

Abstract
Gas chromatography coupled to mass spectrometry (GC-MS) is one of the most frequently used tools for profiling primary metabolites. Instruments are mature enough to run large sequences of samples; novel advancements increase the breadth of compounds that can be analyzed, and improved algorithms and databases are employed to capture and utilize biologically relevant information. Around half the published reports on metabolite profiling by GC-MS focus on biological problems rather than on methodological advances. Applications span from comprehensive analysis of volatiles to assessment of metabolic fluxes for bioengineering. Method improvements emphasize extraction procedures, evaluations of quality control of GC-MS in comparison to other techniques and approaches to data processing. Two major challenges remain: rapid annotation of unknown peaks; and, integration of biological background knowledge aiding data interpretation.
Keywords: Data processing, Flux analysis, Gas chromatography, GC-MS, Mass spectrometry, Metabolite profiling, Metabolomics, Metabonomics, Quality control, Statistics, Time-of-flight
1. Introduction
The terms and the idea of metabolomics were introduced less than 10 years ago [1] focusing on an improved understanding of biological networks by systematic and comprehensive analysis of metabolism. However, comprehensive quantitative analysis of metabolites using gas chromatography coupled to mass spectrometry (GC-MS) had already been advocated in the 1970s [2] and subsequent decades with a focus on diagnostic purposes in clinical [3] and plant biological settings [4]. The advent of faster computers, better algorithms for spectra deconvolution [5] and improved statistical software packages facilitated exploiting GC-MS data files in an unbiased way compared to the classic procedure of pre-selecting analytical target molecules. The complement of detected peaks could now be analyzed by multivariate statistics to yield separation between classes of biological study designs, such as mutant and wild type plants [6], mammalian systems [7] or microorganisms [8]
These tools were subsequently used for proof-of-principle studies in a variety of species and applications. Importantly, minimum requirements to report metabolomics studies have recently been proposed, including standards for chemical analysis [9]. Such standards became necessary because metabolomics involves a variety of convoluted procedures, and, without standardized reporting, results and final interpretations become hardly comparable between studies.
When searching the ISI literature database for GC applications to metabolomics or metabolite profiling, 285 reports were counted until 2006, of which 80% were original research articles (Fig. 1). There has been large increase in the number of reports since 2002, which has continued in 2007. Based on the journal titles and the definition of science disciplines within the ISI database, more than half of these papers focused on application of techniques rather than method improvement (Table 1). This trend indicates a level of maturity of GC-MS that lends itself to be used for a large variety of biological questions.
Figure 1.
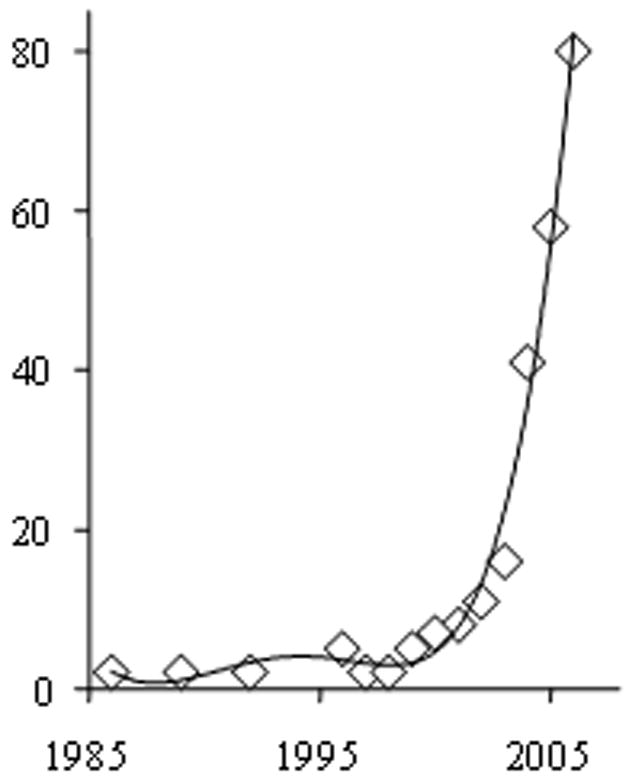
Number of publications published until 2006, querying the ISI database with the key words (metabon* OR metabolom* OR “metabol* profil*”) AND (gas chromatogr* OR GC).
Table 1.
Analysis of query results of the ISI database for GC-MS-based metabolomics. Numbers do not sum up to the total count because *) 148 journals reported at least one paper using this technique and because * scientific reports were often associated to more than one scientific discipline
| Country | Journal *) | Scientific discipline* | |||
|---|---|---|---|---|---|
| U.S.A. | 81 | Anal. Chem. | 15 | Plant Sci. | 65 |
| Germany | 76 | Phytochem. | 11 | Anal. Chem. | 63 |
| England | 32 | Plant Phys. | 11 | Biochem. Mol. Biol. | 50 |
| Japan | 24 | J. Agric. Food Chem. | 10 | Biotechnol. & Appl. Microbiol. | 43 |
| Canada | 21 | Metabolomics | 9 | Biochem. Res. Methods | 38 |
| Netherlands | 17 | J. Chromatogr. B | 8 | Spectroscopy | 22 |
| P.R. China | 14 | Bioinformatics | 6 | Food Sci. Technol. | 17 |
| Australia | 12 | Mass Spectrom. Rev. | 6 | Pharmacology | 16 |
| Denmark | 9 | J. Exp. Bot. | 6 | Appl. Chem. | 15 |
| France | 9 | Plant Cell Physiol. | 6 | Endocrinology | 15 |
| Sweden | 9 | J. Chromatogr. A | 5 | Microbiology | 14 |
| Switzerland | 9 | J. Mass Spectrom. | 5 | Agriculture | 13 |
| Austria | 8 | J. Proteome Res. | 5 | Cell Biol. | 13 |
| Wales | 6 | Rapid Comm. Mass Spectrom. | 5 | Genetics & Heredity | 10 |
| Belgium | 5 | Planta | 5 | Biophysics | 9 |
| Israel | 5 | Analyst (Cambridge, U. K.) | 4 | Statistics | 8 |
| Spain | 5 | Anal. Biochem. | 4 | Medicinal Chem. | 6 |
| South Korea | 5 | Clin. Chem. | 4 | Chemistry | 6 |
| Greece | 3 | Plant J. | 4 | Comp. Sci. | 6 |
| Italy | 3 | Biotechnol. Lett. | 3 | Math. Comp. Biol. | 6 |
| New Zealand | 3 | J. Chromatogr. | 3 | Org. Chem. | 5 |
Compounds screened by GC-MS profiling cover large parts of primary metabolism that are conserved across species, facilitating comparative studies between model organisms (e.g., mouse or rat) and humans. A variety of tools support use of GC-MS-based studies on primary metabolism, from mass-spectral libraries to advances in sample-introduction techniques and MS and interpretation of results by mapping results to known biochemical pathways.
However, compared to biomedical research or microbiology, plant-science papers still form the majority of published papers of GC-MS metabolite profiling. This trend can in part be explained by historical coincidences, but in part also because (in first approximation) all metabolites detected in plants are generated by the plant biochemical machinery itself, whereas metabolites in animals originate from both dietary catabolism and from intracellular anabolic biosynthesis. Biochemical interpretations of metabolite profiles in mammalian organisms are therefore less straightforward than in plants, which is also due to the high metabolic activity of many different organs in animals. Any human metabolome catalog that is generated by genome-based associations rather than analytical chemistry findings is necessarily limited to surveying endogenous anabolic activities or general catabolic pathways. Conversely, an increase in nutrition-related studies can be foreseen for analyzing gene-diet interactions in animals and humans (including a focus on the gut microbiome), which will also probably utilize GC-MS metabolite-profiling methods.
The current dominance of plant-science applications of GC-MS is further due to the availability of an excellent model organism, the small eudicotyledon plant Arabidopsis thaliana. Arabidopsis was the first higher organism with a fully sequenced genome, and generation of knockout and transgenic plants was comparatively easier than for mutant animals. Starting in 1998, metabolite profiling by GC-MS was applied to discover functions of plant genes in industrial research. Today, GC-MS-based metabolite profiling in plants is regarded as a standard tool in plant research and is routinely applied in a variety of laboratories. Applications span from genotype x environment studies, genetic studies of complex traits, and plant-pathogen interactions to agricultural and food-quality investigations, such as the substantial equivalence of genetically-modified food to classic bred cultivars.
In the subsequent sections, I further investigate technological developments and gaps that may foster further fields of applications and that could lead to increased acceptance in other fields of biology, specifically, for mammalian studies.
2. Sample preparation for GC-MS metabolite profiling
Early GC-MS studies on metabolite profiling emphasized the number of peaks that were detected from a specific tissue sample or the number of samples that could be handled per day. Such reports were important to distinguish the technique from transcriptomics or quantitative proteomics projects that are still some 10-fold more expensive on a per-sample basis. However, it had also been recognized that the number of identified peaks and the quality of quantifications ultimately limited the usability of metabolite profiles for comparative studies. Consequently, efforts were undertaken to decrease the degree of technical errors associated with quantifying chemically diverse compounds from complex matrices. While it is accepted that compromises have to be taken with respect to the quantitative accuracy in metabolomics, the number of studies focusing on harvesting of samples and extraction protocols suggest that this area is seen as a confounding factor for quality metabolomics by many research groups. Reducing technical errors during sample preparation might be more important than instrument-related parameters or the actual choice of analytical technique.
A careful design of experiments was suggested for extraction of blood plasma [10], which is particularly difficult due to the amount of proteins in the samples which need to be separated from metabolite pools that may be present in free form or bound to carrier proteins. The pitfalls of any development of novel protocols are to rank the tested parameters with respect to the impact that variation of parameters would incur (such as solvent types or time and temperature during the extraction procedures) and, often, the lack of accepted benchmark data. Once the most important parameters are selected, design of experiments extends to varying these parameters by taking extreme positions with a few intermittent steps in order to approach global method optima. If wrong parameters or inadequate choices for extreme values are taken (e.g., for extraction solvent compositions), obviously even careful study designs will not achieve optimal conditions for sample preparations. There is a lack of certified reference materials that could serve for interlaboratory ring tests or comparisons of technical errors and metabolic coverage between individual studies. At least, it should become accepted reporting standard to include readily available model samples in protocol developments (e.g., standard laboratory animals from commercial vendors instead of laboratory-specific inbred lines) and to report molar concentrations for a small number of critical target metabolites instead of statistics on overall number of peaks and arbitrary units for metabolite levels. For the case of blood plasma, low-abundant sex hormones were shown to be recovered only if proteins were precipitated in a very slow manner at +8°C but not under fast precipitation using colder methods [11].
Applications in microbial biology often focus on metabolic engineering with emphasis on primary metabolism. Interestingly, different protocols for microbial sample preparations were suggested with respect to the optimal temperature required to achieve fast quenching of metabolism and efficient metabolite extraction. A very convincing recent report suggested simplifying and accelerating the overall sample-preparation procedure for microbial samples by using hot temperature [12] instead of infusing cultures into cold methanol solutions [8,13,14]. In the deviating protocol using hot temperatures [12], the quenching and extraction steps already take place during sample transfer from the bioreactor. This method enabled rapid sampling and inactivation within 200–500 ms, which compared well with other sampling systems reported so far.
Protocols on plant tissues focused on integration of metabolite levels with protein and transcript data using ternary solvent compositions under cold temperatures [15]. Similar parameters were also reported in an independent study [16]. A range of applications utilize these or very similar protocols to study biological questions, mostly focused on primary metabolism. It is beyond the scope of this survey to report on the individual findings.
Compared to reports on extraction protocols, relatively little work has been performed on improving derivatization reactions for GC-MS-based metabolite profiling. Most commonly, trimethylsilylation (TMS) is used to exchange acidic protons and thus increase volatility of (polar) metabolites [17] although some amino acids may require special attention with respect to the peak ratio corresponding to the different derivatization status of primary amine groups [18]. As alternative to the mild and universal silylation reaction, derivatization by ethylchloroformate, has been suggested for urine analysis [19] yielding quantitative precision better than 10% RSD for metabolites tested in standard addition series. Two disadvantages of silylation protocols are the relatively high mass that is added by the derivatization agents and the readily neutral loss of TMS-OH groups during MS fragmentation. In order to develop protocols to better assess the positional enrichment of isotopes during labeling studies in carbohydrate research, a variety of derivatization reactions were compared, suggesting a new reagent that would overcome current limitations in flux analysis of sugars [20].
Sample preparation for volatile analysis is relatively straightforward. It can safely be stated that unbiased volatile analysis has become a significant trend in metabolite profiling with a focus on diagnostic purposes, such as recognition of human diseases by breath analysis with adsorbent columns [21], or urine analysis using solid-phase microextractions [22] or headspace techniques [23]. Another area of volatile metabolomics is aroma analysis for plant science [24] (e.g., by using headspace, solid-phase microextractions [25], vapor-phase extraction [26] or a combination of techniques [27]). Similarly to human biomedical applications, profiling of volatiles is a major field for current active research in recognition and discrimination of plant pathogenic infections [28]. These reports indicate the need to combine different analytical techniques to approach truly metabolomic surveys and that volatiles have to be included in these efforts. While it may be argued if liquid chromatography (LC) or nuclear magnetic resonance (NMR)-based techniques might deliver more reliable quantitative data for some classes of compounds, it is beyond doubt that volatiles are best analyzed by GC-based separation.
3. Advancing techniques in GC-MS
Metabolite profiling poses a variety of challenges due to the complexity of the matrix, even if non-volatile material (such as membrane lipids, waxes, proteins and polysaccharides) is removed prior to injection. Peak apexes need to be physically separated, at least to some extent, to be correctly assigned to unique metabolites; otherwise, co-elution of peaks will lead to false-negative peak detections and to spectra that result from the combinatory contributions of two or more compounds. The most abundant metabolites suffer least from spectral contamination, but low-abundant or novel metabolites require efficient separation for positive detection and structural characterization. 50,000–200,000 theoretical plates are regularly achieved in one-dimensional chromatographic separations. However, depending on the complexity of the sample origin, more than 1000 peaks may be present at detectable abundances in a given sample. Average mass-spectral purity for such a number of peaks is dramatically improved if two-dimensional GC is used for separation. Some reports have been published on the use of GCxGC-time-of-flight (TOF)-MS for metabolomic purposes [29]; however, a range of practical problems remain before comprehensive GCxGC separations may become routine applications for metabolically complex samples. For example, modulation-period times inevitably reduce some of the chromatographic resolution that achieved in the first dimension, so first-dimension retention times are less well defined than in truly one-dimensional separations.
In addition to chromatographic separation, quality of analysis was improved by using automated liner exchange systems and on-line derivatizations [30]. Two commercial vendors offer such solutions that facilitate metabolite profiling of unfractionated samples including free fatty acids and other lipophilic metabolites, despite the presence of a non-volatile matrix, such as membrane lipids. The basic concept is that accumulation of non-volatile material is inhibited by constantly exchanging GC liners and by cold injection of samples with subsequent heat ramping. In addition, a timed online derivatization of samples may result in better quantitative results for pairs of compounds that interconvert under room-temperature conditions during waiting times in autosamplers, such as glutamate and its cyclization product, oxoproline.
Other advances in GC-MS have emphasized the usefulness of stable isotopes for quantitative accuracy, either using isotope-dilution techniques [31] or in vivo labeling [32]. Both methods have great value for comparison of results between laboratories; however, complete in vivo labeling is especially difficult to achieve for animal studies.
4. Quality control and method comparisons
Although GC-MS is a mature technique and has proved to be successfully applied to a variety of biological problems, performances of different analytical techniques are rarely compared. Many metabolites need to be derivatized in order to increase volatility before analysis by GC-MS. Amino acids belong to the more problematic classes of compounds to be analyzed by GC-MS, especially when derivatized by the standard procedure using TMS. The standard method for amino-acid analysis, LC with fluorescence detection, proved to be superior with respect to reproducibility of quantitative analyses compared to TMS derivatives and GC-MS analysis [33]. Nevertheless, both techniques yielded the same biological conclusions on environment-induced changes in plant amino-acid levels. TMS derivatives with GC-MS analysis of amino acids was further compared to capillary electrophoresis coupled to MS, demonstrating that both techniques obtain similar results [34].
In principle, other derivatization techniques could be used (e.g., tertiary-butyldimethylsilylation (TBS)). TBS derivatives of amino acids are more stable and yield better precision and accuracy in GC-MS profiling than TMS derivatives [6]; however, TBS derivatives are also bulkier and thus do not enable analysis of carbohydrates by GC-MS. In many derivatization schemes, and specifically for TMS-based silylations, reactions are not stopped by adding quenching chemicals or fractionation schemes but may continue over hours. This fact has raised concerns that drifts over time need to be captured and corrected for [18]. Apart from the obvious necessity to completely randomize analytical sequences, it has been suggested to use technical replicates from pooled samples as baseline allowing comparisons even for unknown peaks and as a general measure to ensure minimal quality control [35].
The use of GC-MS and GC coupled to flame-ionization detection (GC-FID) was compared to NMR-based metabonomics for urine analysis [36]. It was concluded that there was a large overlap of detectable compounds between the GC-techniques and NMR analysis and that similar chemometric results were obtained for a proof-of-principle experiments. However, GC-MS was found to be more versatile with respect to peak identification and the number of independent metabolic signals that were detected. While NMR is regarded as being relatively robust and stable with respect to stability of signal intensities, mass spectrometers and GC-MS usually requires regular calibration curves using internal standards for long-time comparisons of quantifications. There are too many peaks in GC-MS-based metabolomics to be quantified using thorough calibrations. Instead, a combination of internal standards and sum parameters was found to be more suitable than internal normalizations or single-standard corrections in order to correct for instrumental drifts [37]. Such methods may well be suited to improve the stability and the quality of quantitative GC-MS metabolite-profiling results.
GC-MS was further tested in comparison to direct-infusion MS (DIMS) for metabolic footprinting [38] (i.e. analysis of excreted metabolites in microbial cultures). In accordance with the technical characteristics of both analytical techniques, it was concluded that GC-MS was superior for functional analysis of yeast mutants impaired in amino-acid pathways, whereas DIMS performed better for characterization of mutants involved in biosynthesis of polar lipids.
5. Flux analysis
All other approaches mentioned so far focus on identifying and quantifying metabolites based on concentrations or relative levels comparing two or more different biological conditions. A very different and complementary approach to metabolite profiling by GC-MS is taken by employing stable-isotope labeling for flux analysis. Flux analysis emphasizes the turnover of molecules through a number of enzymes, especially investigating cases for branched biochemical pathways that can alter relative flux ratios depending on conditions or disease states. In most cases, stable-isotope glucose is used and is differentially labeled at one or more atom positions within the molecule. Flux through glycolysis or adjacent pathways (pentose-phosphate pathway, TCA cycle and gluconeogenesis) is then estimated by analyzing relative enrichments in isotopes of metabolites comprised in these pathways in a dynamic way [39] or by analyzing isotope enrichments in metabolic sinks, preferably proteins, by complete hydrolysis and investigation of positional isotope enrichments in the corresponding amino acids [40]. Although most flux estimates have focused on microbiology applications [41], often with the aim of metabolic engineering in order to foster direction of fluxes towards a desired product [42], flux analysis by GC-MS has also been shown to be useful to characterize the impact of pancreatic tumors on metabolic fluxes in different organs by use of a rat model [43]. Although a range of different fluxes can be assessed using a single substrate molecule, such as glucose, a comprehensive analysis of fluxes through the metabolic network has not been achieved. Instead, other substrates need to be utilized to assess biochemical mechanisms, such as labeled succinate dimethylester that was used for a more detailed investigation of gluconeogenesis in perfused rat liver [7].
6. Data processing
Metabolite profiling by GC-MS and statistical analysis relies on efficient data-processing procedures, and minimum reporting requirements have recently been suggested [9,44]. The most straightforward way is to utilize retention indices that supposedly should be commonly used in GC-MS. Such retention-index alignment of chromatograms reduces chromatographic shifts to a large extent and enables setting up peak finding and peak matching algorithms that are less dependent on column aging or column cuts. More importantly, unambiguous peak annotations become independent from the sample origins, enabling queries across studies and across samples for which large metabolic alterations are observed or for prolonged series of chromatograms.
Two models can be distinguished: multitarget profiling; or, unbiased (non-targeted) profiling. Multitarget profiling is an extension of classic analytical chemistry by defining compound spectra, search retention-index windows, quantification ions and spectra-similarity thresholds for multiple (known) metabolites (e.g., using directly the corresponding GC-MS instrument software). In an extension of this approach, open source JAVA applets have been published that utilize instrument-independent NetCDF file formats to quantify pre-defined metabolites [45]. Such multitarget-profiling methods have the undisputable advantage that accurate quantifications can be achieved, based on internal or external calibration curves. Reporting absolute (molar) concentrations instead of relative peak intensities render studies more comparable between laboratories or across different studies.
However, unidentified compounds and potentially novel biomarkers remain undetected, unless unbiased (non-targeted) data-processing tools are employed. For such approaches, a variety of methods have been proposed. Popular and stand-alone open-access software are MZmine [46] and XCMS [47], which work on NetCDF files and have actually been first employed for LC-MS data. These tools offer peak-picking and alignment capabilities, but do not comprise further mass-spectral deconvolution. Automated mass-spectral deconvolution is achieved by the freely available AMDIS software [5]. This software has been used to deploy a new service, MSconnect [48], which aligns batches of AMDIS export files from related chromatograms and filters peaks that are not consistently detected in these batch comparisons. Such filtering systems are essential to remove spurious peaks and false positive peak detections (AMDIS deconvolution errors) (see Fig. 2). AMDIS does not support fast spectra acquisitions, such as those used in some TOF instruments, but, nevertheless, users can convert files to the NetCDF formats and upload to AMDIS. As other software tools, AMDIS enables users to select a range of parameters, upon which the number of peak detections will change dramatically. Ultimately, false-positive peak detections (e.g., assigning too many peaks based on minute differences in ion-trace peak shapes) is as detrimental to obtaining high-quality results as false-negative peak detections (i.e. failure to discriminate closely co-eluting peaks or to detect low-abundant signals, specifically in proximity to high-abundant peaks). Thorough investigations of the ratio and the number of false positive/false negative peak detections are therefore as crucial as developing filter systems that eliminate spurious peak counts.
Figure 2.
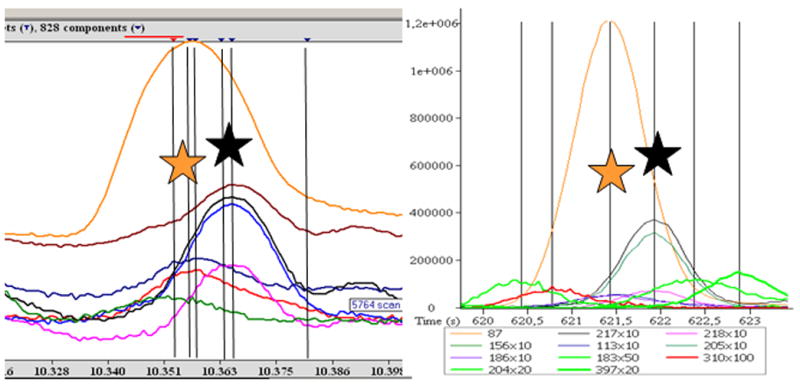
Processing of GC-TOF metabolite profiles using two different software packages. Left panel: Depending on the parameter settings, processing GC-TOF netCDF files with the freely available AMDIS software may yield a high number of false-positive peak detections (indicated by black and orange stars) and a high number of false-negative (undetected) peaks. Right panel: Processing the same chromatogram with the instrument-specific ChromaTOF 2.32 version software does not miss peaks or report false deconvolutions in the retention-time window exemplified here. Note: The AMDIS report is visualized by a log-scaled intensity axis whereas the ChromaTOF ion traces are multiplied by factors ranging from 1x to 100x.
Other data-processing approaches have developed tools implemented in the Matlab software, again using NetCDF-formatted GC-MS chromatograms as input files. An interesting approach was taken by first binning retention-time sections into binned spectra, aligning batches of chromatograms and then obtaining deconvoluted spectra for statistically significant different biomarkers by multicurve fitting, exploiting the different relative contributions of co-eluting compounds to the binned spectra [49].
A different proposed implementation of Matlab based the output of a peak-picking or peak-matching algorithm on the frequency, how often detected peaks were found in a given batch of chromatograms [50]. 965 samples were processed using this method, but it remained unclear how unique the mass spectra eventually obtained were for identification of metabolites.
None of these data-processing methods lends itself directly to constructing unambiguous metabolomics databases. The main reason is that the concept of retention indices has been largely dismissed in the development of the data-processing algorithms, which instead relied on overall similarity of chromatograms or peak-detection counts. Alternatively, databases have been constructed that utilize the output of retention indices and instrument-specific mass-spectral deconvolution software [51,52]. Both databases (and the corresponding libraries of unique spectra/retention-index data sets [53] take advantage of the high data-acquisition rate and spectral continuity of TOF mass spectrometers. A multiple filter system proposed for annotating and automatically adding novel metabolite spectra [51] relied on a range of additional mass-spectral metadata, such as peak purity, signal-to-noise ratios, apex masses and unique ions. Thresholds, such as mass-spectral similarity, were then conditionally set based on these metadata to ensure that even minor peaks next to very abundant compounds were correctly annotated. This database is supported by a study-design database [54] to ensure compliance with minimal reporting requirements proposed by the MSI working groups. Unfortunately, none of these databases are yet supported by open-source-code software and extensive documentation, and both mass-spectral databases rely on instrument-specific chromatogram files, so the utility of these databases is so far limited to the host research institutions but not available to the general public.
Very little progress has been reported for the identification of unknown peaks in GC-MS [55]. Eventually, the usefulness of GC-MS-based metabolomics will be determined by the reliability to detect and store unknown peaks, to identify the corresponding chemical structures and to link statistical patterns of metabolic regulations with potential physiological and biochemical implications. These gaps have to be closed before GC-MS can fully leverage its potentials in metabolism research.
First, existing metabolite libraries have to be extended by obtaining accurate retention-index information and mass spectra for all commercially available metabolites. Nevertheless, many peaks will remain in GC-MS chromatograms that cannot be assigned to identified chemical structures. For these peaks, novel algorithms will have to be developed that derive as much physicochemical information as possible from both the retention times and the mass spectrum. These data could subsequently be used to constrain hit lists obtained from queries of large chemical databases, such as public repository PubChem. Fig. 3 shows an example of how improved algorithms can help annotation of GC-MS peaks by substructure recognition. The electron-impact mass spectrum of trimethylsilylated cytosine was submitted to the NIST substructure algorithm to validate the general idea of exploiting the information comprised in mass spectra in an automated way to guide annotations of unknown compounds. For the trimethylsilylated cytosine spectrum, a high number of substructures were predicted to be present or absent with a high probability. Such information would clearly be useful to constrain candidate structures if integrated in an automated compound-characterization scheme. Fig. 4 shows an example of identification of uncommon metabolites that were found as novel compounds in transgenic potato tubers [56]. Using different chemical derivatizations, substructure recognition, interpretation of mass spectra and, eventually, receiving authentic standards from plant researchers, these compounds were identified as fructosyl-fructoses (inulobiose and levanbiose). Without validation by authentic standards, an automated algorithm can deliver only annotation scores and not definite identifications. It is therefore important that authors keep a clear separation of nomenclature between unambiguous identifications and mere characterizations or annotations of GC-MS spectra.
Figure 3.
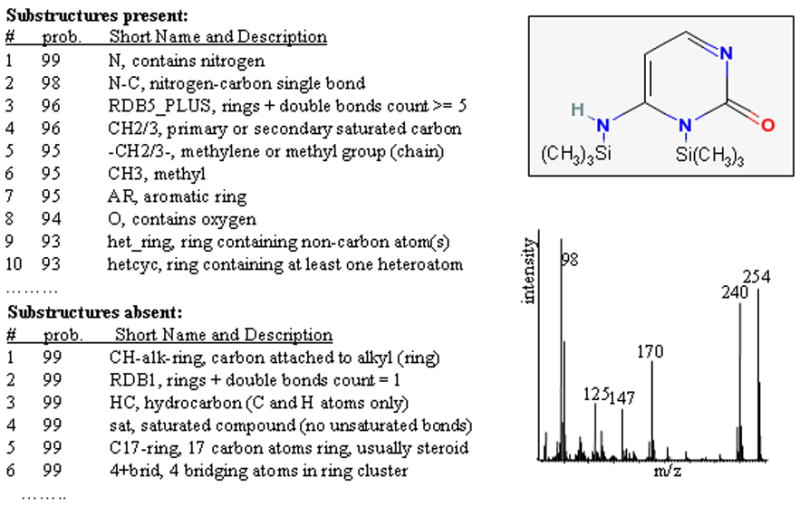
Substructure information (left panel) generated from mass spectrum (right lower panel) for bis(trimethylsilyl)-cytosine using the NIST substructure-recognition algorithm.
Figure 4. Example of identification of uncommon plant disaccharides inulobiose and levanbiose in transgenic potato tubers using derivatization and GC-MS.
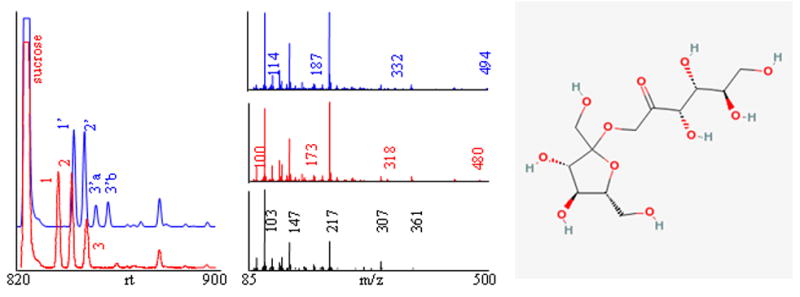
Left panel: Three unidentified peaks (1–3) were detected under methoximation and subsequent trimethylsilylation (TMS) with retention times close to that of sucrose (red ion trace, m/z 217). When using ethoxyamine instead of methoxyamine for the first derivatization step, compounds bearing keto- or aldehyde carbonyl moieties shifted to longer retention times (blue ion trace, peaks 1′–3′, m/z 217), whereas metabolites without such groups (such as sucrose) do not shift. Middle panel: Identical EI mass spectra were observed for peaks 1 and 2 (red spectrum), indicating that these may represent the syn/antiforms of a single methoximated compound. Best library hits and substructure recognition pointed to carbohydrates, specifically, fructose (black spectrum). Under ethoximation/TMS, some ion fragments shifted for 14 amu (blue spectrum), but the most abundant generic carbohydrate ions remained unaltered. Right panel: After database query, diverse authentic standards for fructosyl-fructoses were donated from plant researchers. Peaks 1 and 2 matched retention times and mass spectra for inulobiose (chemical structure in non-derivatized form), peaks 3 and 4 matched levanbiose.
7. Conclusions
A range of novel processes and method improvements have been published for GC-MS-based metabolite profiling. The technical maturity of GC-MS and the existence of commercial and publicly-available spectral libraries render GC-MS an indispensable tool for metabolomic applications. Metabolome coverage, data quality and data processing have been specific areas of research activity. Advanced public databases and repositories are still needed to facilitate data exchange between laboratories and to annotate the structures and the biochemical characteristics of the wealth of novel compounds that are being discovered by unbiased GC-MS analysis of biological specimens.
Acknowledgments
The work presented here and the MS-based metabolomics work in the author’s laboratory have been funded by research grant 5R01ES13932 of the U.S. National Institute of Environmental Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Oliver SG, Winson MK, Kell DB, Baganz F. Trends Biotechnol. 1998;16:373. doi: 10.1016/s0167-7799(98)01214-1. [DOI] [PubMed] [Google Scholar]
- 2.Thompson JA, Markey SP. Anal Chem. 1975;47:1313. doi: 10.1021/ac60358a074. [DOI] [PubMed] [Google Scholar]
- 3.Niwa T. J Chromatogr. 1986;379:313. doi: 10.1016/s0378-4347(00)80688-x. [DOI] [PubMed] [Google Scholar]
- 4.Sauter H, Lauer M, Fritsch H. ACS Symp Ser. 1991;443:288. [Google Scholar]
- 5.Stein SE. J Am Soc Mass Spectrom. 1999;10:770. [Google Scholar]
- 6.Glassop D, Roessner U, Bacic A, Bonnett GD. Plant Cell Physiol. 2007;48:573. doi: 10.1093/pcp/pcm027. [DOI] [PubMed] [Google Scholar]
- 7.Yang L, Kasumov T, Yu L, Jobbins KA, David F, Previs S, Kelleher JK, Brunengraber H. Metabolomics. 2006;2:85. [Google Scholar]
- 8.Villas-Boas SG, Hojer-Pedersen J, Akesson M, Smedsgaard J, Nielsen J. Yeast. 2005;22:1155. doi: 10.1002/yea.1308. [DOI] [PubMed] [Google Scholar]
- 9.Sumner LW, Amberg A, Barrett D, Beger R, Beale MH, Daykin C, Fan T, Fiehn O, Goodacre R, Griffin JL, Higashi R, Kopka J, Lindon JC, Lane AN, Marriott P, Nicholls AW, Reily MD, Viant M. Metabolomics. 2007;3:211. doi: 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiye A, Trygg J, Gullberg J, Johansson AI, Jonsson P, Antti H, Marklund SL, Moritz T. Anal Chem. 2005;77:8086. doi: 10.1021/ac051211v. [DOI] [PubMed] [Google Scholar]
- 11.Boernsen KO, Gatzek S, Imbert G. Anal Chem. 2005;77:7255. doi: 10.1021/ac0508604. [DOI] [PubMed] [Google Scholar]
- 12.Schaub J, Schiesling C, Reuss M, Dauner M. Biotechnol Progr. 2006;22:1434. doi: 10.1021/bp050381q. [DOI] [PubMed] [Google Scholar]
- 13.Buchholz A, Hurlebaus J, Wandrey C, Takors R. Biomol Eng. 2002;19:5. doi: 10.1016/s1389-0344(02)00003-5. [DOI] [PubMed] [Google Scholar]
- 14.Koek MM, Muilwijk B, van der Werf MJ, Hankemeier T. Anal Chem. 2006;78:1272. doi: 10.1021/ac051683+. [DOI] [PubMed] [Google Scholar]
- 15.Weckwerth W, Wenzel K, Fiehn O. Proteomics. 2004;4:78. doi: 10.1002/pmic.200200500. [DOI] [PubMed] [Google Scholar]
- 16.Gullberg J, Jonsson P, Nordstrom A, Sjostrom M, Moritz T. Anal Biochem. 2004;331:283. doi: 10.1016/j.ab.2004.04.037. [DOI] [PubMed] [Google Scholar]
- 17.Halket JM, Waterman D, Przyborowska AM, Patel RKP, Fraser PD, Bramley PM. J Exp Bot. 2005;56:219. doi: 10.1093/jxb/eri069. [DOI] [PubMed] [Google Scholar]
- 18.Kanani HH, Klapa MI. Metabol Eng. 2007;9:39. doi: 10.1016/j.ymben.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Qiu Y, Su M, Liu Y, Chen M, Gu J, Zhang J, Jia W. Anal Chim Acta. 2007;583:277. doi: 10.1016/j.aca.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 20.Price NPJ. Anal Chem. 2004;76:6566. doi: 10.1021/ac049198m. [DOI] [PubMed] [Google Scholar]
- 21.Basanta M, Koimtzis T, Singh D, Wilson I, Thomas CLP. Analyst (Cambridge, U K) 2007;132:153. doi: 10.1039/b608608j. [DOI] [PubMed] [Google Scholar]
- 22.Mills GA, Walker V. J Chromatogr. 2001;B 753:259. doi: 10.1016/s0378-4347(00)00554-5. [DOI] [PubMed] [Google Scholar]
- 23.Wahl HG, Hoffmann A, Luft D, Liebich HM. J Chromatogr. 1999;A 847:117. doi: 10.1016/s0021-9673(99)00017-5. [DOI] [PubMed] [Google Scholar]
- 24.Tikunov Y, Lommen A, de Vos CHR, Verhoeven HA, Bino RJ, Hall RD, Bovy AG. Plant Physiol. 2005;139:1125. doi: 10.1104/pp.105.068130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farag MA, Ryu CM, Sumner LW, Pare PW. Phytochemistry. 2006;67:2262. doi: 10.1016/j.phytochem.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 26.Schmelz EA, Engelberth J, Tumlinson JH, Block A, Alborn HT. Plant J. 2004;39:790. doi: 10.1111/j.1365-313X.2004.02168.x. [DOI] [PubMed] [Google Scholar]
- 27.Zhang ZM, Li GK. Microchem J. 2007;86:29. [Google Scholar]
- 28.Vikram A, Hamzehzarghani H, Kushalappa AC. Can J Plant Path - Revue Canadienne Phytopathol. 2005;27:194. [Google Scholar]
- 29.Welthagen W, Shellie RA, Spranger J, Ristow M, Zimmermann R, Fiehn O. Metabolomics. 2005;1:57. doi: 10.1016/j.chroma.2005.05.088. [DOI] [PubMed] [Google Scholar]
- 30.Denkert C, Budczies J, Kind T, Weichert W, Tablack P, Sehouli J, Niesporek S, Konsgen D, Dietel M, Fiehn O. Cancer Res. 2006;66:10795. doi: 10.1158/0008-5472.CAN-06-0755. [DOI] [PubMed] [Google Scholar]
- 31.Adlercreutz H, Kiuru P, Rasku S, Wahala K, Fotsis T. J Steroid Biochem Mol Biol. 2004;92:399. doi: 10.1016/j.jsbmb.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 32.Birkemeyer C, Luedemann A, Wagner C, Erban A, Kopka J. Trends Biotechnol. 2005;23:28. doi: 10.1016/j.tibtech.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 33.Noctor G, Bergot GL, Mauve C, Thominet D, Lelarge-Trouverie C, Prioul JL. Metabolomics. 2007;3:16. [Google Scholar]
- 34.Williams BJ, Cameron CJ, Workman R, Broeckling CD, Sumner LW, Smith JT. Electrophoresis. 2007;28:1371. doi: 10.1002/elps.200600609. [DOI] [PubMed] [Google Scholar]
- 35.Sangster T, Major H, Plumb R, Wilson AJ, Wilson ID. Analyst (Cambridge, U K) 2006;131:1075. doi: 10.1039/b604498k. [DOI] [PubMed] [Google Scholar]
- 36.Fancy SA, Beckonert O, Darbon G, Yabsley W, Walley R, Baker D, Perkins GL, Pullen FS, Rumpel K. Rapid Comm Mass Spectrom. 2006;20:2271. doi: 10.1002/rcm.2583. [DOI] [PubMed] [Google Scholar]
- 37.Deport C, Ratel J, Berdague JL, Engel E. J Chromatogr. 2006;A 1116:248. doi: 10.1016/j.chroma.2006.03.092. [DOI] [PubMed] [Google Scholar]
- 38.Mas S, Villas-Boas SG, Hansen ME, Akesson M, Nielsen J. Biotechnol Bioeng. 2007;96:1014. doi: 10.1002/bit.21194. [DOI] [PubMed] [Google Scholar]
- 39.Marin S, Lee WNP, Bassilian S, Lim S, Boros LG, Centelles JJ, Fernandez-Novell JM, Guinovart JJ, Cascante M. Biochem J. 2004;381:287. doi: 10.1042/BJ20031737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer E, Sauer U. Eur J Biochem. 2003;270:880. doi: 10.1046/j.1432-1033.2003.03448.x. [DOI] [PubMed] [Google Scholar]
- 41.Raghevendran V, Gombert AK, Christensen B, Kotter P, Nielsen J. Yeast. 2004;21:769. doi: 10.1002/yea.1136. [DOI] [PubMed] [Google Scholar]
- 42.Yang TH, Wittmann C, Heinzle E. Metabol Eng. 2006;8:432. doi: 10.1016/j.ymben.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 43.Boros LG, Lerner MR, Morgan DL, Taylor SL, Smith BJ, Postier RG, Brackett DJ. Pancreas. 2005;31:337. doi: 10.1097/01.mpa.0000186524.53253.fb. [DOI] [PubMed] [Google Scholar]
- 44.Goodacre R, Broadhurst D, Smilde AK, Kristal BS, Baker JD, Beger R, Bessant C, Connor S, Capuani G, Craig A, Ebbels T, Kell DB, Manetti C, Newton J, Paternostro G, Sjoestroem M, Trygg J, Wulfert F. Metabolomics. 2007;3:231. [Google Scholar]
- 45.Bunk B, Kucklick M, Jonas R, Munch R, Schobert M, Jahn D, Hiller K. Bioinformatics. 2006;22:2962. doi: 10.1093/bioinformatics/btl526. [DOI] [PubMed] [Google Scholar]
- 46.Katajamaa M, Oresic M. BMC Bioinformatics. 2005;6:179. doi: 10.1186/1471-2105-6-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith CA, Want EJ, O’Maille G, Abagyan R, Siuzdak F. Anal Chem. 2006;78:779. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 48.Styczynski MP, Moxley JF, Tong LV, Walther JL, Jensen KL, Stephanopoulos GN. Anal Chem. 2007;79:966. doi: 10.1021/ac0614846. [DOI] [PubMed] [Google Scholar]
- 49.Jonsson P, Johansson AI, Gullberg J, Trygg J, Grung B, Marklund BS, Sjostrom M, Antti H, Moritz T. Anal Chem. 2005;77:5635. doi: 10.1021/ac050601e. [DOI] [PubMed] [Google Scholar]
- 50.Dixon SJ, Brereton RG, Soini HA, Novotny MV, Penn DJ. J Chemometr. 2006;20:325. [Google Scholar]
- 51.Fiehn O, Wohlgemuth G, Scholz M. Proc Lect Notes Bioinformatics. 2005;3615:224. [Google Scholar]
- 52.Kopka J, Schauer N, Krueger S, Birkemeyer C, Usadel B, Bergmuller E, Dormann P, Weckwerth W, Gibon Y, Stitt M, Willmitzer L, Fernie AR, Steinhauser D. Bioinformatics. 2005;21:1635. doi: 10.1093/bioinformatics/bti236. [DOI] [PubMed] [Google Scholar]
- 53.Schauer N, Steinhauser D, Strelkov S, Schomburg D, Allison G, Moritz T, Lundgren K, Roessner-Tunali U, Forbes MG, Willmitzer L, Fernie AR, Kopka J. FEBS Lett. 2005;589:1332. doi: 10.1016/j.febslet.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 54.Scholz M, Fiehn O. Pacific Symp Biocomp. 2007;12:169. [PubMed] [Google Scholar]
- 55.Boroczky K, Laatsch H, Wagner-Dobler I, Stritzke K, Schulz S. Chem Biodiv. 2006;3:622. doi: 10.1002/cbdv.200690065. [DOI] [PubMed] [Google Scholar]
- 56.Catchpole G, Beckmann M, Enot DP, Mondhe M, Zywicki B, Taylor J, Hardy N, Smith A, King RD, Kell DB, Fiehn O. J Draper Proc Natl Acad Sci USA. 2005;102:14458. doi: 10.1073/pnas.0503955102. [DOI] [PMC free article] [PubMed] [Google Scholar]