proteolipids by comparing them to the NtpK subunit of the V-Type (Na+)-ATPase from Enterococcus hirae (27). If the connecting loop between the fused proteins is too short it may not allow the two halves of the protein to adopt a native conformation or insert properly into the membrane. However, if the connecting loop is too long it could allow for another c subunit to insert between the two halves of the fusion construct. Using a model of the c12 ring from the E. coli F-ATPase and the c10 ring from E. hirae V-ATPase (15,27), we estimate that a loop length of 10–14 amino acids is sufficient to connect adjacent subunits without allowing an additional copy of subunit c to insert between them. Therefore all proteolipid fusions were constructed with a linker length of 10–14 amino acids.
In vivo function and stability of V-ATPase containing proteolipid subunit fusions
To address the functional role of different fusions, we first tested the ability of these constructs to complement the phenotype of yeast strains from which the various proteolipid genes have been deleted. It has been shown previously that yeast lacking any of the V-ATPase proteolipid genes display a conditional lethal phenotype (vma−) characterized by an inability to grow at pH 7.5 but retaining the ability to grow at pH 5.5 (28). Data from our lab and others have previously shown that plasmid-borne VMA3, VMA11, VMA16, or VMA16ΔTM1 were able to complement their own disruption (25,26). The c-c′(ΔTM1), c′(ΔTM1)-c′ and c′-c fusion proteins were all able to confer wild-type growth on YEPD media buffered at pH 7.5, while the c-c′, c′-c′(ΔTM1) and c′(ΔTM1)-c constructs could not (Fig. 3). We also observed that the c-c′(ΔTM1) fusion construct did not support growth at pH 7.5 of the vma3Δ, vma16Δ double deletion strain while the c′-c fusion construct did not support growth at pH 7.5 of the vma3Δ, vma11Δ double deletion strain (data not shown). This latter result is consistent with the need for multiple copies of subunit c, which cannot be provided by the fusion constructs alone in the double deletion strains.
Figure 3. The ability of proteolipid fusion proteins to complement the vma− phenotype in proteolipid deletion strains.

Serial dilutions of yeast strains carrying plasmid-borne proteolipids and proteolipid fusions were spotted onto YEPD agar plates at either pH 5.5 or pH 7.5 and grown overnight at 30°C. Five μl of yeast culture grown to an absorbance at 600 nm of 0.20 was spotted onto the plate at the far left hand position for each pH and the spots to the right of this position correspond to five μl of serial 10-fold dilutions of the original culture.
Next the expression and stability of the fusion proteins as well as their ability to assemble into a V-ATPase complex was tested. Western blot analysis was performed on whole cell lysates using the monoclonal antibody 10D7 for subunit a, 8B1-F3 for subunit A, and 3F10 for the HA tag. Since there is currently no available antibody raised against any of the proteolipid subunits, an HA-epitope tag was added at the C-terminal end of all constructs. Three HA tags were added to the C-terminus of c′(ΔTM1)-c′ because of the weak signal observed with only one tag. All the proteolipid fusions had apparent molecular masses of 32–36 kDa, whereas the monomers (c, c′ or c′(ΔTM1)) had a molecular mass of 17 kDa (Fig. 4). The lower level of antibody staining of the c′(ΔTM1)-c construct in whole cell lysate may reflect altered reactivity of the HA epitope in this construct or the low expression or degradation of the fusion protein in this mutant strain. These results demonstrate that five of the six fusion proteins are expressed and are stable.
Figure 4. The presence of V1 and V0 subunits in whole cell lysates.
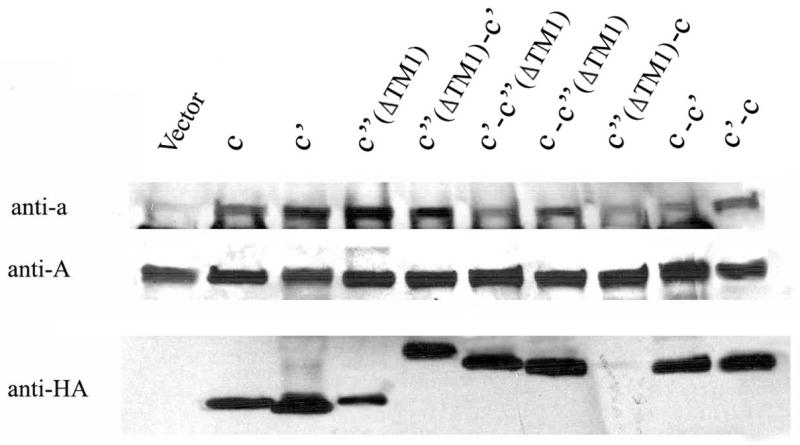
Whole cell lysates were isolated from strains expressing proteolipid subunits and proteolipid fusion constructs, subjected to SDS-PAGE, transferred to nitrocellulose, and probed with anti-a (10D7), anti-A (8B1–F3), or anti-HA (3F10) antibodies as described under Experimental Procedures.
Previous studies have shown that the loss of subunit c or subunit d results in reduced levels of subunit a (29), likely due to reduced subunit a stability when not assembled into V0. In contrast, because V1 assembles independently of V0 (29,30), V1 subunits are unaffected by the deletion of V0 subunits. To determine if the proteolipid fusion constructs were able to stabilize subunit a, Western blotting was performed on whole cell lysates. As expected, lysates from all strains, including that carrying the empty vector, showed normal levels of subunit A (Fig. 4). Lysates isolated from strains expressing c-c′(ΔTM1), c′(ΔTM1)-c′, or c′-c showed almost the same level of subunit a relative to strains carrying the c, c′ or c′(ΔTM1) controls. Strains carrying the c-c′, c′-c′(ΔTM1) or c′(ΔTM1)-c showed reduced levels of subunit a, suggesting that these proteolipid fusions do not assemble properly.
To confirm this assembly defect and to test for proper targeting of the V-ATPase subunits to the vacuolar membrane, partially purified vacuoles were subjected to SDS-PAGE and Western blot analysis was performed. As can be seen in Fig. 5, Western blots of isolated vacuoles carrying c-c′(ΔTM1), c′(ΔTM1)-c′ or c′-c showed slightly reduced levels of subunits a and A relative to vacuoles isolated from the strains expressing the monomer of the three proteolipid subunits, whereas the c-c′, c′-c′(ΔTM1) and c′(ΔTM1)-c constructs showed dramatically reduced levels of subunits a and A, showing that the latter three constructs were unable to assemble into either a stable V0 or V1V0 complex. These results suggest that the fusions c-c′, c′-c′(ΔTM1) and c′(ΔTM1)-c cannot form the proper arrangement of proteolipid subunits in the V0 domain while the c-c′(ΔTM1), c′(ΔTM1)-c′, c′-c are able to do so.
Figure 5. Assembly and targeting of V-ATPases subunits to the vacuole.
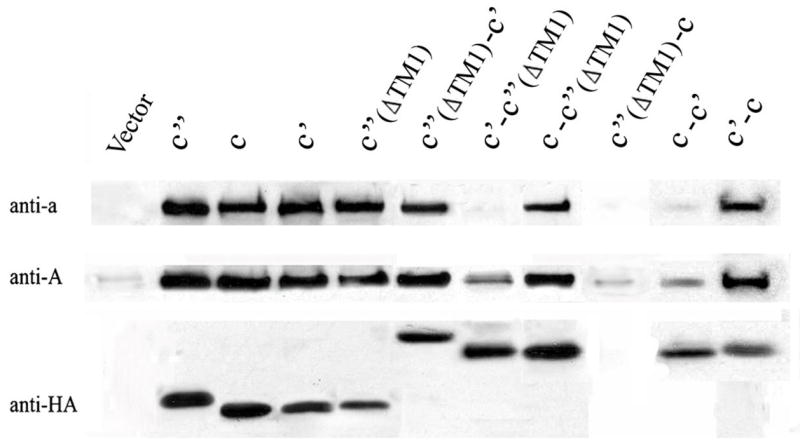
To assess the ability of the fusion constructs to assemble into V-ATPase complexes, vacuolar membrane fractions were isolated from strains expressing plasmid-borne proteolipids, and 15 μg were subjected to SDS-PAGE, followed by Western blotting using the monoclonal antibodies against subunit a, subunit A and the HA epitope tag, as described in the legend to Fig. 4.
ATPase and proton transport activity of V-ATPase complexes containing proteolipid fusion proteins
To determine the effect of the fusion constructs on activity of the V-ATPase, both concanamycin-sensitive ATPase activity and ATP-dependent proton transport (as assessed by quenching of ACMA fluorescence) were measured in isolated vacuoles as described under Experimental Procedures. The data shown in Fig. 6 represents the ATPase and proton transport activities sensitive to 1 μM concanamycin. The activities are expressed relative to those measured for vacuolar membranes isolated from a yeast strain expressing wild type subunit c″. For ATPase activity this corresponds to 0.51 μmol ATP/min/mg protein. Representative traces for ATP-dependent ACMA quenching as a measure of proton transport are shown in Fig. 7. Previous results have suggested that retention of 20% of wild type V-ATPase activity is sufficient to confer on cells a wild type growth phenotype (31,32). As expected, vacuolar membranes from strains expressing the c-c′(ΔTM1), c′(ΔTM1)-c′ or c′-c fusion proteins had more than 25% of the wild type ATPase and proton pumping activities whereas vacuole membranes from cells expressing the c-c′, c′-c′(ΔTM1) or c′(ΔTM1)-c fusions showed almost no activity. The activities observed for the functional constructs are due to V-ATPase since both ATPase and proton transport activities are inhibitable by the specific V-ATPase inhibitor concanamycin. These data indicate that, although the c-c′(ΔTM1), c′(ΔTM1)-c′ or c′-c fusion constructs do not give fully active complexes, the resultant complexes do possess significant V-ATPase activity.
Figure 6. Concanamycin A-sensitive ATPase activity and ATP-dependent proton transport of vacuolar membranes containing wild type and proteolipid fusion constructs.
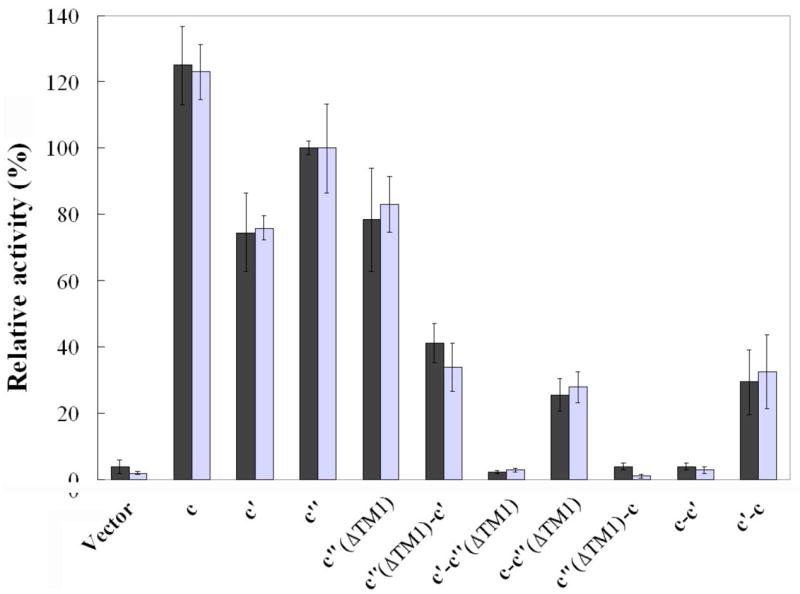
Vacuolar membranes were isolated from cells expressing plasmid-borne proteolipids or proteolipid fusion constructs, and the ATPase activity and ATP-dependent proton transport sensitive to 1 μM concanamycin A were measured as described under Experimental Procedures. Solid bars represent the concanamycin-sensitive ATPase activities and are expressed relative to vacuolar membranes isolated from cells expressing wild type subunit c′, which had a specific activity of 0.51 μmol ATP/min/mg protein. Concanamycin-sensitive ATP-dependent proton transport (open bars) was measured as the initial rate of ATP-dependent fluorescence quenching using the fluorescent dye ACMA. Values represent the average of the three measurements on two independent vacuole preparations plus or minus standard deviation.
Figure 7. ATP-dependent quenching of ACMA fluorescence by vacuolar membranes isolated from cells expressing wild type and fusion constructs.
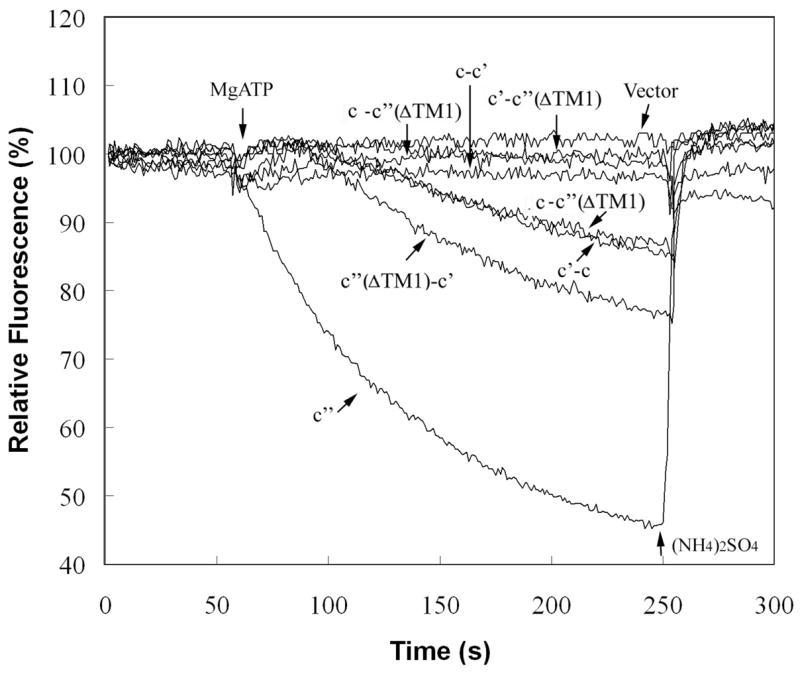
Vacuolar membranes (4 μg protein) isolated from cells expressing plasmid-borne proteolipids or proteolipid fusion constructs were assayed for ATP-dependent quenching of ACMA fluorescence as a measure of proton transport by addition of 1 mM MgATP and measurement of fluorescence intensity at 490 nm (excitation at 410 nm) as described under Experimental Procedures. The trace indicted by c″ corresponds to the activity observed for a vma16Δ strain expressing an HA-tagged form of subunit c″, whereas the remaining traces correspond to the activities measured for the fusion constructs expressed in the deletions strains indicated in Fig. 3. The activities were quantitated from the slopes of the traces and the values shown in Fig. 6 were corrected for any activity observed in the presence of 1 μM concanamycin, which was generally almost indistinguishable from the vector alone control. The slopes are proportional to the proton transport activity as indicated by the nearly linear relationship of the slope to the amount of vacuolar protein added (data not shown). At the indicated point, 5 mM (NH4)2SO4 was added to dissipate the pH gradient generated. It should be noted that the mutant strains give traces showing a significant lag before quenching following addition of MgATP, although the basis for this lag is not understood.
DISCUSSION
The results presented in the current study suggest that the three proteolipid subunits of the yeast V-ATPase adopt a particular arrangement in the proteolipid ring. Thus, of the five possible arrangements of proteolipid subunits described in Fig. 2, only that described by model B is consistent with the data obtained (Table III). In this model, subunit c′ is adjacent to and counterclockwise from subunit c″ as viewed from the luminal side of the membrane, with the four copies of subunit c making up the remainder of the proteolipid ring. Moreover, although the results do not rule out other possible arrangements of transmembrane segments within each proteolipid subunit, they do place some constraints on the relative arrangements in adjacent subunits (see below).
The proteolipid ring of the V-ATPases plays a crucial role in proton transport. The V-ATPases, like the F-ATPases, operate by a rotary mechanism (33,34). ATP hydrolysis in the V1 domain drives rotation of a rotary complex that includes the proteolipid ring of V0 (34). Rotation of the proteolipid ring relative to subunit a within the V0 domain drives unidirectional proton transport from the cytoplasmic to the luminal side of the membrane, as originally proposed for the F-ATPases (35,36). Subunit a is held fixed relative to the catalytic head of V1 by peripheral stalks or stators (37,38) and is thought to provide access channels (hemichannels) for protons to reach and leave the critical carboxyl groups on the proteolipid ring (1). Evidence for such hemi-channels has been obtained for the F-ATPase a subunit (39). Subunit a also contains a critical arginine residue in TM7 which interacts with the carboxyl groups on the proteolipid ring (40–42), thereby displacing protons from these sites into the luminal hemichannel following rotation. By functioning as the proton acceptors and donors during rotary catalysis, the proteolipids serve an essential function in proton transport.
Because of its importance in proton transport, it is of interest to determine the structure of the proteolipid ring of the V-ATPases. A 3.9-Å resolution structure has been reported for the proteolipid ring of the F-ATPase from yeast mitochondria (43). The structure reveals a 10-membered ring with the outer part of the ring composed of TM2 of subunit c and the inner portion composed of TM1 (43). Individual c subunits have the TM1/TM2 loop facing the cytoplasmic side of the membrane, where it can interact with other subunits of the rotor. The critical acidic residues are located near the middle of TM2, although the structure is not of sufficiently high resolution to visualize the orientation of these groups. Modeling studies of the F-ATPase c ring based on the NMR structure of subunit c as well as cross-linking studies suggest that the buried carboxyl group is oriented inward towards a pocket formed between adjacent c subunits (15).
A 2.1-Å resolution structure has also been reported for the proteolipid ring of the (Na+)-V-ATPase from the archaebacteria E. hirae (27). Like the eukaryotic V-ATPase c and c′ subunits, the archaebacterial protein is composed of four TMs with the critical carboxyl group located in TM4. Because the E. hirae ring contains 10 copies of this protein, the ring is composed of forty transmembrane helices, twice as many as the mitochondrial c ring. Nevertheless, the buried carboxyl groups (which are bound to Na+) are located near the middle of TM4 and are oriented towards a common pocket formed by TM2, TM3 and TM4.
Unlike the proteolipid rings from both the F-ATPase and the (Na+)-V-ATPase, which contain only a single species of c subunit, the eukaryotic V-ATPases contain either two (for mammals) or three (for fungi) distinct proteolipid subunits. Moreover, mutagenesis studies in yeast indicate that all three proteolipid subunits are essential to form a functional V-ATPase complex, and do not replace one another upon gene disruption (14). Subunit c″, which is common to all eukaryotic V-ATPases, is unique both in having an additional transmembrane helix at the N-terminus and in containing an additional (non-essential) buried carboxyl group in TM5. By placing the essential carboxyl group in TM3 rather than TM5, an irregularity is introduced in the spacing of the carboxyl groups around the ring (Fig. 2). This irregularity may affect the ability of the free V0 domain to passively conduct protons. In fact, one important difference between V0 and F0 is that free V0 does not carry out passive proton conduction (44). This property is important since an essential mechanism of regulating V-ATPase activity in vivo involves reversible dissociation of the complex into its component V1 and V0 domains (45). The data in the present paper support the relative orientation of subunits c, c″ and c′ shown in model B. In this model, TM4 of subunit c is in proximity to TM2 of subunit c″ and TM5 of subunit c″ is in proximity to TM1 of subunit c′. As a result, helices containing essential glutamic acid residues are spaced evenly around the proteolipid ring except for the border of subunits c and c″, where they are in adjacent helices, and subunits c″ and c′, where there is a gap lacking any critical acidic residue. The proteolipid ring of the V0 domain from eukaryotes also appears to differ from that in E. hirae in size. Early measurements of subunit stoichiometry using quantitative amino acid analysis suggested 5–6 copies of c plus c′ (not distinguishable by SDS-PAGE) and a single copy of subunit c″ (16). Later studies employing epitope tagged subunits demonstrated that yeast V-ATPase complexes contain single copies of subunits c′ and c″, but multiple copies of subunit c (24). The current work shows that the c″-c′ fusion is expressed, stable and functional, further confirming that there are only single copies of the c′ and c″ subunits present in the V0 proteolipid ring. Thus the eukaryotic proteolipid ring appears to be both more complex and significantly smaller than that from archaebacteria.
Attempts were made by our lab and the Stevens lab (University of Oregon) to construct homo-dimers of subunit c in order to establish whether the functional complex contains an even or odd number of c subunits. Tandem repeat DNA sequences of subunit c were constructed in plasmids and amplified in E. coli deficient in homologous recombination by mutation of the recA gene. Introduction into wild type or yeast lacking the VMA3 gene resulted in immediate recombination of the tandem repeat DNA sequence to efficiently and accurately produce a single copy of the VMA3 gene. Several yeast strains were constructed that contained disruptions of various genes in the RAD gene family in an attempt to stabilize the tandem repeat DNA sequences of subunit c (L.A. Graham and T.H. Stevens, personal communication). Unfortunately none of the mutations adequately stabilized the plasmid-borne c-c fusion gene constructs and at best only slowed the conversion by recombination to the subunit c monomer gene, and these results were confirmed both at the DNA level and protein level. Thus it has not been possible to stably maintain the subunit c homo-dimer gene in yeast to test the various models for subunit c stoichiometry.
Given the unique properties of the eukaryotic proteolipid subunits and V0 domain, it was of interest to observe that these subunits appear to adopt a unique arrangement in the proteolipid ring of functional V-ATPase complexes. Because of the close similarity of subunit c and c′, it is somewhat surprising that subunit c cannot replace subunit c′ adjacent to and counterclockwise from subunit c″. One possible reason for this may be related to the role that subunit c′ plays in assembly of the V-ATPase complex. Subunit c′ has been shown to serve as the binding site for Vma21p, which is one of three dedicated chaperone proteins (the others being Vma12p and Vma22p) that are localized to the endoplasmic reticulum and that are required for assembly of the V0 domain (46). Vma21p appears to promote assembly of the proteolipid subunits into a ring and the association of the ring with subunit d, which sits in the middle of the cytoplasmic side of the ring (47). Perhaps, by virtue of this interaction with the assembly factor Vma21p, subunit c′ facilitates insertion and association of subunit c″ with the remainder of the ring. Subunit c″ may pose unique problems in this regard because of the presence of the additional transmembrane helix at the amino terminus. It is also possible that subunit c′ may need to be in contact with both subunit c and subunit c″ in order for Vma21p to mediate assembly of the proteolipid ring. If this is the case, the actual helical contacts between these subunits also appear to be important, since the protelipid subunits cannot assemble into a functional complex when constrained to adopt the arrangement shown in Fig. 2, model A. Because mammalian cells contain no subunit c′, subunit c presumably assumes this role, suggesting a difference in the binding properties of the mammalian counterpart to Vma21p.
It should be noted that, from the data in Fig. 5, the chimeras that fail to complement the vma- phenotype (c′-c″ (ΔTM1), c″ (ΔTM1)-c and c-c′) do not allow assembly of intact V-ATPase complexes in the vacuolar membrane. It is therefore not possible to determine from our data whether complexes that did contain these alternate arrangements would be functional in ATP-dependent proton transport or not.
In summary, we have shown that the three proteolipid subunits of the yeast V-ATPase must adopt a unique arrangement in order to assemble functional complexes in vivo.
Acknowledgments
We thank Drs. Tom Stevens and Laurie Graham (University of Oregon) for the kind gift of the vma3Δ, vma11Δ and vma3Δ, vma16Δ double deletion strains and for sharing their information concerning homologous recombination of c-c dimer constructs. We also thank Drs. Ayana Hinton and Takao Inoue as well as Jie Qi, Kevin Jeffries and Sarah Bond for many helpful discussions and Kathleen Forgac for careful reading of our manuscript.
Footnotes
This work was supported by National Institutes of Health Grant GM34478 (to M.F.), a Postdoctoral Fellowship from the Northeast Affiliate of the American Heart Association. (to Y.W.) and a Postdoctoral Fellowship from the Canadian Institutes of Health Research (to D.J.C). E.coli strains were provided through NIH Grant DK34928.
The abbreviations used are: V-ATPase, vacuolar proton-translocating adenosine triphosphatase; F-ATPase, F1F0 ATP synthase; HA, influenza hemagglutinin; TM, transmembrane segment; ACMA, 9-amino-6-chloro-2-methoxyacridine; YEPD, yeast extract peptone dextrose; DMSO, dimethylsulfoxide; PAGE, polyacrylamide gel electrophoresis
References
- 1.Nishi T, Forgac M. Nat Rev Mol Cell Biol. 2002;3:94–103. doi: 10.1038/nrm729. [DOI] [PubMed] [Google Scholar]
- 2.Kane PM. Microbiol Mol Biol Rev. 2006;70:177–191. doi: 10.1128/MMBR.70.1.177-191.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson N. J Bioenerg Biomembr. 2003;35:281–289. doi: 10.1023/a:1025768529677. [DOI] [PubMed] [Google Scholar]
- 4.Wagner CA, Finberg KE, Breton S, Marshansky V, Brown D, Geibel JP. Physiol Rev. 2004;84:1263–1314. doi: 10.1152/physrev.00045.2003. [DOI] [PubMed] [Google Scholar]
- 5.Sun-Wada GH, Wada Y, Futai M. Biochim Biophys Acta. 2004;1658:106–114. doi: 10.1016/j.bbabio.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 6.Inoue T, Wang Y, Jefferies K, Qi J, Hinton A, Forgac M. J Bioenerg Biomembr. 2005;37:393–398. doi: 10.1007/s10863-005-9478-8. [DOI] [PubMed] [Google Scholar]
- 7.Abrami L, Lindsay M, Parton RG, Leppla SH, van der Goot FG. J Cell Biol. 2004;166:645–651. doi: 10.1083/jcb.200312072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pietrement C, Sun-Wada GH, Silva ND, McKee M, Marshansky V, Brown D, Futai M, Breton S. Biol Reprod. 2006;74:185–194. doi: 10.1095/biolreprod.105.043752. [DOI] [PubMed] [Google Scholar]
- 9.Li YP, Chen W, Liang Y, Li E, Stashenko P. Nat Genet. 1999;23:447–451. doi: 10.1038/70563. [DOI] [PubMed] [Google Scholar]
- 10.Sennoune SR, Bakunts K, Martinez GM, Chua-Tuan JL, Kebir Y, Attaya MN, Martinez-Zaguilan R. Am J Physiol Cell Physiol. 2004;286:C1443–1452. doi: 10.1152/ajpcell.00407.2003. [DOI] [PubMed] [Google Scholar]
- 11.Sambade M, Kane PM. J Biol Chem. 2004;279:17361–17365. doi: 10.1074/jbc.M314104200. [DOI] [PubMed] [Google Scholar]
- 12.Supek F, Supekova L, Mandiyan S, Pan YC, Nelson H, Nelson N. J Biol Chem. 1994;269:24102–24106. [PubMed] [Google Scholar]
- 13.Mandel M, Moriyama Y, Hulmes JD, Pan YC, Nelson H, Nelson N. Proc Natl Acad Sci U S A. 1988;85:5521–5524. doi: 10.1073/pnas.85.15.5521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirata R, Graham LA, Takatsuki A, Stevens TH, Anraku Y. J Biol Chem. 1997;272:4795–4803. doi: 10.1074/jbc.272.8.4795. [DOI] [PubMed] [Google Scholar]
- 15.Dmitriev OY, Jones PC, Fillingame RH. Proc Natl Acad Sci U S A. 1999;96:7785–7790. doi: 10.1073/pnas.96.14.7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arai H, Terres G, Pink S, Forgac M. J Biol Chem. 1988;263:8796–8802. [PubMed] [Google Scholar]
- 17.Gietz D, St Jean A, Woods RA, Schiestl RH. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfeifer K, Arcangioli B, Guarente L. Cell. 1987;49:9–18. doi: 10.1016/0092-8674(87)90750-1. [DOI] [PubMed] [Google Scholar]
- 19.Uchida E, Ohsumi Y, Anraku Y. J Biol Chem. 1985;260:1090–1095. [PubMed] [Google Scholar]
- 20.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Arata Y, Baleja JD, Forgac M. Biochemistry. 2002;41:11301–11307. doi: 10.1021/bi0262449. [DOI] [PubMed] [Google Scholar]
- 22.Arata Y, Baleja JD, Forgac M. J Biol Chem. 2002;277:3357–3363. doi: 10.1074/jbc.M109967200. [DOI] [PubMed] [Google Scholar]
- 23.Vasilyeva E, Liu Q, MacLeod KJ, Baleja JD, Forgac M. J Biol Chem. 2000;275:255–260. doi: 10.1074/jbc.275.1.255. [DOI] [PubMed] [Google Scholar]
- 24.Powell B, Graham LA, Stevens TH. J Biol Chem. 2000;275:23654–23660. doi: 10.1074/jbc.M004440200. [DOI] [PubMed] [Google Scholar]
- 25.Flannery AR, Graham LA, Stevens TH. J Biol Chem. 2004;279:39856–39862. doi: 10.1074/jbc.M406767200. [DOI] [PubMed] [Google Scholar]
- 26.Nishi T, Kawasaki-Nishi S, Forgac M. J Biol Chem. 2003;278:5821–5827. doi: 10.1074/jbc.M209875200. [DOI] [PubMed] [Google Scholar]
- 27.Murata T, Yamato I, Kakinuma Y, Leslie AG, Walker JE. Science. 2005;308:654–659. doi: 10.1126/science.1110064. [DOI] [PubMed] [Google Scholar]
- 28.Nelson H, Nelson N. Proc Natl Acad Sci U S A. 1990;87:3503–3507. doi: 10.1073/pnas.87.9.3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bauerle C, Ho MN, Lindorfer MA, Stevens TH. J Biol Chem. 1993;268:12749–12757. [PubMed] [Google Scholar]
- 30.Doherty RD, Kane PM. J Biol Chem. 1993;268:16845–16851. [PubMed] [Google Scholar]
- 31.Liu J, Kane PM. Biochemistry. 1996;35:10938–10948. doi: 10.1021/bi9608065. [DOI] [PubMed] [Google Scholar]
- 32.MacLeod KJ, Vasilyeva E, Baleja JD, Forgac M. J Biol Chem. 1998;273:150–156. doi: 10.1074/jbc.273.1.150. [DOI] [PubMed] [Google Scholar]
- 33.Imamura H, Nakano M, Noji H, Muneyuki E, Ohkuma S, Yoshida M, Yokoyama K. Proc Natl Acad Sci U S A. 2003;100:2312–2315. doi: 10.1073/pnas.0436796100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hirata T, Iwamoto-Kihara A, Sun-Wada GH, Okajima T, Wada Y, Futai M. J Biol Chem. 2003;278:23714–23719. doi: 10.1074/jbc.M302756200. [DOI] [PubMed] [Google Scholar]
- 35.Vik SB, Antonio BJ. J Biol Chem. 1994;269:30364–30369. [PubMed] [Google Scholar]
- 36.Junge W, Sabbert D, Engelbrecht S. Ber Bunsenges Phys Chem. 1996;100:2014–2019. [Google Scholar]
- 37.Boekema EJ, van Breemen JF, Brisson A, Ubbink-Kok T, Konings WN, Lolkema JS. Nature. 1999;401:37–38. doi: 10.1038/43369. [DOI] [PubMed] [Google Scholar]
- 38.Wilkens S, Inoue T, Forgac M. J Biol Chem. 2004;279:41942–41949. doi: 10.1074/jbc.M407821200. [DOI] [PubMed] [Google Scholar]
- 39.Fillingame RH, Angevine CM, Dmitriev OY. Biochim Biophys Acta. 2002;1555:29–36. doi: 10.1016/s0005-2728(02)00250-5. [DOI] [PubMed] [Google Scholar]
- 40.Kawasaki-Nishi S, Nishi T, Forgac M. Proc Natl Acad Sci U S A. 2001;98:12397–12402. doi: 10.1073/pnas.221291798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawasaki-Nishi S, Nishi T, Forgac M. J Biol Chem. 2003;278:41908–41913. doi: 10.1074/jbc.M308026200. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Inoue T, Forgac M. J Biol Chem. 2004;279:44628–44638. doi: 10.1074/jbc.M407345200. [DOI] [PubMed] [Google Scholar]
- 43.Stock D, Leslie AG, Walker JE. Science. 1999;286:1700–1705. doi: 10.1126/science.286.5445.1700. [DOI] [PubMed] [Google Scholar]
- 44.Zhang J, Myers M, Forgac M. J Biol Chem. 1992;267:9773–9778. [PubMed] [Google Scholar]
- 45.Kane PM. J Biol Chem. 1995;270:17025–17032. [PubMed] [Google Scholar]
- 46.Malkus P, Graham LA, Stevens TH, Schekman R. Mol Biol Cell. 2004;15:5075–5091. doi: 10.1091/mbc.E04-06-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwata M, Imamura H, Stambouli E, Ikeda C, Tamakoshi M, Nagata K, Makyio H, Hankamer B, Barber J, Yoshida M, Yokoyama K, Iwata S. Proc Natl Acad Sci U S A. 2004;101:59–64. doi: 10.1073/pnas.0305165101. [DOI] [PMC free article] [PubMed] [Google Scholar]