

Abstract
Increased iron levels and iron-mediated oxidative stress play an important role in the pathogenesis of many neurodegenerative diseases. The finding that mutations in the ferritin light polypeptide (FTL) gene cause a neurodegenerative disease known as neuroferritinopathy or hereditary ferritinopathy (HF) provided a direct connection between abnormal brain iron storage and neurodegeneration. HF is characterized by a severe movement disorder and by the presence of nuclear and cytoplasmic ferritin inclusion bodies in glia and neurons throughout the CNS and in tissues of multiple organ systems. Here we report that the expression in transgenic mice of a human FTL cDNA carrying a thymidine and cytidine insertion at position 498 (FTL498–499InsTC) leads to the formation of nuclear and cytoplasmic ferritin inclusion bodies. As in HF, ferritin inclusions are seen in glia and neurons throughout the CNS as well as in cells of other organ systems. Our studies show histological, immunohistochemical, and biochemical similarities between ferritin inclusion bodies found in transgenic mice and in individuals with HF. Expression of the transgene in mice leads to a significant decrease in motor performance and a shorter life span, formation of ferritin inclusion bodies, misregulation of iron metabolism, accumulation of ubiquitinated proteins, and incorporation of elements of the proteasome into inclusions. This new transgenic mouse represents a relevant model of HF in which to study the pathways that lead to neurodegeneration in HF, to evaluate the role of iron mismanagement in neurodegenerative disorders, and to evaluate potential therapies for HF and related neurodegenerative diseases.
Keywords: neurodegeneration, hereditary ferritinopathy, nuclear inclusion, CNS, iron, ferritin
Introduction
Mammalian ferritins are heteropolymers composed of 24 polypeptide subunits that contain variable proportions of the light (FTL) and heavy (FTH1) polypeptide subunits (Harrison and Arosio, 1996). The FTH1 subunit has a potent ferroxidase activity that catalyzes the oxidation of ferrous iron, whereas the FTL subunit plays a role in iron nucleation and protein stability, giving ferritin the dual functions of iron detoxification and iron reserve. Iron is an essential element needed for vital processes such as neuronal development, myelination, synthesis and catabolism of neurotransmitters, electron transport, and iron–sulfur cluster synthesis (Beard and Connor, 2003). Because of its ability to participate directly as a donor or acceptor in electron-transfer reactions, iron may become toxic by the generation of highly reactive free radicals that cause lipid peroxidation, DNA strand breaks, and protein modifications. For that reason, the iron storage protein ferritin plays a key role to reduce the bio-availability of iron to stem uncontrolled production of reactive oxygen radicals that may result in cell death (Harrison and Arosio, 1996).
Recently, nucleotide insertions in the coding sequence of the ferritin light polypeptide (FTL) gene have been found associated with the development of a novel autosomal dominant neurodegenerative disease (Curtis et al., 2001; Vidal et al., 2004a; Mancuso et al., 2005) or neuroferritinopathy (Curtis et al., 2001). We described a two-nucleotide (thymidine and cytidine) insertion at position 498–499 of the FTL gene (FTL498–499InsTC) in members of a French kindred (Vidal et al., 2004a) and documented for the first time that pathologic alterations extended beyond the CNS, suggesting the term “hereditary ferritinopathy” (HF) (Vidal et al., 2003). Clinically, HF is characterized by the presence of tremor, cerebellar ataxia, parkinsonism, pyramidal signs, behavioral disturbances, and cognitive decline; the age at onset is in the third decade of life. The disease progresses gradually, affecting first striatal and cerebellar function, then later cortical functions. Magnetic resonance imaging (MRI) studies show cerebral and cerebellar atrophy and abnormal signals in the basal ganglia. Neuropathologically, the disease is characterized by the presence of ferritin nuclear (NI) and cytoplasmic inclusion bodies and iron accumulation in glia and neurons throughout the CNS. To investigate the role of the mutant FTL (mt-FTL) polypeptide in the development of HF, we generated mice transgenic for the FTL498–499InsTC mutation. Our results reveal that the expression of the mutant subunit in mice leads to a significant decrease in motor performance and a shorter life span, misregulation of iron metabolism, formation of ferritin inclusions, accumulation of ubiquitinated proteins, and incorporation of elements of the proteasome into ferritin inclusions. This new transgenic mouse represents a relevant mammalian model to study the pathways that lead to neurodegeneration in HF and to evaluate potential therapies for HF and related neurodegenerative diseases.
Materials and Methods
Construction of the MoPrP-FTL498–499InsTC transgene.
The FTL498–499InsTC mutant human FTL cDNA sequence was cloned by reverse transcription (RT)-PCR (Promega, Madison, WI) using oligonucleotide primers containing an XhoI site, a Kozak consensus sequence, and a stop codon. The 5′-untranslated region containing the iron regulatory element (IRE) sequence (Klausner et al., 1993) was excluded from the construct. The cDNA was inserted into the pBS/MoPrP.Xho vector as described previously (Borchelt et al., 1996), and the orientation was verified by sequencing. The resulting construct was linearized and gel purified before injection into hybrid C3HeB/FeJ mouse embryos. Transgenic lines were established using standard methods at the Indiana University Transgenic and Knock-out Mouse Core Facility. Lines were crossed to nontransgenic C57BL/6J mice for >10 generations and maintained by crossing transgenic animals to nontransgenic C57BL/6J mice. The transgene was detected by PCR amplification of a 533 bp product using the forward primer FTLFE1 and the reverse primer FTLRE4 located in the FTL cDNA sequence (supplemental Table 1, available at www.jneurosci.org as supplemental material). Southern blot analysis was performed using a human FTL cDNA as a probe as described previously (Vidal et al., 1999). For phenotypic analysis, wild-type, heterozygous, and homozygous animals were routinely monitored from birth to death as described previously (Clark et al., 1997). The care and use of animals in this study was in accordance with institutional guidelines.
Rotarod testing.
Four-month-old homozygous FTL-Tg4 were compared with same-age wild-type mice, 13-month-old homozygous FTL-Tg4 were compared with same-age heterozygous FTL-Tg4 and wild-type mice, and 17-month-old heterozygous FTL-Tg4 were compared with same-age wild-type mice. Ten mice for each age and genotype (n = 70 in total) were used in this study. Only naive mice were used. Animals were tested for four trials for 4 consecutive days using a Rotarod device (Columbus Instruments International, Columbus, OH) as described previously (Clark et al., 1997). Each trial lasted a maximum of 10 min, during which time the rotating rod underwent a linear acceleration from 4 to 40 rpm over the first 5 min of the trial and then remained at maximum speed for the remaining 5 min. Animals were scored for their latency to fall (in seconds) for each trial. Animals were rested a minimum of 10 min between trials to avoid fatigue and exhaustion. The results were analyzed by a two-way repeated-measures ANOVA that factors genotype, day, and trial using SAS PROC MIXED version 9.1.3 (SAS Institute, Cary, NC).
Western blot analysis.
Mutant and wild-type FTL polypeptides were detected using polyclonal antibodies (Abs) 1283 and 1277, respectively, as described previously (Vidal et al., 2004a). Mouse Ftl and Fth1 were detected using rabbit antisera (Dako, Carpinteria, CA) and goat antisera (Y-16; Santa Cruz Biotechnology, Santa Cruz, CA), respectively. Ubiquitin was detected using rabbit antisera (Dako). Mouse β-actin was detected using a monoclonal Ab (Sigma, St. Louis, MO). Abs were used at a dilution of 1:1000. To detect the presence of the transgene, cerebral cortex was homogenized in radioimmunoprecipitation assay buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm Na3VO4, 50 mm NaF, 1.0% Triton X-100, 0.5% deoxycholate, and 0.1% SDS; all from Sigma), with a protease inhibitor mixture (Complete; Roche Applied Science, Indianapolis, IN). Lysates containing equal amounts of protein were loaded. Human wild-type and mt-FTL recombinant polypeptides were overexpressed using the pET expression system (EMD Chemicals, San Diego, CA), purified, and used as controls. Proteins were run on 16% acryalamide Tris-tricine-SDS PAGE or 4–12% NuPAGE Novex Bis-Tris Gels (Invitrogen, Carlsbad, CA) and transferred to Immobilon-P (Millipore, Billerica, MA) as described previously (Vidal et al., 1999, 2004a).
Solubility of ferritin.
Frozen cerebral cortex (0.1 g wet weight) was homogenized in 1000 μl of 50 mm Tris, pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% deoxycholate, and 1 mm 2-mercaptoethanol (all from Sigma) with protease inhibitors (Complete). Homogenates were extracted with a 5% SDS solution, sonicated for 20 s, and centrifuged at 150,000 × g for 30 min. The supernatant (SDS soluble) was removed, and the SDS-insoluble pellet was resuspended in 8 m urea/5% SDS. The corresponding Tris-soluble, SDS-soluble, and the urea-treated, SDS-insoluble pellets were analyzed by Western blot.
Histology and immunohistochemistry.
Wild-type and transgenic animals were studied at different ages. After anesthesia, animals were perfusion fixed with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2 (Sigma), after which brains and organs were removed, embedded in paraffin, and sectioned. Alternatively, animals were decapitated, and organs were removed and placed in 10% formalin. Eight micrometer sections were cut and mounted on poly-l-lysine-coated slides. Sections were stained with hematoxylin and eosin and Perls' method for ferric iron as described previously (Vidal et al., 2004a). Immunohistochemical stainings were performed as described previously (Vidal et al., 2004a) by using primary Abs directed against the following antigens: ferritin (Ab1277, Ab1283, anti-ferritin Abs from Dako and Y-16); ubiquitin (Dako); SUMO1 (Santa Cruz Biotechnology); UCH-L1, 11S, 19S, and 20S (Biomol, Plymouth Meeting, PA); heat shock protein 70 (HSP70; Santa Cruz Biotechnology); and clusterin (Quidel, San Diego, CA).
Quantitative RT-PCR analysis.
Total RNA was extracted from frozen cerebral cortex and from cerebellum using Trizol reagent (Invitrogen) and was treated with RNase-free DNase I (Invitrogen) before purification by RNeasy column (Qiagen, Valencia, CA). Three micrograms of total RNA were used to generate the first-strand cDNA by using Superscript III (Invitrogen) and Oligo(dT) primers. Real-time PCR was performed on a 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). FAM and Tampra dual-labeled probes as well as gene-specific primers were used for quantitative PCR. Mouse Fth1 (Mm00850707_g1) and mouse β-actin (Mm00607939_s1) were from Applied Biosystems. The probe and primers for the mouse Ftl gene and for the human FTL498–499InsTC sequence were designed using the software Primer Express (Applied Biosystems). Primer and probe sequences used for real-time RT-PCR are listed in supplemental Table 1 (available at www.jneurosci.org as supplemental material). PCR was conducted by initial denaturation at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s, 60°C for 1 min. The relative quantification of gene expression between multiple samples was achieved by normalization against β-actin using the ΔCT method for quantification. Samples were run by triplicates in each experiment. Statistical analysis was done using one-way ANOVA with GraphPad Prism version 4.03 (GraphPad Software, San Diego, CA).
MRI.
Two-dimensional T2-weighted images were obtained using a 9.4 tesla 31 cm horizontal bore magnet (Varian, Palo Alto, CA) equipped with a 12-cm-diameter shielded gradient set. A loop-gap resonator volume coil (20 mm in diameter and 15 mm in depth) tuned to (1H) proton frequency was used. A total of 10 mice (5 FTL-Tg4 homozygous mice and 5 wild-type mice) between 8 and 9 months of age were studied. Mice were anesthetized with 0.75% isoflurance at a rate of 1 L/min. The T2-weighted multislice spin-echo imaging technique was used with the following parameters: repetition time, 2 s; echo time, 40 ms; two-signal averaging; 30 slices of 0.4 mm thickness; 256 × 256 data points over a field of view 20 × 20 mm resulting in the voxel dimensions of 78 ×78 μm, respectively. The scan time was 17 min, 10 s. The time domain data were zero-filled to 1024 × 1024 in both directions and Fourier transformed. The processed data were transferred to a Linux system equipped with IVA, a graphical user interface (INDYPET image processing software), for visualization.
Electron microscopy.
After anesthesia, mice were perfused intracardially with 4% paraformaldehyde and subsequently with 5% glutaraldehyde. Brains were postfixed with Dalton's chrome osmium, dehydrated in graded ethanols, cleared in propylene oxide, and embedded in Epon. One-micrometer-thick sections were stained with toluidine blue. Ultrathin sections were contrasted with uranyl acetate and lead citrate. Processing of human tissue was performed as described previously (Vidal et al., 2004a). Photographs were obtained using a Philips EM 300 electron microscope.
Results
Ferritin transgenic mice show a clinical phenotype and have a shorter life span
To create a model in which to study the pathway(s) that leads to HF, we generated transgenic mice in which expression of the mt-FTL polypeptide (Fig. 1a) is regulated by the mouse prion protein (MoPrP) promoter (Borchelt et al., 1996) (Fig. 1b). Seven independent transgenic lines were obtained, four of which (FTL-Tg1–4) produced stable lines of mice that express the transgene at different levels. Southern blot analyses indicate a unique integration site of the transgene in each line. Densitometric scanning of the autoradiographs estimate the following number of copies: FTL-Tg1, 40–50 copies; FTL-Tg2, 5–10 copies; FTL-Tg3, 1–5 copies; FTL-Tg4, 1–5 copies. Western blot analysis and quantitative real-time RT-PCR show expression of the transgene in the cerebral cortex and in the cerebellum of the transgenic lines, with FTL-Tg1 showing the highest level of expression (Fig. 1c,d). To emulate the human disease and avoid possible artifacts on ferritin assembly attributable to the overexpression of the transgene, we decided to study further the transgenic line (FTL-Tg4) that expresses relatively low levels of the mt-FTL protein. FTL-Tg mice display a progressive behavioral phenotype, consisting of abnormal grooming, tremor, and incoordination. Feet clasping was observed after suspension of the mice by their tails (Fig. 1e). Transgenic mice would flex their front and hindlimbs inward, with paws clasped together and drawn in toward the body. In contrast, wild-type mice demonstrate normal limb posture when suspended by their tails. Toward the end of the life span, the mice display a hunchbacked posture when walking (Fig. 1f). Mice show a very low body position with some degree of lordosis and later a shuffle gait with dragging hindlimbs. Transgenic mice have a shorter life span. Survival is reduced with statistical significance (p = 0.0048, log rank test) by 15% in heterozygous mice and by 36% in homozygous mice (Fig. 1g). Analysis of motor performance of transgenic mice on a rotating rod reveals a significant decrease in performance at the earliest age tested, 4 months (Fig. 1h), before obvious clinical phenotype is observed. Wild-type animals perform significantly better than transgenic animals at 13 months of age, with heterozygous mice performing better than homozygous mice (Fig. 1i). At 17 months of age, both the level of performance and improvement in performance of heterozygous mice did not show any statistically significant difference with wild-type animals (p = 0.0607).
Figure 1.
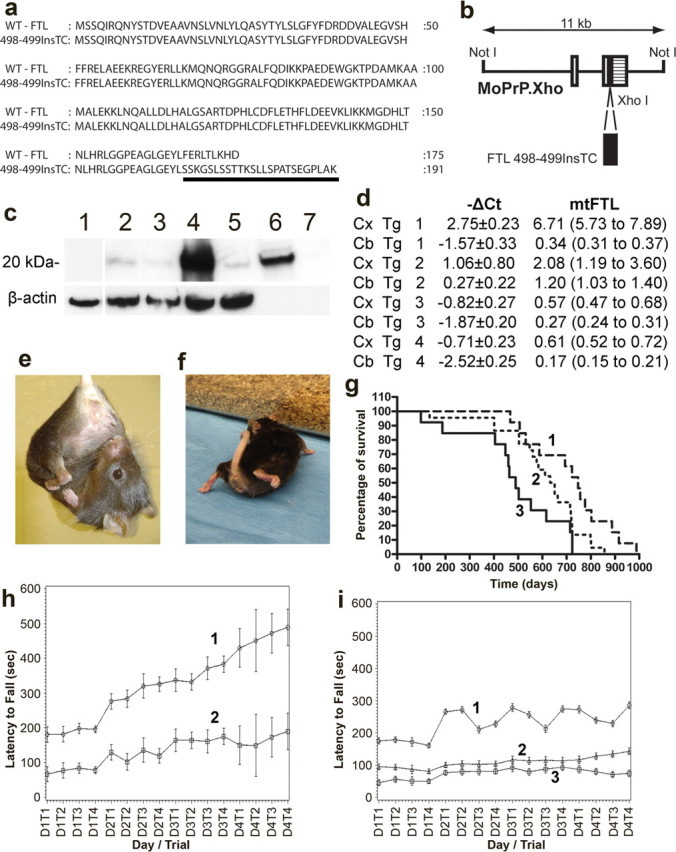
Generation and characterization of transgenic mice. a, The wild-type FTL polypeptide (WT-FTL) consists of 175 amino acids. The FTL498–499InsTC mutant polypeptide, expressed in FTL-Tg mice, has 191 amino acids and a different C terminus (underlined). b, Expression plasmid MoPrP.Xho containing a cDNA sequence carrying the FTL498–499InsTC mutation. Open boxes, Noncoding 5′ sequences; crosshatched box, noncoding 3′ sequences; filled box, FTL498–499InsTC coding sequence. c, Immunoblot analysis of transgene-derived expression in the cerebral cortex of normal control mice (lane 1), FTL-Tg4 (lane 2), FTL-Tg2 (lane 3), FTL-Tg1 (lane 4), and FTL-Tg3 (lane 5) using Ab 1283. Recombinant mt-FTL (lane 6) and WT-FTL (lane 7) were run as controls. The blot was reprobed with anti-mouse β-actin. d, Relative quantification of transgenic expression by quantitative RT-PCR in the cerebral cortex (Cx) and in the cerebellum (Cb) using the ΔCT method. Transgene mRNA levels were determined by evaluating the expression 2−ΔCT with ΔCT + s and ΔCT − s, where s is the SD of the ΔCT value. No transgene expression was detected in normal littermates (ΔCt = 0). e, f, Representative photographs of a homozygous FTL-Tg1 mouse at 12 month of age showing clasping of the hindlimb (e) and a very low body position (from ∼14–15 months of age; f). g, The life expectancies of wild-type (1; n = 13), FTL-Tg4 heterozygous (2; n = 22), and homozygous (3; n = 13) mice range between 467 and 988, 134 and 865, and 98 and 723 d, respectively. The median survival was 750 d for wild-type mice, 640 d for heterozygous mice, and 480 d for homozygous mice. Statistical analysis was done using GraphPad Prism version 4.03. h, Performance of wild-type (1; n = 10) and FTL-Tg4 homozygous (2; n = 10) animals on an accelerating rotating rod apparatus at 4 months of age. FTL-Tg4 homozygous mice failed to improve their performance compared with age-matched wild-type animals (p < 0.0001). Differences in daily performances between wild-type and FTL-Tg4 homozygous animals approach statistical significance on day 1. i, Performance of wild-type (1; n = 10), FTL-Tg4 heterozygous (2; n = 10), and FTL-Tg4 homozygous (3; n = 10) animals on an accelerating rotating rod apparatus at 13 months of age. Differences in daily performances between wild-type and FTL-Tg4 transgenic animals approach statistical significance on day 1; wild-type animals perform significantly better than FTL-Tg animals (p < 0.0001). Transgenic mice failed to improve their performance compared with age-matched wild-type animals. Heterozygous mice performed better than homozygous mice with statistical significance (p < 0.0001).
Pathological analysis of ferritin deposits in transgenic mice
Histological and immunohistochemical analysis of FTL-Tg mice reveal that the mt-FTL polypeptide was ubiquitously expressed in the CNS, with highest levels in forebrain, hindbrain, and spinal cord. The most significant neuropathological finding in FTL-Tg mice was the presence of NIs in neurons and glia (Fig. 2). The distribution of NIs in the CNS is shown in supplemental Table 2 (available at www.jneurosci.org as supplemental material). In addition to NIs, cytoplasmic ferritin inclusion bodies were also observed in some of the cells containing NIs. Normal control aged-matched littermates did not show the presence of NIs or cytoplasmic ferritin aggregates. FTL-Tg mice show an age-dependent increase in the size of NIs and in the number of cells with NIs. In the cerebral cortex, nuclei of many small to medium-sized neurons contain nuclear and cytoplasmic inclusion bodies (Fig. 2a–c). The olfactory bulb is severely affected, showing in addition to NIs the presence of large intracytoplasmic ferritin inclusions (Fig. 2d). Ferritin immunopositive nuclei are present in the CA4, CA1, and fascia dentata of the hippocampus (Fig. 2e). NIs are observed in the thalami and other cortical deep gray matter regions. In the striatum, large neurons are in a more advanced stage of atrophy than small neurons, showing the presence of large NIs and cytoplasmic ferritin aggregates and vacuoles. In the cerebellum, granule cells are most affected, and immunopositive nuclei are also found throughout the molecular layer (Fig. 2f,g). Purkinje cell nuclei are also positive (Fig. 2f). Loss of Purkinje cell bodies is evident, particularly at the crown of the cerebellar folia. NIs are also seen in the spinal cord (Fig. 2h) and in epithelial cells of the choroid plexuses (Fig. 2i). Inclusion bodies are also found in the nuclei of endothelial cells of arteries and veins as well as in cells of the adventitia (data not shown). Ferritin inclusions are also observed in non-CNS tissues (see supplemental Table 3 and supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Immunohistochemical and biochemical analyses show the presence of NIs in postmitotic cells of a number of tissues, including skeletal and cardiac muscle, liver, kidney, the grastrointestinal tract, adrenal glands, and skin. In all cases, inclusions were readily apparent by 8 weeks by hematoxylin and eosin staining. The finding of inclusions in skin, mucosal cells, and skeletal muscle is particularly important because these are tissues accessible by biopsy that could have an important application in monitoring clinical trials. Transgene expression in the brain of homozygous mice is approximately two times higher than in heterozygous mice (Fig. 3a). At 1 month of age, homozygous transgenic mice from the low transgene expression line FTL-Tg4 show a diffuse cytoplasmic ferritin staining and the presence of small NIs in cortical neurons (Fig. 3b). Thalamic neurons show a homogeneous nuclear staining and some small NIs. Large neurons of the striatum show the presence of NIs and a diffuse nuclear staining for ferritin, whereas small neurons are not affected. In the cerebellum, small NIs are seen in neurons of the granule cell layer and in Purkinje cells, which also show a diffuse nuclear staining (Fig. 3c). Similar distribution and degree of ferritin deposition is seen in 15-d-old homozygous FTL-Tg4 mice (Fig. 3d–f). In electron micrographs, NIs appear to be composed of granular electron-dense particles with no limiting membrane separating the NI from the nucleoplasm (Fig. 4). As observed in individuals with HF, the NI occupies almost completely the nucleus and, as a result, displaces the chromatin up against the nuclear membrane. By electron microscopy, NIs in HF and FTL-Tg mice show a comparable granular morphological appearance and chromatin rearrangement (Fig. 4e,f).
Figure 2.

Ferritin inclusions in FTL-Tg mice. Histological and immunohistochemical studies shown are from paraffin-embedded sections of a perfused 343-d-old FTL-Tg4 heterozygous mouse. a, Numerous eosin-positive intraneuronal inclusions in the cerebral cortex. b, Intranuclear and intracytoplasmic ferritin deposits in the cerebral cortex immunolabeled by Abs against the Ftl polypeptide. c, Ferritin deposits in the cerebral cortex stained for the Fth1 polypeptide. d, Ferritin deposits in the olfactory lobe, where the MoPrP promoter is very strong. e, Ferritin inclusions are present in all areas of the hippocampus, in particular in the granular layer of the dentate gyrus and CA4. f, g, Intranuclear and intracytoplasmic ferritin inclusion bodies in Purkinje cells and NIs in granule cells of the cerebellum. h, NIs in neurons of the gray matter of the spinal cord. i, NIs in simple cuboidal epithelial cells of the choroid plexus. NIs are indicated with arrows, and cytoplasmic inclusions are indicated with arrowheads. Hematoxylin and eosin stain was used (a). Immunohistochemistry was done using Abs 1277 (b, h, i), Y16 (c), 1283 (d, f), and Dako anti-FTL (e, g). Scale bars: a, 20 μm; b, c, f–i, 10 μm; d, 30 μm; e, 100 μm.
Figure 3.

Expression of ferritin in young FTL-Tg homozygous mice. a, Relative quantification of transgenic expression in the cerebral cortex and in the cerebellum of 30-d-old FTL-Tg4 mice. Transgene expression was normalized to the endogenous control gene mouse β-actin. A twofold difference between heterozygous and homozygous mice is seen. Error bars indicate SEM. b, c, Intranuclear and intracytoplasmic ferritin deposits in the cerebral cortex (b) and in the cerebellum (c) of 30-d-old homozygous FTL-Tg4 mice. d–f, Small NIs and cytoplasmic aggregates of ferritin in the amygdala (d), in the cerebral cortex (e), and in Purkinje cells and granule cells of the cerebellum (f) of a 15-d-old homozygous FTL-Tg4 mouse. NIs are indicated with arrows, and cytoplasmic inclusions are indicated with arrowheads. Immunohistochemistry was done using Abs 1283 (b, d) and Dako anti-ferritin light chain polypeptide (c, e, f). Scale bars: b–f, 10 μm.
Figure 4.
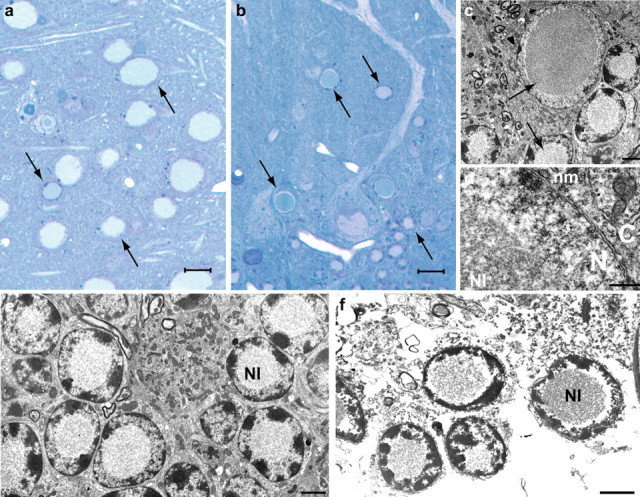
Ultrastructural studies on a 340-d-old heterozygous FTL-Tg1 mouse. a, b, Ferritin inclusion bodies in the nuclei of neurons of the striatum (a) and cerebellum (b) appear homogeneously blue in toluidine blue-stained sections. NIs are indicated with arrows. c–f, Electron micrographs (EMs) of cerebellar sections show that the NI occupies most of the nucleoplasm and is not limited by a membrane. The chromatin appears to be centrifugally displaced to varying degrees, often forming a thin layer adjacent to the nuclear membrane (d). Degenerating Purkinje cells (c) and granule cells (e) are shown. An EM of a section obtained from postmortem brain tissue of an individual carrying the FTL498–499InsTC mutation is shown for comparison (f). Scale bars: a, b, 10 μm; c–f, 2 μm. N, nucleus; C, cytoplasm; nm, nuclear membrane.
Iron accumulation in the CNS
We detected ferric iron accumulation by Perls' staining throughout the CNS of transgenic mice (Fig. 5a,b). Iron deposition is seen colocalizing with NIs, although cytoplasmic staining may also be seen in cells with cytoplasmic ferritin inclusions. The finding of iron deposition following a pattern similar to the pattern observed for ferritin deposition suggests that the ferric iron detected is probably sequestered within ferritin inclusions. Although high levels of intracellular iron and ferritin deposition are observed, iron accumulation is not seen in association with an increase in the levels of endogenous mouse Ftl and Fth1 mRNAs (Fig. 5c,d). Brain iron deposition was also studied in vivo using T2-weighted MRI scans. In MRI, decreased signal intensity on T2-weighted images provides an accurate in vivo map of the normal distribution of brain iron (Drayer et al., 1986). T2-weighted images obtained at 9.4 T show a significant difference in signal intensities between FTL-Tg mice and wild-type mice (Fig. 5e,f), consistent with the anatomical distribution of ferritin and ferric iron seen by immunohistochemistry and Perls'.
Figure 5.

Ferric iron accumulates in ferritin inclusions. a, b, Prussian blue staining of sections of the substantia nigra of a 270-d-old homozygous FTL-Tg4 mouse (a) and the striatum of a 495-d-old homozygous FTL-Tg1 mouse (b). Real-time RT-PCR analysis of cerebral cortex and cerebellum of wild-type, heterozygous, and homozygous FTL-Tg4 mice at 4 months of age. c, d, No significant change in the expression of endogenous mouse Ftl (c) and Fth1 (d) genes was observed. MR transaxial images of mouse brain obtained at 9.4 T. e, f, The T2-weighted images acquired at a TE of 40 ms showed an excellent and detectable contrast between 8-month-old transgenic (homozygous FTL-Tg4) (e) and age-matched wild-type (f) mice. Compared with wild-type mice, FTL-Tg mice show hypo-intense signals (low T2 values) in several areas of the brain, such as the hippocampus, thalamus, globulus padillus, and substantia nigra. Scale bars: c, 25 μm; d, 5 μm.
Characterization of ferritin deposits in transgenic mice
Western blot analysis of cerebral inclusions isolated from an individual member of the French kindred carrying the FTL498–499InsTC mutation (Vidal et al., 2004a) and from FTL-Tg mice shows immunoreactive bands of ∼20 kDa using Abs 1277 and 1283 (Fig. 6a,b) raised against the wild-type Ftl and mt-FTL polypeptides, respectively. Immunoreactivity is also observed using Y-16, an Ab raised against the Fth1 polypeptide (data not shown). We investigated the solubility of the ferritin deposits in HF and in transgenic mice. SDS insolubility was seen in association with ferritin deposition in both cases. In contrast, ferritin from normal human controls or endogenous mouse ferritin from normal littermate mice are not present in the detergent-insoluble fraction (Fig. 6a,b). Detergent-insoluble ferritin containing the mutant and wild-type murine ferritin polypeptides was also seen in young transgenic animals. The quantity of insoluble ferritin was observed to increase as a function of age. Western blot analysis (Fig. 6c) and immunohistochemical studies (Fig. 6d,e, supplemental Fig.1c, available at www.jneurosci.org as supplemental material) showed accumulation of ubiquitinated proteins in FTL-Tg mice and in HF. In FTL-Tg mice, ubiquitination appears to occur shortly after ferritin aggregates are formed (Fig. 6c,e). In addition, different elements of the proteasome are redistributed to the sites of ferritin protein aggregation. Abs against the 20S proteasome (Fig. 6f) and the ubiquitin C-terminal hydrolase (UCH-L1) (data not shown) showed a diffuse nuclear and cytoplasmic staining of the cells containing inclusions. Abs against the 11S (PA28α) proteasome regulator protein (Fig. 6g–i) and the 19S regulator ATPase subunit 6b (Tbp7) (not shown) immunostained NIs and diffuse cytoplasmic deposits. No evidence of SUMOylation was seen by immunohistochemistry in HF and FTL-Tg mice. No evidence of HSP70, an inducible member of the heat shock protein family of molecular weight ∼70 kDa, was observed in HF and FTL-Tg brain sections by immunohistochemistry, suggesting that ferritin NIs do not elicit the stress response necessary to increase the expression of HSP70. In contrast, the chaperon protein clusterin, which has been shown to be much more efficient than α-crystallin in preventing the precipitation from solution of stressed target proteins (Poon et al., 2002), was found in ferritin deposits in HF (data not shown).
Figure 6.

Western blot and immunohistochemical analysis of ferritin deposits. a, Western blot analysis using Ab 1283 of SDS-soluble (lanes 1, 3, 6, 8, 10, 12) and SDS-insoluble (lanes 2, 4, 5, 7, 9, 11) fractions. The SDS-insoluble fractions from an individual with the FTL498–499InsTC mutation (lane 4), a 4.5-month-old heterozygous FTL-Tg4 mouse (lane 5), and 15-d-old (lane 9) and 30-d-old (lane 11) homozygous FTL-Tg4 mice show immunoreactivity for the transgene. Normal human (lanes 1, 2) and nontransgenic (lanes 7, 8) age-matched controls were not immunoreactive. b, Western blot analysis using Ab 1277 of SDS-soluble (lanes 1, 3, 5, 7, 9, 11) and SDS-insoluble (lanes 2, 4, 6, 8, 10, 12) fractions. The SDS-insoluble fractions from an individual with the FTL498–499InsTC mutation (lane 2), a 4.5-month-old heterozygous FTL-Tg4 mouse (lane 12), and 15-d-old (lane 8) and 30-d-old (lane 6) homozygous FTL-Tg4 mice show immunoreactivity for the light chain subunit. Normal human (lanes 3, 4) and nontransgenic (lanes 9, 10) age-matched controls were not immunoreactive. c, Western blot analysis using anti-ubiquitin Abs shows accumulation of ubiquitinated proteins in HF and FTL-Tg mice compared with controls. Fractions are as in b. d–i, Representative sections from the frontal cortex of an individual carrier of the FTL498–499InsTC mutation (d, f, g) and a 15-d-old homozygous FTL-Tg4 mouse (e, h) and from the substantia nigra of a 30-d-old homozygous FTL-Tg4 mouse (i). Immunohistochemistry was performed using Abs against ubiquitin (d, e), 20S subunit (f), and 11S subunit (g–i). Scale bars: d, e, 40 μm; f, h, i, 10 μm; g, 20 μm. MW, Molecular weight.
Discussion
We generated transgenic mice expressing a human cDNA carrying the FTL498–499InsTC mutation driven by the MoPrP.Xho vector. In FTL-Tg mice, transgene expression was seen throughout the CNS and led to the deposition of ferritin as intranuclear and intracytoplasmic inclusion bodies in glia and neurons. In addition, ferritin deposition was seen in tissues of multiple organ systems. The histological, immunohistochemical, and biochemical data suggest that ferritin deposition in FTL-Tg mice is comparable to that observed in HF. Expression of the transgene in FTL-Tg mice resulted in a marked alteration in the intracellular localization of ferritin at the earliest ages examined here, followed by an age-dependent progressive intracellular accumulation of ferritin. Expression of the transgene causes a behavioral and motor dysfunction phenotype, leading to a shorter life span of the mice. FTL-Tg mice misregulate iron metabolism, accumulate ubiquitinated proteins, and incorporate several elements of the proteasome into ferritin inclusions.
In FTL-Tg mice, the distribution of cerebral NIs and cytoplasmic ferritin inclusions is widespread, as typically seen in HF (Vidal et al., 2004a; Mancuso et al., 2005), with inclusions prominent not only in the putamen, globus pallidus, thalamus, and the cerebellum but also in the hippocampus and the olfactory bulb. Compared with the neuropathology seen in HF, a more prominent neuronal involvement is observed, particularly in the cerebral cortex of transgenic mice. These differences may be attributable to the fact that expression of the transgene is driven not by the FTL promoter but by the MoPrP promoter, by the absence of the IRE sequence (Klausner et al., 1993) in the transgene, or because mice and humans have different neuronal vulnerability to mutant human ferritin. In HF and FTL-Tg mice, lesions extend beyond the CNS (Vidal et al., 2004a; Mancuso et al., 2005). Transgene expression in various organ systems in animal models of neurodegenerative diseases has been reported previously to occur using different promoters, including the MoPrP promoter vector (Borchelt et al., 1996; Sathasivam et al., 1999). The study of the formation of NIs in non-CNS tissues may be important for the understanding of early, non-neurological, symptoms of the disease as well as for monitoring therapeutic treatments.
The FTL498–499InsTC mutation leads to the formation of a polypeptide of 191 amino acids, with the last 9 amino acid residues of the wild-type polypeptide being replaced by a novel sequence of 25 amino acids (Vidal et al., 2004a,b). Two other nucleotide insertions in exon 4 of the FTL gene, an adenine insertion at position 460–461 (Curtis et al., 2001) and a cytidine insertion at position 646–647 (Mancuso et al., 2005), also alter the C-terminal sequence of the protein. These mutations are predicted to disrupt the C terminus of the FTL polypeptide and may have a significant impact on the tertiary structure and stability of FTL. In HF, ferritin deposits contain the wild-type FTL and FTH1 polypeptides and the mt-FTL polypeptide (Vidal et al., 2004a). The human and mouse FTL and FTH1 polypeptides are interchangeable and able to form ferritin particles (Rucker et al., 1996), and our biochemical and immunohistochemical data indicate that in FTL-Tg mice, the human mt-FTL polypeptide is able to form heteropolymers with the endogenous mouse Ftl and Fth1 polypeptides. Once formed, heteropolymers containing the mt-FTL subunit may have a decreased stability and precipitate. The presence of SDS-insoluble ferritin in HF and FTL-Tg mice strongly suggests that ferritin may be deposited in an insoluble state or forming an irreversible aggregate. Thus, FTL-Tg mice provide an in vivo model that exhibits two important aspects of HF, namely the association within the mutant and wild-type ferritin subunits and the selective insolubility of ferritin deposits. Moreover, the finding of abnormal ubiquitination and the presence of elements of the proteasome in ferritin deposits suggest that ferritin aggregates are recognized as misfolded proteins in HF and in transgenic mice. Abnormal protein ubiquitination and degradation by the proteasome, and recruitment of molecular chaperones such as HSP70 to protein inclusions, is a feature of many neurodegenerative diseases (Cummings et al., 1998; Rockwell et al., 2000; Bonini, 2002; Schmitt, 2006). The study of FTL-Tg mice may provide important information for our understanding of the role of these molecules in the early pathogenesis of HF and in other, more common, neurodegenerative diseases.
Ferritin is known to have a cytoplasmic distribution, although small quantities of ferritin are present in human serum and CSF (Harrison and Arosio, 1996). Little is known about the presence of ferritin in the cell nucleus. Intranuclear aggregates of ferritin have been found in animal models of iron overload (Smith et al., 1990; Kondo et al., 1998), and nuclear ferritin has been reported in avian and human cells (Cai et al., 1997; Thompson et al., 2002). The exact role of ferritin in the nucleus is not known, but it has been postulated that ferritin may have a role in transcriptional regulation (Broyles et al., 2001). The passage of ferritin through the nuclear pore into the nucleus is not dependent on known nuclear localization signals (Thompson et al., 2002) and does not require supramolecular assembly of ferritin or the presence of the E-helical domain (Cai and Linsenmayer, 2001), which is mutated in HF. Intranuclear inclusions in HF and FTL-Tg mice occupy almost completely the nucleus and as a result displace the chromatin up against the nuclear membrane. NIs may physically interfere with normal gene transcription or affect the turnover of transcription factors, because the accumulation of ubiquitinated proteins and elements of the proteasome in NIs suggests abnormal proteasomal activity. The study of FTL-Tg mice may bring some light on the understanding of the mechanism(s) involved in nuclear transport and accumulation of ferritin during normal homeostasis, iron overload, and in HF.
The pathogenic mechanism(s) of ferritin-mediated neurodegeneration in HF remains to be determined. Future studies using light and electron microscopy in young animals may establish whether neuronal degeneration is apoptotic, necrotic, autophagic, or cytoplasmic (Przedborski et al., 2003). Abnormal brain iron homeostasis has been observed in common neurodegenerative diseases, such as Alzheimer disease and Parkinson disease, in which increased brain iron levels and iron-mediated oxidative stress seem to play an important role in the disease (Connor et al., 1992; Perry et al., 2002; Berg and Youdim, 2006). A more direct link between abnormal iron metabolism and neurodegeneration is observed in diseases such as Friedreich ataxia (FRDA), aceruloplasminemia and neurodegeneration with brain iron accumulation (NBIA) (Beard and Connor, 2003). FRDA, the most frequent autosomal recessive cerebellar ataxia, is characterized by iron accumulation in several organs, including the brain. FRDA is caused by a deficiency of the mitochondrial protein frataxin an iron chaperone involved in the mitochondrial assembly of Fe-S clusters. Aceruloplasminemia, an autosomal recessive disorder, is caused by loss-of-function mutations in the ceruloplasmin gene leading to misregulation of both systemic and CNS iron trafficking. NBIA comprises a clinically and genetically heterogeneous group of disorders with high basal ganglia iron and includes pantothenate kinase-associated neurodegeneration, an autosomal recessive disease caused by mutations in the pantothenate kinase 2 (PANK2) gene. PANK2 is essential for the synthesis of coenzyme A from pantothenate, and the defect in PANK2 is predicted to cause the accumulation of cysteine, which binds iron and causes oxidative stress in the globus pallidus (Beard and Connor, 2003; Berg and Youdim, 2006). In HF and FTL-Tg mice, a primary defect in cerebral iron homeostasis is the pathophysiology underlying the disease rather than iron accumulation as a consequence of a different insult. In both cases, the presence of the mt-FTL subunit in ferritin particles appears to compromise the normal iron-storage function of ferritin (Vidal et al., 2004b). Iron accumulation is one of the prominent pathological findings in HF and in the transgenic animal model, and MRI studies show the presence of abnormal signals in brain areas affected by ferritin and iron deposition. Excess free iron detected by the cell may lead to an increase in the translation of ferritin mRNAs (Klausner et al., 1993), ferritin deposition, and neurodegeneration (Berg and Youdim, 2006). The FTL-Tg model will allow a comprehensive study of the consequences of disrupting cellular iron metabolism in the CNS and in other organ systems and will be a valuable model in which to test therapeutic interventions.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant R01 NS050227 (R.V.) and National Institute on Aging Grant P30 AG10133 (B.G.). We are grateful to Debra Lucas, Rose Richardson, Brenda Dupree, Urs Kuederli, Connie Alyea, and Yingbin Su for technical assistance.
References
- Beard JL, Connor JR. Iron status and neural functioning. Annu Rev Nutr. 2003;23:41–58. doi: 10.1146/annurev.nutr.23.020102.075739. [DOI] [PubMed] [Google Scholar]
- Berg D, Youdim MB. Role of iron in neurodegenerative disorders. Top Magn Reson Imaging. 2006;17:5–17. doi: 10.1097/01.rmr.0000245461.90406.ad. [DOI] [PubMed] [Google Scholar]
- Bonini NM. Chaperoning brain degeneration. Proc Natl Acad Sci USA. 2002;99:16407–16411. doi: 10.1073/pnas.152330499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Broyles RH, Belegu V, DeWitt CR, Shah SN, Stewart CA, Pye QN, Floyd RA. Specific repression of beta-globin promoter activity by nuclear ferritin. Proc Natl Acad Sci USA. 2001;98:9145–9150. doi: 10.1073/pnas.151147098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai CX, Linsenmayer TF. Nuclear translocation of ferritin in corneal epithelial cells. J Cell Sci. 2001;114:2327–2334. doi: 10.1242/jcs.114.12.2327. [DOI] [PubMed] [Google Scholar]
- Cai CX, Birk DE, Linsenmayer TF. Ferritin is a developmentally regulated nuclear protein of avian corneal epithelial cells. J Biol Chem. 1997;272:12831–12839. doi: 10.1074/jbc.272.19.12831. [DOI] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT. Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci. 1997;17:7385–7395. doi: 10.1523/JNEUROSCI.17-19-07385.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Menzies SL, St Martin SM, Mufson EJ. A histochemical study of iron, transferrin, and ferritin in Alzheimer's diseased brains. J Neurosci Res. 1992;31:75–83. doi: 10.1002/jnr.490310111. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Curtis AR, Fey C, Morris CM, Bindoff LA, Ince PG, Chinnery PF, Coulthard A, Jackson MJ, Jackson AP, McHale DP, Hay D, Barker WA, Markham AF, Bates D, Curtis A, Burn J. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet. 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- Drayer B, Burger P, Darwin R, Riederer S, Herfkens R, Johnson GA. MRI of brain iron. Am J Roentgenol. 1986;147:103–110. doi: 10.2214/ajr.147.1.103. [DOI] [PubMed] [Google Scholar]
- Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- Klausner RD, Rouault TA, Harford JB. Regulating the fate of mRNA: the control of cellular iron metabolism. Cell. 1993;72:19–28. doi: 10.1016/0092-8674(93)90046-s. [DOI] [PubMed] [Google Scholar]
- Kondo A, Deguchi J, Okada S. Intranuclear iron deposition in hepatocytes and renal tubular cells in mice treated with ferric nitrilotriacetate. Virchows Arch. 1998;433:543–548. doi: 10.1007/s004280050287. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Davidzon G, Kurlan RM, Tawil R, Bonilla E, Di Mauro S, Powers JM. Hereditary ferritinopathy: a novel mutation, its cellular pathology, and pathogenetic insights. J Neuropathol Exp Neurol. 2005;64:280–294. doi: 10.1093/jnen/64.4.280. [DOI] [PubMed] [Google Scholar]
- Perry G, Sayre LM, Atwood CS, Castellani RJ, Cash AD, Rottkamp CA, Smith MA. The role of iron and copper in the aetiology of neurodegenerative disorders: therapeutic implications. CNS Drugs. 2002;16:339–352. doi: 10.2165/00023210-200216050-00006. [DOI] [PubMed] [Google Scholar]
- Poon S, Treweek TM, Wilson MR, Easterbrook-Smith SB, Carver JA. Clusterin is an extracellular chaperone that specifically interacts with slowly aggregating proteins on their off-folding pathway. FEBS Lett. 2002;513:259–266. doi: 10.1016/s0014-5793(02)02326-8. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Vila M, Jackson-Lewis V. Neurodegeneration: what is it and where are we? J Clin Invest. 2003;111:3–10. doi: 10.1172/JCI17522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwell P, Yuan H, Magnusson R, Figueiredo-Pereira ME. Proteasome inhibition in neuronal cells induces a proinflammatory response manifested by upregulation of cyclooxygenase-2, its accumulation as ubiquitin conjugates, and production of the prostaglandin PGE(2) Arch Biochem Biophys. 2000;374:325–333. doi: 10.1006/abbi.1999.1646. [DOI] [PubMed] [Google Scholar]
- Rucker P, Torti FM, Torti SV. Role of H and L subunits in mouse ferritin. J Biol Chem. 1996;271:33352–33357. doi: 10.1074/jbc.271.52.33352. [DOI] [PubMed] [Google Scholar]
- Sathasivam K, Hobbs C, Turmaine M, Mangiarini L, Mahal A, Bertaux F, Wanker EE, Doherty P, Davies SW, Bates GP. Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet. 1999;8:813–822. doi: 10.1093/hmg/8.5.813. [DOI] [PubMed] [Google Scholar]
- Schmitt HP. Protein ubiquitination, degradation and the proteasome in neuro-degenerative disorders: no clear evidence for a significant pathogenetic role of proteasome failure in Alzheimer disease and related disorders. Med Hypotheses. 2006;67:311–317. doi: 10.1016/j.mehy.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Smith AG, Carthew P, Francis JE, Edwards RE, Dinsdale D. Characterization and accumulation of ferritin in hepatocyte nuclei of mice with iron overload. Hepatology. 1990;12:1399–1405. doi: 10.1002/hep.1840120622. [DOI] [PubMed] [Google Scholar]
- Thompson KJ, Fried MG, Ye Z, Boyer P, Connor JR. Regulation, mechanisms and proposed function of ferritin translocation to cell nuclei. J Cell Sci. 2002;115:2165–2177. doi: 10.1242/jcs.115.10.2165. [DOI] [PubMed] [Google Scholar]
- Vidal R, Frangione B, Rostagno A, Mead S, Revesz T, Plant G, Ghiso J. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;399:776–781. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- Vidal R, Delisle MB, Rascol O, Ghetti B. Hereditary ferritinopathy. J Neurol Sci. 2003;207:110–111. doi: 10.1016/s0022-510x(02)00435-5. [DOI] [PubMed] [Google Scholar]
- Vidal R, Ghetti B, Takao M, Brefel-Courbon C, Uro-Coste E, Glazier BS, Siani V, Benson MD, Calvas P, Miravalle L, Rascol O, Delisle MB. Intracellular ferritin accumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the ferritin light polypeptide gene. J Neuropathol Exp Neurol. 2004a;63:363–380. doi: 10.1093/jnen/63.4.363. [DOI] [PubMed] [Google Scholar]
- Vidal R, Delisle MB, Ghetti B. Neurodegeneration caused by proteins with an aberrant carboxyl-terminus. J Neuropathol Exp Neurol. 2004b;63:787–800. doi: 10.1093/jnen/63.8.787. [DOI] [PubMed] [Google Scholar]