

Abstract
Transmissible Spongiform Encephalopathies are fatal neurodegenerative disorders of humans and animals that are familial, sporadic, and infectious in nature. Familial disorders of humans include Gerstmann–Straussler–Scheinker disease (GSS), familial Creutzfeldt–Jakob disease (CJD), and fatal familial insomnia, and result from point mutations in the prion protein gene. Although neurotoxicity in familial cases is believed to result from a spontaneous change in conformation of mutant prion protein (PrP) to the pathogenic PrP-scrapie (PrPSc) form, emerging evidence indicates otherwise. We have investigated the processing and metabolism of mutant PrP D202N (PrP202N) in cell models to elucidate possible mechanisms of cytotoxicity. In this report, we demonstrate that PrP202N expressed in human neuroblastoma cells fails to achieve a mature conformation following synthesis and accumulates in the endoplasmic reticulum as ‘curly’ aggregates. In addition, PrP202N cells show increased sensitivity to free radicals, indicating that neuronal susceptibility to oxidative damage may account for the neurotoxicity observed in cases of GSS resulting from PrP D202N mutation.
Keywords: GSS D202N, Oxidative stress, Curly aggregates, Inherited disease, Prion disorders
Introduction
Transmissible Spongiform Encephalopathies or prion disorders are fatal neurodegenerative diseases of humans and animals that are infectious, familial, and sporadic in nature. The common underlying event in all prion disorders is a change in conformation of normal cell surface glycoprotein or prion protein (PrPC) from a mainly α-helical conformation to a β-sheet rich isoform or PrP-scrapie (PrPSc). PrPSc is the principal constituent of the ‘prion infectious particle’ and is also believed to be the main cause of neuronal death in prion disorders, regardless of their origin. Thus, exposure to PrPSc, accidental or experimental, of otherwise healthy humans and animals leads to infectious disorders, while familial disorders result from point mutations in the prion protein gene (PRNP), and sporadic disorders occur as a random event where PrPC undergoes a spontaneous change in conformation to PrPSc (Aguzzi and Polymendiou 2004; Prusiner 1998). Although a wealth of data implicate PrPSc in prion disease associated neurotoxicity, other reports demonstrate prion specific neuropathology in the absence of detectable PrPSc, indicating the presence of additional or alternative pathways of neurotoxicity (Harris and True 2006; Lasmezas et al. 1997). The identity of these neurotoxic signals, however, remains unclear.
In an attempt to identify additional or alternative factors contributing to prion disease associated neurotoxicity, we have investigated the less common familial prion disorders that are easier to model experimentally and have historically provided useful information about the more common but difficult to model sporadic disorders. Familial prion disorders comprise ∼15% of all cases, and are inherited as an autosomal dominant trait with almost ∼90% penetrance. These disorders exhibit a high degree of phenotypic variability and are classified as familial Creutzfeldt–Jakob disease (CJD), Gerstmann–Straussler–Scheinker (GSS) syndrome, and fatal familial insomnia (FFI), based mainly on the phenotypic presentation. More than 30 different point mutations and octapeptide repeat insertions in PRNP segregate with these disorders, with little correlation between the mutation and the associated phenotype. Point mutations at codons P102L, P105L, A117V, G131V, Y145stop, F198S, D202N, Q217R, M232T, and insertions of octapeptide repeat sequences segregate with GSS, whereas mutations at codons D178N, V180N/M232R, T183A, E200K, R208H, V210I, M232R are associated with CJD (Aguzzi and Polymendiou 2004). Although neuronal damage in most familial cases has been attributed to a spontaneous change in conformation of mutant PrP to PrPSc, the correlation is less than perfect (Harris and True 2006; Lasmezas et al. 1997). Recent evidence suggests that misfolding and consequent misprocessing of mutant PrP is an important underlying metabolic defect, and may induce neurotoxicity by altering cellular function in the absence of detectable PrPSc (Ma and Lindquist 2001; Ma et al. 2002; Singh et al. 2005).
To evaluate this possibility, we investigated the processing and metabolism of mutant PrP D202N (PrP202N) associated with GSS in human neuroblastoma cells (M17). For comparison, cells expressing PrP200K, the most common form of familial CJD (Aguzzi and Polymendiou 2004; Capellari et al. 2000), and PrPC-expressing cells were evaluated in parallel. We report that majority of PrP202N synthesized in M17 cells does not acquire a mature conformation and is not expressed on the plasma membrane of transfected cells. Instead, greater than 60% of PrP202N is retained in the endoplasmic reticulum (ER) where it forms detergent insoluble aggregates. Intracellular accumulation of PrP202N causes upregulation of the ER chaperone calnexin. Surprisingly, aggregated PrP202N forms ‘curly’ or fibrillar deposits, a unique finding that has not been reported for any other mutant PrP form. In addition, cells expressing PrP202N (PrP202N-cells) are more sensitive to oxidative stress when exposed to H2O2, suggesting that improper folding of PrP202N interferes with normal PrP function as an anti-oxidant and increases the vulnerability of cells to oxidative damage (Brown et al. 2002, 1997; White et al. 1999). Additionally, ER stress induced by intracellular PrP202N may further contribute to the above process (Hetz et al. 2005), providing a mechanism by which mutant PrP D202N may induce neuronal death in the absence of detectable PrPSc.
Materials and Methods
Cell Culture and Transfection
Human neuroblastoma cells (M17) expressing PrPC and mutant PrP E200K (PrP200K) and D202N (PrP202N) were generated and cultured as described (Gu and Singh 2004). All cell culture supplies were obtained from Invitrogen, Hoescht from Molecular Probes (Eugene, Oregon), Tran35S-label from ICN (Costa Mesa, CA), and protein A-agarose, endoglycosydase-H (endo-H), and N-glycosidase F (PNGase-F) were from Roche Molecular Biochemicals. Anti-PrP antibodies 3F4 and 8H4 were obtained from Sigma-Aldrich and Drs. Sy and Gambetti (Department of Pathology, Case Western Reserve University), anti-calnexin antibody was from Stressgen.
Cell Staining and Confocal Microscopy
Cells expressing PrPC, PrP200K, and PrP202N were immunostained and evaluated as described in previous reports (Mishra et al. 2002; Singh et al. 1997). For co-immunostaining with anti-calnexin antibody, cells expressing PrPC and PrP202N were immuno-stained with 3F4-anti-mouseFITC, and anti-Calnexin immune serum-anti-rabbit-TRITC. To examine the effect of free radicals, PrPC and PrP202N cells were exposed to 100 μM H2O2 for 30 min, followed by fixation, permeabilization, and staining with 3F4-anti-mouse-FITC and Hoescht. Stained coverslips were mounted on gel-mount (Biomeda Corp., Foster City, CA, USA) and observed using optical fluorescent microscope or a laser scanning confocal microscope.
Immunoblotting
Cells expressing PrPC, PrP200K, or PrP202N were lysed in a non-ionic buffer containing a cocktail of protease inhibitors, and methanol-precipitated proteins were fractionated by SDS-PAGE and immunoblotted with 3F4 (1:40,000) or 8H4 (1:1000) essentially as described (Mishra et al. 2002). To assay detergent insolubility, lysates of PrPC- and PrP202N-cells were centrifuged at low speed to pellet nuclei and cell debris, and the pellet was kept aside as P1 fraction. Clarified supernatant or S1 fraction was further ultra-centrifuged at 100,000 g in a Beckman SW50 rotor for 1 h at 4°C to separate the high-speed pellet (P2) and supernatant (S2) fractions. All four fractions were analyzed by SDS-PAGE followed by western blotting and probed with anti-PrP antibody 3F4 (1:40,000).
Metabolic Labeling and Immunoprecipitation
PrPC- and PrP202N-cells were pre-incubated in methionine/cysteine-free medium containing 5% dialyzed serum for 1 h at 37°C and labeled with 50 μCi/mL of Tran-[35S]-label for 30 min or overnight in the same medium. Subsequently, cells were either washed with cold phosphate-buffered saline (PBS) and lysed as above, or washed with Opti-MEM and chased as indicated. At the end of each time point, cells were washed with cold PBS, and clarified lysates were immunoprecipitated with 3F4 or 8H4. Bound proteins were eluted in boiling sample buffer (2% SDS, 10% glycerol, 5% β-mercaptoethanol) and analyzed by SDS-PAGE-fluorography.
Results and Discussion
Mutant PrP202N is Retained in the Endoplasmic Reticulum of Human Neuroblastoma Cells
To understand the metabolism of PrP202N in transfected cells, steady state distribution of PrPC, PrP200K, and PrP202N was evaluated in human neuroblastoma cells expressing these proteins at comparable levels. PrPC represents the normal control, whereas PrP E200K is a well-studied mutant PrP form associated with familial CJD (Capellari et al. 2000). Thus, cells plated overnight in 10 cm culture dishes at a confluency of 9×106 were lysed in non-ionic detergents, and cellular proteins were precipitated with cold methanol followed by fractionation on SDS-PAGE and Western blotting. Immunoblotting with anti-PrP antibody 3F4 shows the expected glycoforms of PrP including the unglycosylated form of 27 kDa, mono- and di-glycosylated forms of 30 and 32 kDa, and the complex glycosylated form of 37 kDa. PrP200K shows aberrant migration of glycosylated forms as reported earlier (Capellari et al. 2000), whereas PrP202N shows a preponderance of the diglycosylated form with a barely detectable signal for unglycosylated PrP and a significantly reduced signal for the complex glycosylated form (Fig. 1a, lanes 1, 4, and 7).
Figure 1.
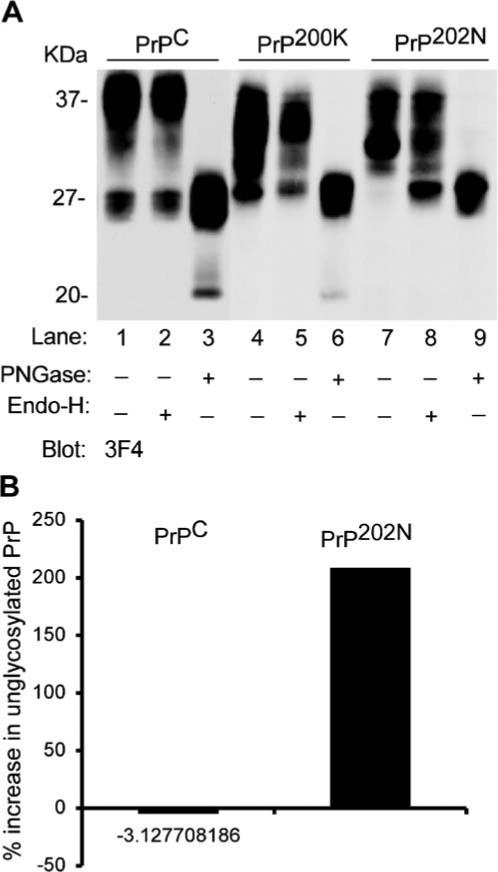
a Mutant PrP202N is retained in the endoplasmic reticulum. Proteins precipitated from lysates of PrPC, PrP200K, and PrP202N-expressing cells were treated with endo-H and PNGase-F, and analyzed by Western blotting. Immunoblotting of untreated samples with 3F4 shows the expected glycoforms of PrPC (lane 1), abnormally glycosylated forms of PrP200K (lane 4), and mainly the diglycosylated form of PrP202N (lane 7). Treatment with endo-H reveals no sensitivity of glycans on PrPC or PrP200K to the treatment, while glycans on PrP202N are cleaved, causing a significant increase in the signal for unglycosylated PrP202N (lanes 2, 5, and 8, respectively). Treatment with PNGase-F cleaves all glycans, and the different glycoforms of PrP migrate at 27 kDa as expected (lanes 3, 6, and 9). b Quantitative estimation of endo-H sensitive PrP202N forms
To evaluate if the reduction in glycosylated forms of PrP202N is due to increased degradation or block of transport at a site proximal to the Golgi, lysates of PrPC, PrP200K, and PrP202N-cells were digested with endoglycosidase-H (endo-H) and the N-glycanase PNGase-F. The enzyme endo-H cleaves core glycans that have not been processed by Golgi enzymes, and indicates that the protein has not been transported past the cis-Golgi. The enzyme PNGase-F, on the other hand, cleaves all N-linked glycans. Following treatment of cell lysates with these enzymes, the glycosylated forms of PrPC are not affected by endo-H, indicating maturation and acquisition of complex glycans in the Golgi region (Fig. 1a, lane 2). A similar observation is noted for PrP200K (Fig. 1a, lane 5), indicating that even though the glycans of this mutant PrP form are abnormal, these have been modified by Golgi enzymes and have therefore exited the ER. In contrast, PrP202N shows partial but significant sensitivity to endo-H treatment, indicating that this protein is not transported beyond the ER/cis-Golgi region (Fig. 1a, lane 8, Fig. 1b). Treatment with PNGase-F cleaves glycans of all three PrP forms, and the unglycosylated polypeptide migrates at 27 kDa as expected (Fig. 1a, lanes 3, 6, and 9).
Biogenesis of PrP202N at early time points following synthesis was evaluated by labeling PrPC and PrP202N-cells with trans-[35S]-methionine for 30 min followed by chase in normal medium for 2, 4, and 6 h, respectively (Fig. 2). At the end of each time point, cells were washed with PBS, lysed in non-ionic detergents, and lysates were subjected to immunoprecipitation with anti-PrP antibodies 8H4 and 3F4 (Fig. 2, upper and lower panels, respectively). Following the pulse, unglycosylated and two immature glycoforms of PrPC are detected. Only a small amount of PrPC acquires complex glycans, indicating that most of the newly synthesized PrPC is still within the ER after 30 min of synthesis (Fig. 2, lane 1). Following the chase, as expected, PrPC glycoforms exit the ER and are transported along the secretory path, acquiring complex glycans in the Golgi on their way to the plasma membrane (Fig. 2, lanes 1−4). PrP202N, on the other hand, does not acquire mature glycans and appears to undergo degradation within 2 h of chase (Fig. 2, lanes 5−8).
Figure 2.
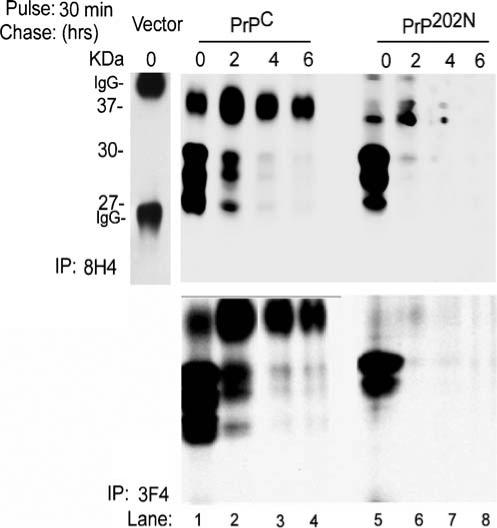
PrP202N undergoes degradation within 2 h of synthesis: PrPC and PrP202N-cells metabolically labeled with Tran35S-methionine for 30 min were chased for 2, 4, and 6 h, and cell lysates were subjected to immunoprecipitation with 8H4 and 3F4. Radiolabeled PrP was visualized following SDS-PAGE-fluorography. Immediately following the pulse, the unglycosylated and core glycosylated forms of PrPC are detected with both antibodies (lane 1). PrP202N lysates show a similar glycoform profile, though the unglycosylated form is under-represented (lane 5). Following the chase, PrPC forms mature and acquire complex glycans, whereas PrP202N degrades rapidly with little or no signal detected after 2 h of chase (lanes 1−4 and 5−8, respectively). Control cells do not show detectable PrP when immunoprecipitated with 8H4 (vector lane)
Overnight labeling of PrPC, PrP200K, and PrP202N-cells with tran-[35S]-methionine followed by immunoprecipitation with 3F4 and 8H4 as above shows the expected glycoforms of PrPC (Fig. 3, lanes 1 and 4), and abnormally glycosylated forms of PrP200K as reported previously (Fig. 3, lanes 2 and 5; Capellari et al. 2000). In contrast, PrP202N shows a prominent diglycosylated form and less prominent mono- and complex glycosylated forms when immunoprecipitated with 3F4 (Fig. 3, lane 3), and minimal reactivity with 8H4 (Fig. 3, lane 6).
Figure 3.
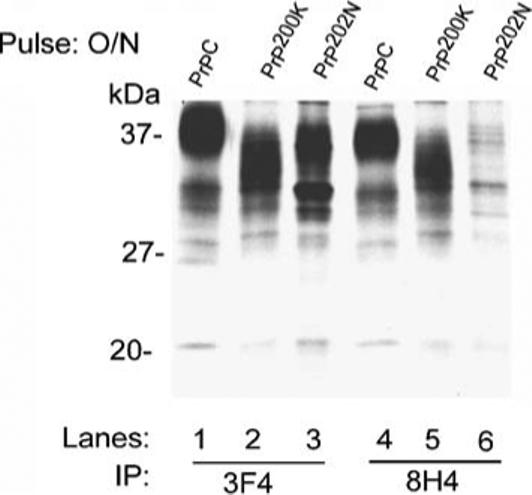
PrP202N is misfolded. Following an overnight pulse with Tran35S-methionine, normal glycoforms of PrPC are immunoprecipitated with 3F4 and 8H4 (lanes 1 and 4). PrP202N, on the other hand, shows a prominent diglycosylated form with 3F4, and no signal with 8H4 (lanes 3 and 6). Similar evaluation of PrP200K lysates shows abnormally glycosylated forms as expected (lanes 2 and 5)
Together, the above results show that in contrast to PrPC and PrP200K, PrP202N is retained in the ER of transfected cells. Although a small amount escapes the ER and acquires endo-H resistant glycans (Fig. 1a, lane 8 and Fig. 1b), majority of the protein remains in the ER following synthesis. Non-reactivity of PrP202N with anti-PrP antibody 8H4 suggests that the epitope for this antibody is unavailable due to misfolding of the protein as reported for another mutant PrP form (Gu and Singh 2004).
Mutant PrP202N Forms Detergent-Insoluble Aggregates in Cells
To determine if PrP202N forms detergent insoluble aggregates as noted for certain other mutant PrP forms (Singh et al. 2005), PrPC and PrP202N-cells were lysed in non-ionic detergents and subjected to differential centrifugation. Under these conditions, normal PrPC partitions in the detergent soluble low speed (S1) and high speed supernatant fractions (S2), whereas detergent insoluble PrP is detected in the high speed pellet fraction (P2). Thus, cell lysates were centrifuged at low speed to pellet nuclei and cell debris (P1), and clarified supernatant fractions were divided into two parts. One was kept aside (S1), whereas the other was ultracentrifuged at 100,000 g for 1 h to isolate the high speed detergent soluble supernatant (S2) and detergent insoluble pellet (P2) fractions. Proteins from all fractions were precipitated with cold methanol and resolved by SDS-PAGE followed by immunoblotting with anti-PrP antibody 3F4. As expected, most of PrPC is detected in the detergent-soluble fractions S1 and S2, with minimal amounts in the P2 fraction (Fig. 4, lanes 1−3). In contrast, most of PrP202N fractionates in the detergent-insoluble P2 fraction (Fig. 4, lanes 4−6).
Figure 4.
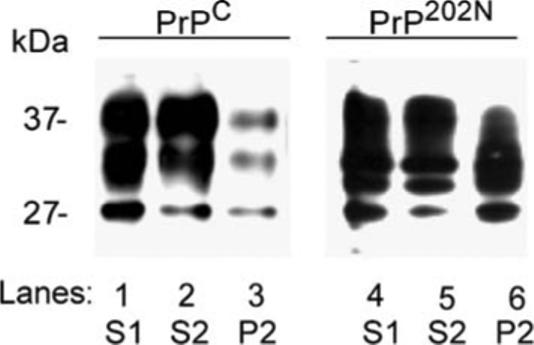
PrP202N is aggregated. Clarified lysates (S1) of PrPC and PrP202N-cells were subjected to differential centrifugation to isolate the low speed pellet (P1) and high speed detergent soluble (S2) and detergent insoluble pellet (P2) fractions. Proteins precipitated from each fraction were immunoblotted with 3F4. As expected, most of PrPC partitions in the detergent soluble S1 and S2 fractions (lanes 1−3), whereas a significant amount of PrP202N partitions in the detergent insoluble P2 fraction (lanes 4−6)
Thus, PrP202N retained in the ER assumes an aggregated form, much like disease-associated PrPSc. However, unlike PrPSc, PrP202N is not resistant to digestion by proteinase K (data not shown). Intracellular accumulation of PrP202N results in upregulation of the ER chaperone calnexin (Fig. 5), indicating an abortive attempt on the part of the cell to fold PrP202N.
Figure 5.
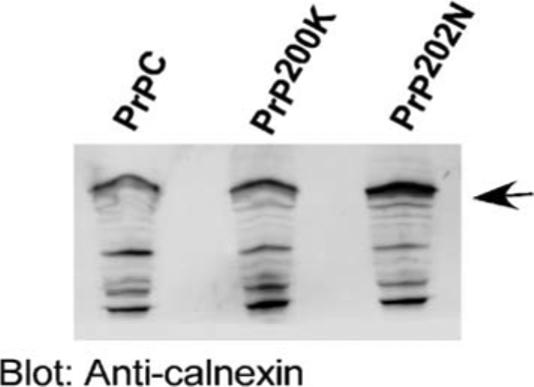
ER chaperone calnexin is upregulated in PrP202N cells. Lysates prepared from PrPC and PrP202N-cells were immunoblotted with anti-calnexin antibody. As compared to PrPC and PrP200K lysates, the expression of calnexin is significantly increased in PrP202N lysates
Intracellular PrP202N Forms Curly Aggregates in the ER
Morphological confirmation of intracellular localization of PrP202N was obtained by immunostaining PrPC and PrP202N cells with anti-PrP antibodies 3F4 and 8H4. Immuno-reaction of Triton permeabilized PrPC-cells with 3F4 and 8H4 shows typical reaction of PrPC on the plasma membrane and in the Golgi area (Fig. 6a, panels 1 and 2). In PrP202N-cells, on the other hand, ‘curly’ deposits of PrP that immunoreact with 3F4 and 8H4 are detected in the perinuclear region, suggestive of ER localization (Fig. 6a, panels 3, 4, and 4 inset). To confirm ER localization of PrP202N, Triton permeabilized PrP200K and PrP202N-cells were co-immunostained for PrP and the ER specific protein calnexin (Fig. 6b). While PrP200K cells do not show co-localization of PrP and calnexin signals (Fig. 6b, panels 1−3), PrP202N-cells show co-immunostaining of PrP and calnexin when immunoreacted with either 3F4 or 8H4 followed by anti-calnexin (Fig. 6b, panels 4−6 and 7−9, respectively). As noted in Fig. 6a, PrP immunoreactive material shows a curly appearance with both 3F4 and 8H4 antibodies (Fig. 6b, panels 4 and 7).
Figure 6.
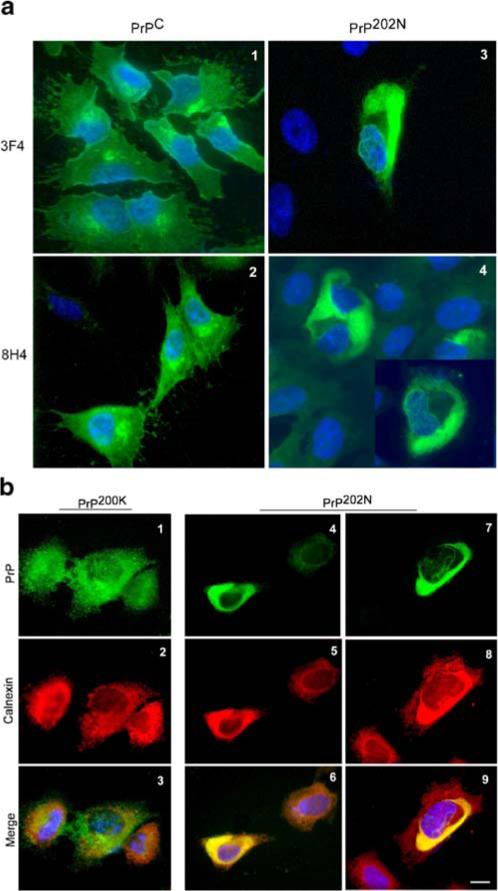
a PrP202N forms ‘curly’ aggregates in intracellular compartments. PrPC and PrP202N-cells were fixed, permeabilized, and immunostained with anti-PrP antibodies 3F4 and 8H4. PrP immunoreactivity in PrPC-cells is distributed on the plasma membrane and in the Golgi region as expected (panels 1 and 2). In PrP202N-cells, on the other hand, PrP is distributed in the perinuclear region as ‘curly’ fibrils (panels 3, 4, and 4 inset). Bar: 10 μm. b PrP202N accumulates in the ER of transfected cells. PrP200K and PrP202N cells were permeabilized and immunoreacted with 3F4-anti-mouse-FITC followed by anti-calnexin immune serum-anti-rabbit TRITC. No co-localization of PrP and calnexin is noted in PrP200K-cells (panels 1−3), whereas in PrP202N-cells PrP and calnexin co-localize when PrP is immunostained with either 3F4 (panels 4−6) or 8H4 (panels 7−9). Bar: 10 μm
Together, the above results show that PrP202N aggregates in the ER and assumes a ‘curly’ or a fibrillar appearance.
Cells Expressing PrP202N are Sensitive to Oxidative Stress
To evaluate if intracellular PrP202N aggregates increase the susceptibility of cells to oxidative stress, PrPC and PrP202N cells were exposed to 100 μM H2O2 for 30 min, fixed, permeabilized, and immunoreacted with 3F4-anti-mouse-FITC followed by Hoescht. In contrast to PrPC-cells that show minimal cell death following this treatment, ∼80% of PrP202N-transfected cells show signs of apoptosis (Fig. 7, arrows). Thus, PrP202N expressing cells are more susceptible to oxidative damage than PrPC-expressing and nontransfected M17 cells.
Figure 7.
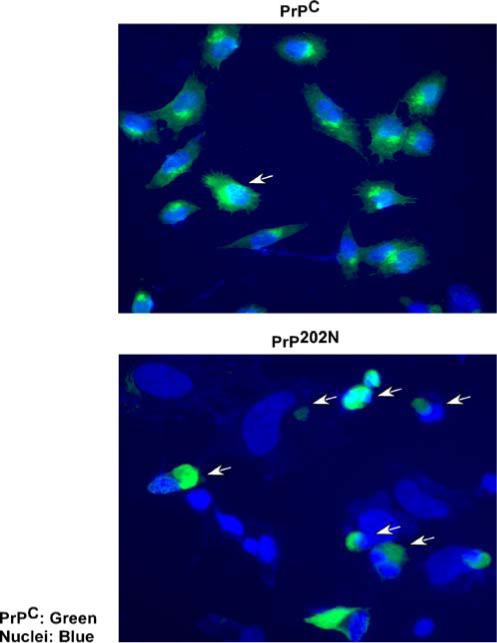
PrP202N-cells are more vulnerable to toxicity by H2O2. PrPC and PrP202N-cells were exposed to 100 μM of H2O2 for 30 min, fixed, and immunoreacted with 3F4-anti-mouse-FITC. The nuclei were visualized by staining with Hoescht. As opposed to PrPC-cells, a significant number of PrP202N-expressing cells show apoptotic nuclei when exposed to H2O2 (arrows). Bar: 10 μm
Together, the above data demonstrate that mutant PrP202N is unable to fold normally following synthesis and is retained in the ER by the cellular quality control. Accumulated PrP202N forms ‘curly’ or fibrillar aggregates within the ER that are insoluble in non-ionic detergents and induce upregulation of the ER chaperone calnexin. Intracellular aggregation of PrP202N sensitizes the cells to oxidative stress, indicating that loss of anti-oxidant function of PrPC, gain of toxic function by aggregated PrP202N, or a state of metabolic stress induced by intracellular PrP202N aggregates increases the vulnerability of cells to free radical damage. This conclusion is supported by previous reports implicating PrPC as a superoxide dismutase (Brown et al. 1997, 2002). Accumulation of PrP202N in the ER would compromise this function and induce ER stress, adding to the already compromised ability of the cell to deal with free radicals (Hetz et al. 2005). Although our data is specific to PrP202N, several other mutant PrP forms share similar features of abnormal processing and loss of normal function, suggesting that compromised ability of cells expressing mutant PrP forms to cope with free radical induced damage may represent a more generalized pathway of neurotoxicity in familial prion disorders than previously believed.
Acknowledgments
We thank Kathleen Murphy and Juliet Holmes (Hathaway Brown School, Shaker Heights, Ohio) for help with certain experimental procedures, and Yogarshi Mondal (Case Western Reserve University) for help in the preparation of the manuscript. This study was supported by NIH grants NS39089, NS35962, and a generous donation from Joseph Schnieders Foundation to NS.
Footnotes
Yaping Gu and Susamma Verghese contributed equally.
References
- Aguzzi A, Polymendiou M. Mammalian prion biology: One century of evolving concepts. Cell. 2004;116:313–327. doi: 10.1016/s0092-8674(03)01031-6. [DOI] [PubMed] [Google Scholar]
- Brown DR, Nicholas RS, Canevari L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. Journal of Neuroscience Research. 2002;67:211–224. doi: 10.1002/jnr.10118. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schulz-Schaeffer WJ, Schmidt B, Kretzschmar HA. Prion protein-deficient cells show altered response to oxidative stress due to decreased SOD-1 activity. Experimental Neurology. 1997;146:104–112. doi: 10.1006/exnr.1997.6505. [DOI] [PubMed] [Google Scholar]
- Capellari S, Parchi P, Russo CM, Sanford J, Sy M-S, Gambetti P, et al. Effect of the E200K mutation on prion protein metabolism. American Journal of Pathology. 2000;157:613–622. doi: 10.1016/S0002-9440(10)64572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Singh N. Doxycycline and protein folding agents rescue the abnormal phenotype of CJD H187R in a cell model. Brain Research: Molecular Brain Research. 2004;123:37–44. doi: 10.1016/j.molbrainres.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Harris DA, True HL. New insights into prion structure and toxicity. Neuron. 2006;50:353–357. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Hetz C, Russelakis-Carneiro M, Walchli S, Carboni S, Vial-Knecht E, Maundrell K, et al. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. Journal of Neuroscience. 2005;25:2793–2802. doi: 10.1523/JNEUROSCI.4090-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasmezas CI, Deslys JP, Robain O, Jaegly A, Beringue V, Peyrin JM, et al. Transmission of the BSE agent to mice in the absence of detectable abnormal prion protein. Science. 1997;275:402–405. doi: 10.1126/science.275.5298.402. [DOI] [PubMed] [Google Scholar]
- Ma J, Lindquist S. Wild-type PrP and a mutant associated with prion disease are subject to retrograde transport and proteasome degradation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:14955–14960. doi: 10.1073/pnas.011578098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Wollmann R, Lindquist S. Neurotoxicity and neurodegeneration when PrP accumulates in the cytosol. Science. 2002;298:1781–1785. doi: 10.1126/science.1073725. [DOI] [PubMed] [Google Scholar]
- Mishra RS, Gu Y, Bose S, Verghese S, Kalepu S, Singh N, et al. Cell surface accumulation of a truncated transmembrane prion protein in Gerstmann–Straussler–Scheinker disease P102L. Journal of Biological Chemistry. 2002;277:24554–24561. doi: 10.1074/jbc.M200213200. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3363–3383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Gu Y, Bose S, Basu S, Luo X, Mishra R, et al. Processing and mis-processing of the prion protein: Insights into the pathogenesis of familial prion disorders. In: Brown D, editor. Neurodegeneration and prion disease. Life Sciences, Springer; New York, NY: 2005. pp. 299–318. [Google Scholar]
- Singh N, Zanusso G, Chen SG, Fujioka H, Richardson S, Gambetti P, et al. Prion protein aggregation reverted by low temperature in transfected cells carrying a prion protein gene mutation. Journal of Biological Chemistry. 1997;272:28461–28470. doi: 10.1074/jbc.272.45.28461. [DOI] [PubMed] [Google Scholar]
- White AR, Collins SJ, Maher F, Jobling MF, Stewart LR, Thyer JM, et al. Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. American Journal of Pathology. 1999;155:1723–1730. doi: 10.1016/S0002-9440(10)65487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]