

Abstract
DNA damage generated by oxidant byproducts of cellular metabolism has been proposed as a key factor in cancer and aging. Oxygen free radicals cause predominantly base damage in DNA, and the most frequent mutagenic base lesion is 7,8-dihydro-8-oxoguanine (8-oxoG). This altered base can pair with A as well as C residues, leading to a greatly increased frequency of spontaneous G·C→T·A transversion mutations in repair-deficient bacterial and yeast cells. Eukaryotic cells use a specific DNA glycosylase, the product of the OGG1 gene, to excise 8-oxoG from DNA. To assess the role of the mammalian enzyme in repair of DNA damage and prevention of carcinogenesis, we have generated homozygous ogg1−/− null mice. These animals are viable but accumulate abnormal levels of 8-oxoG in their genomes. Despite this increase in potentially miscoding DNA lesions, OGG1-deficient mice exhibit only a moderately, but significantly, elevated spontaneous mutation rate in nonproliferative tissues, do not develop malignancies, and show no marked pathological changes. Extracts of ogg1 null mouse tissues cannot excise the damaged base, but there is significant slow removal in vivo from proliferating cells. These findings suggest that in the absence of the DNA glycosylase, and in apparent contrast to bacterial and yeast cells, an alternative repair pathway functions to minimize the effects of an increased load of 8-oxoG in the genome and maintain a low endogenous mutation frequency.
DNA bases are susceptible to damage in vivo as a consequence of exposure to endogenous and environmental mutagens, including the endogenous production of reactive oxygen species during aerobic cellular metabolism. Oxidative DNA damage has been implicated in the etiology of many degenerative diseases, aging, and cancer, and a diet high in fruit and vegetables, a major source of antioxidants, is associated with a lowered cancer risk (1, 2). Oxygen free radicals induce a variety of lesions in DNA, including oxidized bases, abasic (AP) sites, and DNA strand breaks. A major site of attack is at the 8 position of guanine to produce 7,8-dihydro-8-oxoguanine (8-oxoG) (3, 4). 8-OxoG is strongly mutagenic, having the propensity to mispair with A residues, leading to an increased frequency of spontaneous G·C→T·A transversion mutations in repair-deficient bacterial and yeast cells (5, 6). However, because direct measurements of the level of this mutagenic oxidative base lesion in vivo are compromised by the limitations of current methodologies and variations between different cell types and compartments, its biological significance remains a matter of some debate (1, 2, 7–10).
In bacterial cells, three enzymes act to prevent spontaneous mutagenesis because of 8-oxoG (5); the Fpg (MutM) DNA glycosylase/AP lyase removes the oxidized base from 8-oxoG:C base pairs in duplex DNA, the MutY DNA glycosylase specifically excises A misincorporated opposite unrepaired 8-oxoG during replication, and MutT is an 8-oxo-dGTPase preventing incorporation of 8-oxo-dGMP into nascent DNA. MutY and MutT homologues have been identified in mammalian cells (11, 12) but are not apparent in the Saccharomyces cerevisiae genome sequence. Both mammalian and yeast cells use a distinct DNA glycosylase, the product of the OGG1 gene, to excise 8-oxoG from DNA; the cloned human and mouse cDNAs encode distinct nuclear and mitochondrial forms of the enzyme generated by alternative RNA splicing (13–20). Although there is little sequence similarity between yeast/human OGG1 and bacterial Fpg, OGG1 is a functional homologue of the Escherichia coli enzyme with high specificity for 8-oxoG paired with C (13, 15, 17–21). The ubiquitous existence throughout evolution of a specific repair enzyme to excise 8-oxoG from DNA underscores the importance of removing this mutagenic lesion from the genome.
Here we have generated homozygous ogg1−/− null mice to assess the role of the mammalian enzyme in the repair of oxidative DNA base damage, the extent and physiological relevance of such damage in vivo, and the contribution of OGG1 to counteracting the mutagenic and carcinogenic potential of 8-oxoG.
Materials and Methods
Construction of the Targeting Vector and Generation of ogg1 Mutant Mice and Cell Lines.
Procedures for generating gene-targeted knockout mice were essentially as described (22). Briefly, a partial murine OGG1 cDNA clone was isolated by reverse transcription–PCR and used to screen a mouse 129SV lambda genomic library. Two partially overlapping genomic clones were obtained that spanned ≈30.5 kb, and sequences encoding a highly conserved helix–hairpin–helix motif required for DNA glycosylase/AP lyase activity (15, 17) were located to the overlapping region by Southern hybridization analysis with an oligonucleotide probe (Fig. 1a). Approximately 4.6 kb of genomic DNA including this motif were replaced by the neomycin-resistance gene/polyadenylation signal (Neo) in the targeting construct by cloning a flanking 3.1-kb XhoI fragment (5′) into the XhoI site of pSK-MC1-NEO-poly(A) and a flanking 4.7-kb HindIII fragment (3′) into the BamHI site by means of double-stranded oligonucleotide linkers; HindIII sites were not regenerated during this process (Fig. 1a). The targeting construct was verified by restriction enzyme digest and propagated in the “SURE” E. coli strain (Stratagene). NotI-linearized targeting vector was electroporated into GK129 embryonic stem (ES) cells from 129SV mice. Genomic DNA was isolated from G418-resistant colonies transferred to 96-well plates, digested with EcoRI, and analyzed by Southern hybridization analysis (Fig. 1b). An ES clone in which homologous recombination had occurred in one allele of the ogg1 gene was expanded from a duplicate plate and injected into blastocysts from C57BL/6J mice. Resultant chimeras were mated and agouti F1 progeny were screened by tail biopsy. Mice heterozygous for the targeted ogg1 allele (+/−) were interbred, and F2 progeny were genotyped as above. Detailed intron–exon structure was not determined here but subsequently has been published (23).
Figure 1.

Targeted disruption of the murine OGG1 locus. (a) Physical map of the genomic DNA containing the OGG1 gene. The location of the helix–hairpin–helix motif is indicated; amino acids identical in yeast, human, and murine OGG1 are boxed; the murine amino acid sequence shown is identical to the human sequence except for the two residues underlined; the Lys and Asp residues associated with DNA glycosylase/AP lyase activity are shown with an arrow and a circle, respectively (15, 17). Genomic fragments were subcloned on either side of the Neo gene to generate a construct that deleted ≈4.6 kb including this motif at the targeted locus. Genomic DNA is shown by boxes; vector sequences are shown by a line. Restriction digest with EcoRI gives rise to a ≈12-kb fragment at the wild-type locus and a ≈5-kb fragment at the targeted locus that are detected by hybridization with a 5′ flanking probe (striped box). (b) Representative wild-type (+/+), heterozygous (+/−), and ogg1 null (−/−) live-born F2 progeny genotyped by EcoRI digestion of tailsnip DNA and hybridization with the 5′ probe.
Primary mouse embryo fibroblast (MEF) cultures were established by standard procedures from individual embryonic day 13.5 (E13.5) embryos derived from heterozygous matings. Cells were cultured in DMEM/Ham’s F-12 (3:1) with 10% FBS and genotyped by Southern hybridization as above, and permanent cell lines were established from transformed clones arising spontaneously after repeated passage in culture (24).
Preparation of Nuclear Extracts and Enzyme Assays.
Organs were removed, quick-frozen in liquid nitrogen, and stored at −80°C until use. Scissor-macerated tissue was passed through a 19-gauge, 1.5-inch needle. Cell suspensions were washed in hypotonic buffer A (10 mM Hepes⋅KOH, pH 7.7/0.5 mM MgCl2/10 mM KCl/1 mM DTT/0.2 mM PMSF), and cells were lysed by incubation in 2 vol of buffer A for 15 min. Nuclei were recovered by centrifugation at 2,000 × g for 10 min and extracted with 2 vol of buffer B (20 mM Hepes⋅KOH, pH 7.7/0.5 mM MgCl2/0.42 M NaCl/0.2 mM EDTA/1 mM DTT/0.2 mM PMSF/25% glycerol). After centrifugation at 14,000 × g for 10 min, the supernatant was recovered and briefly dialyzed against buffer C (25 mM Hepes⋅KOH, pH 7.7/50 mM KCl/2 mM DTT), aliquoted, and quick-frozen in liquid nitrogen. All steps were carried out at 0°C. Enzyme assays were carried out essentially as described (19). Assays were repeated with organs from two different mice and gave consistent results (see Figs. 2–4). Briefly, a 49-mer oligonucleotide containing a single, centrally placed 8-oxoG residue (Fig. 2b) was 32P-labeled at the 5′ terminus and annealed to a complementary oligonucleotide with a C residue opposite the 8-oxoG. All oligonucleotides included phosphorothioate linkages at the ultimate and penultimate 5′ and 3′ residues to reduce exonucleolytic attack. Similarly, a series of 22-bp double-stranded oligonucleotide substrates was used to compare 8-oxoG repair opposite A/C/G/T (ref. 15; see Fig. 4). Standard reaction mixtures (refs. 15 and 19) contained 150 fmol double-stranded oligonucleotide substrate and 2 μg nuclear extract (unless otherwise stated). Oligonucleotides were recovered (15, 19), resolved by denaturing 20% PAGE, and visualized and quantified by using a PhosphorImager.
Figure 2.

Absence of OGG1 activity in extracts of organs from ogg1 null mice. (a) Nuclear extracts were prepared of the organs indicated from wild-type (+/+), heterozygous (+/−), and ogg1 null (−/−) mice and incubated with a double-stranded oligonucleotide substrate (b) with a 8-oxoG:C mismatch and 32P-labeled at the 5′ end of the 8-oxoG-containing strand. Reaction products were analyzed by PhosphorImager after denaturing PAGE. OGG1 activity excises the 8-oxoG and cleaves the 32P-labeled, 8-oxoG-containing strand (49 nt) 3′ of the lesion; subsequent cleavage of the terminal sugar phosphate by HAP1 endonuclease (36) in the extract produces a 21-nt, 32P-labeled species. Lower molecular weight bands result from exonuclease degradation at the exposed 3′ residues of the 21-nt cleavage product. Levels of two control enzyme activities, uracil-DNA glycosylase and hNth1 (36), were normal in all extracts.
Figure 4.

Comparative activity on different 8-oxoG-containing mismatches in extracts from ogg1 null mice. Nuclear extracts prepared from testes of wild-type (+/+), heterozygous (+/−), and ogg1 null (−/−) mice were incubated with double-stranded oligonucleotide substrates containing different residues opposite 8-oxoG (as indicated). The assay was as in Fig. 2, except, in this case, a 22-bp substrate was used and OGG1 activity is indicated by cleavage of the 32P-labeled 8-oxoG-containing strand to produce a 9-nt species.
DNA glycosylase activity removing the imidazole ring-opened guanine lesion, 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine (faPy), from a substrate of poly(dG·dC) containing [3H]faPy (5,000 dpm/μg) was assayed as described (15) in a reaction mixture containing 0.4 μg faPy substrate and 1 μg nuclear extract. Released [3H]faPy was quantitated by scintillation counting after ethanol precipitation of the substrate.
Preparation of Nuclear DNA and Analysis of 8-oxoG by HPLC–Electrochemical Detection (HPLC-ECD).
Male mice were sacrificed at 13–15 weeks; livers were removed, quick-frozen in liquid nitrogen, and stored at −80°C until use. Scissor-macerated liver was passed through a 19-gauge, 1.5-inch needle. Extraction of DNA and hydrolysis to nucleosides by nuclease P1 and alkaline phosphatase were as described (25). To reduce oxidation during the preparation of DNA, TEMPO (2,2,6,6-tetramethylpiperidine-N-oxyl) was added to all solutions at 100 μM immediately before use (25). 8-Hydroxy-2′-deoxyguanosine and 2′-deoxyguanosine were separated by HPLC and analyzed by electrochemical detection (ECD; +300 mV) and UV light (290 nm), and results were expressed as the ratio of 8-oxoG/106 bp in each DNA sample. The following conversions were used: 1 8-oxoG/105 G = 4 8-oxoG/106 bp; 1 8-oxoG/106 bp = 6,000 8-oxoG/diploid genome.
Analysis of Spontaneous and Induced Oxidative DNA Base Damage in the ogg1 Null Cell Line by Means of Fpg Cleavage and Alkaline Elution.
The E. coli Fpg protein excises primarily 8-oxoG and faPy lesions and cleaves the DNA backbone 3′ of the base; thus, incision of the DNA by Fpg at its substrate lesion(s) generates single-strand breaks that can be readily detected by alkaline elution and used to quantitate oxidative DNA base damage (26, 27). The exposure of cells to the photosensitizer Ro19–8022 plus visible light, cell lysis, treatment of DNA bound to polycarbonate filters with Fpg protein (28), and estimation of single-strand DNA breaks after alkaline elution were as described (26, 29). Results were expressed as the number of Fpg-sensitive sites/106 bp (see above). Repair kinetics were determined as described (29); data were corrected for dilution of the originally induced damage through cell division by determining the ratio of the cell number at later time points (8 and 16 h) vs. time 0. The low number of modifications induced by Ro19–8022 plus visible light gave no apparent toxicity in these experiments; the proliferation of both wild-type and ogg1-null fibroblasts was not reduced significantly compared with untreated cells.
Analysis of Spontaneous Mutagenesis in Vivo.
To analyze spontaneous mutation frequency in vivo, a nonexpressed lacI transgene was introduced into the ogg1 null background and mutational response was examined in vivo by using the Big Blue Transgenic Rodent Mutagenesis Assay System (Stratagene). Procedures were carried out essentially according to the manufacturer’s instructions; briefly, homozygous ogg1−/− null mice were bred with homozygous Big Blue mice harboring 40 tandemly integrated copies of the λLIZ shuttle vector on chromosome four. F1 heterozygous progeny were intercrossed to recover OGG1+/+ and ogg1−/− homozygotes, which then were screened to identify those still harboring high-copy-number integrations of the lambda vector by PCR with the Big Blue PCR primer set. Appropriate wild-type and null animals were sacrificed at 10 or 20 weeks of age, genomic DNA was extracted from selected organs by using the RecoverEase DNA isolation kit, and the lambda shuttle vector was recovered and packaged into viable phage particles by using the Transpack packaging extract and used to infect the appropriate E. coli host. Plaque-forming units (250,000–300,000) were plated on medium containing X-Gal (5-bromo-4-chloro-3-indolyl β-d-galactopyranoside) for each organ tested, and mutant (blue) plaques were scored. Mutation frequencies were calculated for each organ sample as the ratio of mutant (blue) to nonmutant (colorless) plaques. Bacteriophages were recovered from representative mutant plaques, the lacI target gene was amplified by PCR (as above), and the mutation spectrum was analyzed by using the BigDye Terminator Cycling DNA Sequencing Kit and an ABI Prism 310 DNA Sequencer (Applied Biosystems).
Results
Generation of ogg1 Null Mice.
Homozygous null mice were generated via targeted disruption of the murine OGG1 gene in embryonic stem cells. Sequences encoding the highly conserved helix–hairpin–helix motif required for enzyme activity (15, 17) were replaced by Neo in the targeting vector (Fig. 1a). A clone in which homologous recombination had occurred in one allele of the OGG1 gene was identified and injected into C57BL/6J blastocysts to generate germ-line chimeras and heterozygous ogg1+/− mice. Genotyping of 45 live-born mice from four intermatings of F1 heterozygotes (Fig. 1b) showed that ogg1 null mice are viable; homozygous −/− mutant mice were recovered in F2 litters at frequencies consistent with Mendelian segregation (data not shown) and were indistinguishable from their +/− heterozygous and +/+ wild-type siblings. Null animals remained viable and apparently healthy into adulthood (oldest mice are 18 months) with no overt phenotype; systematic histopathological examination of two such animals sacrificed at 8 and 11 months of age did not reveal any abnormalities (G. Stamp and D.E.B., unpublished data).
Enzyme Activity in Cell-Free Tissue Extracts from ogg1 Null Mice.
OGG1 enzyme activity was assayed in cell-free extracts of various tissues derived from ogg1−/− null, +/− heterozygous, or +/+ wild-type animals (Fig. 2a). Mammalian OGG1 releases free 8-oxoG from DNA and also cleaves the DNA strand 3′ of the lesion by AP lyase activity, introducing a single-strand interruption in a double-stranded oligonucleotide at an 8-oxoG residue base-paired with cytosine (refs. 13, 15, and 17–21; Fig. 2b). OGG1 activity was detected in wild-type extracts from all tissues examined and was most abundant in testes (Fig. 2a) and brain (data not shown). OGG1 activity was ≈2-fold reduced in heterozygotes; for both heterozygote and wild-type tissue, the observed enzyme activity was proportional to protein concentration (Fig. 3a). No cleavage of the 8-oxoG:C substrate was detected in tissue extracts from two ogg1−/− knockout mice (Figs. 2a, 3a, and 4), indicating that OGG1 is the only mammalian DNA glycosylase that efficiently removes 8-oxoG residues from DNA. Conversely, removal of faPy, which also is excised from DNA by OGG1, was only partially reduced (≈3-fold) in ogg1−/− knockout extracts (Fig. 3b), demonstrating the existence of a second enzyme able to remove this lesion; this is likely to be the hNth1 DNA glycosylase that excises oxidized pyrimidines (L. Luna, M. Bjøras, and E.S., unpublished data). Our results agree with previous data on the substrate specificity of the murine and human OGG1 enzyme (13, 15, 17–21), with no detectable activity on a 8-oxoG:A substrate and only weak activity on 8-oxoG:T or 8-oxoG:G base pairs (Fig. 4). The 8-oxoG:T activity was proportionately reduced/absent in ogg1 heterozygous/homozygous mutants, respectively. However, some low-level cleavage of the 8-oxoG:G substrate persisted in extracts of −/− null tissues and, thus, appears unrelated to the activity of OGG1.
Figure 3.

Comparative activity on 8-oxoG and faPy DNA lesions in extracts from ogg1 null mice. Nuclear extracts prepared from testes of wild-type (WT, ●), heterozygous (Het., ○), and ogg1 knockout (KO, ▴) mice were assayed for either cleavage at 8-oxoG:C (a) or release of faPy lesions (b) by using appropriate DNA substrates.
Accumulation of 8-oxoG in the Nuclear DNA of Liver from ogg1 Null Mice.
The steady-state levels of 8-oxoG in the genome of ogg1 null vs. wild-type animals were measured in 13- to 15-week-old animals by HPLC-ECD. A problem with this method is oxidation of G to 8-oxoG during DNA isolation (2, 7–10). Analysis of enzymatic hydrolysates of nuclear DNA isolated from the liver of five wild-type adult mice, under conditions designed to minimize oxidation during the work-up process (25), yielded results in agreement with previous chromatographic determinations with a mean of 3.2 8-oxoG residues/106 bp (2, 8, 25). At least 90% of this is background that can be ascribed to artificial in vitro oxidation of G residues (Fig. 5a), because the DNA of a mammalian cell contains, at most, ≈400–1,500 8-oxoG lesions (0.07–0.24/106 bp; refs. 2, 8, 9, 26, and 30; see below). Parallel experiments using livers from five ogg1 null mice showed a mean increase over the wild-type value of 1.7-fold, demonstrating the presence above background of ≈15,000 chemically stable 8-oxoG residues per genome in nonproliferating liver of adult ogg1 null mice (Fig. 5a).
Figure 5.
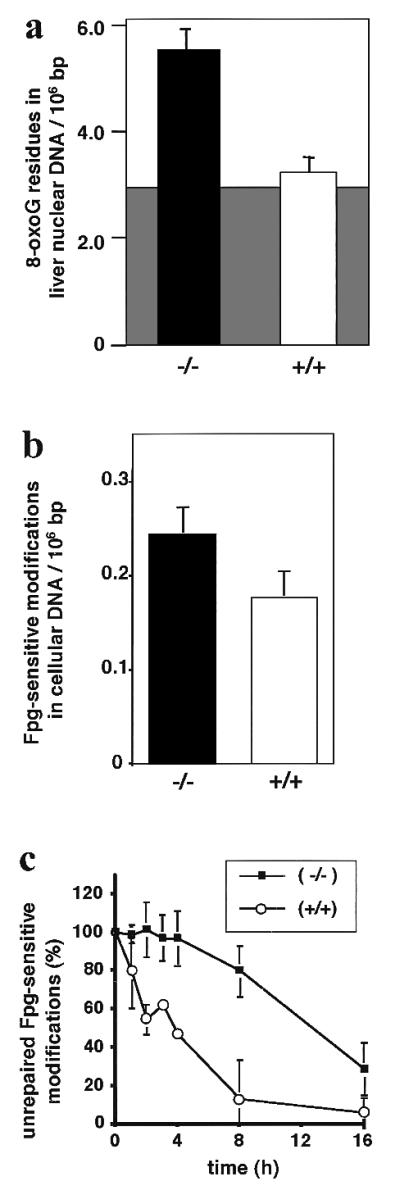
Apparent steady-state levels and delayed repair of oxidative DNA damage in liver (a) and an MEF cell line (b and c) from ogg1 null mice. (a) Nuclear DNA was isolated from the liver of ogg1−/− null (solid bar) or wild-type OGG1+/+ (open bar) adult mice under conditions designed to minimize artificial background oxidation (shaded area) during the extraction and work-up process (see text), and levels of 8-oxoG were quantitated by HPLC-ECD after enzymatic DNA hydrolysis. (b) Total DNA was isolated from cultured ogg1−/− null (solid bar) or wild-type OGG1+/+ (open bar) MEF cell lines, and levels of Fpg-sensitive modifications were quantitated by alkaline elution. (c) Oxidative DNA damage was induced in the ogg1−/− null (■) or wild-type OGG1+/+ (○) cell line by treatment with a photosensitizer and visible light. The persistence of Fpg-sensitive base modifications was measured (as in b) after various recovery times up to 16 h and corrected for dilution of the induced damage by cell proliferation. Error bars are shown for the SD of the mean from five mice (a) and five (b) or three (c) replica experiments. Note the different scale for y axes in a and b.
Accumulation and Repair of 8-oxoG in the DNA of an ogg1 Null MEF Cell Line.
The endogenous steady-state levels of 8-oxoG in the genome of ogg1 null vs. wild-type MEF cell lines was estimated as the number of sites sensitive to cleavage by the Fpg repair enzyme. The steady-state levels of modifications sensitive to Fpg are at least 10-fold lower than levels of 8-oxoG determined by HPLC-ECD (26); this is caused by oxidation of G to 8-oxoG during DNA isolation in the latter method (see above). Thus, the level of Fpg-sensitive modifications is taken as an upper limit for the number of oxidative purine base lesions in the mammalian genome (refs. 2, 8, 9, and 26; Fig. 5a). However, even by this improved but indirect analytical approach, some of the 8-oxoG detected may still reflect oxidation of G during experimentation (see Discussion). Analysis of Fpg-sensitive sites in the genomic DNA of the OGG1+/+ control cell line yielded a value of 0.18/106 bp; this is ≈20-fold lower than the value obtained here by HPLC-ECD with DNA isolated from liver. The present data are in agreement with recent determinations of 0.07–0.24 Fpg-sensitive modifications/106 bp for various cell types (2, 8, 9, 26). Parallel experiments using the ogg1−/− null cell line showed a mean increase over the wild-type value of 1.4-fold (Fig. 5b), demonstrating an increased steady-state level of oxidative base damage because of the absence of OGG1 of ≈400 8-oxoG residues per genome. Although the alkaline elution method gives much lower background values than the HPLC-ECD approach to 8-oxoG measurements, it so far has been applicable only to cell lines rather than solid tissues, for technical reasons.
The relatively small increase in the level of oxidative base damage in the ogg1 null cell line growing under high oxygen tension in culture suggested that some DNA repair of 8-oxoG occurs in dividing cells. To test this directly, we determined the repair kinetics of Fpg-sensitive modifications after oxidative stress induced by a photosensitizer in the ogg1 null cell line vs. the wild-type control. In the control cell line, ≈50% of Fpg-sensitive lesions had been repaired within 2–4 h but there was no detectable repair in ogg1 null cells during this time (Fig. 5c). However, there was substantial repair in the ogg1 null cell line when incubation times were extended; at 16 h, when repair was essentially complete in the wild-type control, ≈70% of oxidative base lesions had been removed in the ogg1 null cells. It is difficult to determine what proportion of the remaining lesions is 8-oxoG, but because there is an enzyme other than OGG1 able to repair faPy lesions in ogg1 null extracts (Fig. 3b), it seems likely that all the unrepaired lesions after 16 h are 8-oxoG; if ≈75% of the originally induced lesions were 8-oxoG (29, 31), these data indicate that 8-oxoG is removed from the DNA of the ogg1 null cell line with a half-life of ≈16 h. Because cell division has been taken into account (see Materials and Methods), this active repair occurs in addition to the passive dilution of the induced modifications by cell division.
Spontaneous Mutation Frequency in ogg1 Null Mice.
OGG1 wild-type +/+ and ogg1−/− null mice were established on a homozygous Big Blue background such that spontaneous mutation frequencies could be determined for the lacI transgene recovered from liver and testis of 10- and 20-week-old animals. The number of lacI mutant plaques, the total number of plaque-forming units plated, and the calculated mutation frequency are presented in Table 1. There was no significant difference in the spontaneous mutation frequency observed in the testis of wild-type vs. ogg1 null animals, either at 10 or 20 weeks of age. The mutation frequency in liver was the same in wild-type animals at 10 and 20 weeks of age, and the observed frequency is in good agreement with previous data (32, 33). However, in ogg1 null animals, the mutation frequency in liver was elevated 2- to 3-fold at 10 weeks compared with wild-type animals; there was no further significant increase in mutation frequency between 10 and 20 weeks. Bacteriophages were recovered from 10 (OGG1+/+) or 20 (ogg1−/−) mutant plaques, and the lacI gene was sequenced to give an indication of the in vivo mutation spectrum in liver in the ogg1 null vs. wild-type background. In the ogg1 knockout, 10 of a total of 16 base substitutions detected represented G→T transversions (the remainder were C→T transitions), compared with only one of seven in the wild type, in which the other six base substitutions all were C→T transitions, the major class of spontaneous mutation (33). This finding is consistent with the elevated in vivo mutation frequency in the liver of ogg1 null animals being a result of mutagenesis by 8-oxoG.
Table 1.
Spontaneous mutation frequencies in Big Blue/OGG1+/+ and Big Blue/ogg1−/− mice
| Organ | Genotype | Age, weeks | pfu plated | Mutants | Frequency, ×10−5 |
|---|---|---|---|---|---|
| Testis | +/+ | 10 | 264,000 | 7 | 2.65 |
| 20 | 299,000 | 5 | 1.67 | ||
| −/− | 10 | 254,000 | 7 | 2.75 | |
| 20 | 281,000 | 6 | 2.13 | ||
| Liver | +/+ | 10 | 287,000 | 11 | 3.83 |
| 20 | 292,000 | 10 | 3.42 | ||
| −/− | 10 | 295,000 | 26 | 8.81 | |
| 20 | 307,000 | 28 | 9.12 |
pfu, plaque-forming units.
In the present work, a lacI transgene-based assay system was used to measure spontaneous mutation frequencies caused by a deficiency in an enzyme that repairs endogenous DNA damage. Studies of mismatch repair-deficient mice lacking MSH2 (34, 35) showed 10- to 20-fold increased spontaneous mutation frequencies of the Big Blue lacI transgene in proliferating tissues but only 2- to 5-fold increases in organs having low levels of cell turnover in the adult animal, as expected for an enzyme that removes DNA polymerase incorporation errors. The results here are entirely different, because significantly increased mutagenesis was observed for an organ with a low level of proliferation, liver, whereas no increased mutagenesis was detected in rapidly proliferating testes cells (Table 1).
Discussion
The unavoidable but potentially mutagenic and carcinogenic DNA alterations resulting from endogenous damage by oxygen free radicals are removed by the base excision-repair (BER) pathway, which is initiated by various DNA glycosylases that each excise a specific subset of aberrant bases (36). We have generated knockout mice deficient in the OGG1 DNA glycosylase that excises the most frequent mutagenic oxidative base lesion, 8-oxoG. Similarly, to knockout mice deficient in the 3-methyladenine-DNA glycosylase (37, 38), mice deficient in OGG1 are viable. In contrast, ablating any subsequent steps of the DNA-repair pathway to generate knockout mice totally deficient in BER is incompatible with life (39). The availability of ogg1 null mice and cell lines facilitates assessments of the incidence and consequences of 8-oxoG formation in the mammalian genome. We show here that (i) 8-oxoG residues accumulate in the genome to substantially increased levels in the nonproliferating liver of ogg1 null mice; (ii) there is no excision of 8-oxoG initiated by a different DNA glycosylase in cell-free tissue extracts, but there is significant slow repair in an ogg1 null cell line; (iii) the in vivo spontaneous mutation frequency is increased (2- to 3-fold) in liver but not in more rapidly proliferating testis; and (iv) ogg1 null mice do not show an increased tumor incidence, although inactivating mutations in the OGG1 gene have been documented in a small number of sporadic human lung, kidney, gastric, and head-and-neck tumors (refs. 40 and 41; G. Herrmann and T.L., unpublished data).
The estimation of 8-oxoG residues in the DNA of cells and tissues not exposed to deliberate oxidative stress has been controversial, with reported values in the literature steadily decreasing over the last few years as analytical procedures have improved (2, 7–10). Still, there is no current method available that totally avoids some artifactual oxidation of G residues in DNA during experimental work-up and analysis. The data reported here on 8-oxoG levels in ogg1−/− knockout mouse cells, combined with data on relative repair rates, allow an approximate determination of the endogenous level of 8-oxoG residues in mammalian cellular DNA. A steady-state level of ≈400 8-oxoG residues over the background seen with OGG1+/+ cells was observed here for an ogg1−/− cell line (Fig. 5b). Because the rate of repair of the lesion was reduced at least 4-fold in the ogg1−/− vs. OGG1+/+ cells (Fig. 5c), it can be estimated that the endogenous level of 8-oxoG in wild-type OGG1+/+ cells is, at most, 100 residues per genome.
The persistence in the genome of substantially increased numbers of potentially miscoding 8-oxoG residues, as observed here for ogg1−/− cells and tissues, would not appear to be compatible with a low, spontaneous mutation rate and absence of tumors. A possible solution to this paradox could be provided by selective repair of the opposite DNA strand, with excision of A misincorporated opposite 8-oxoG in dividing cells, by the human homologue of the MutY DNA glycosylase (5, 11). By removal of the relevant A residue, the cell is given a second opportunity to incorporate either an A, or a correct C, opposite the 8-oxoG residue; this enzyme activity would not have been detected in the in vitro enzyme assays (see Materials and Methods). Although MutY could contribute to neutralizing the mutagenic effect of 8-oxoG in dividing cells, there is also active removal of the 8-oxoG lesion from the ogg1 null genome (Fig. 5c). This “back-up” repair of 8-oxoG might be by the nucleotide excision-repair (NER) pathway (42, 43) and could account for the slow but significant repair observed in the ogg1 null cell line. The increased half-life for 8-oxoG in dividing ogg1 null cells of ≈16 h would mean that this inefficient repair could, together with dilution of DNA damage through cell division, barely suffice to maintain a tolerably low steady-state level of the lesion in proliferating but not nonproliferating tissue. This would account for the elevated steady-state levels of 8-oxoG in DNA in the liver of adult ogg1 null mice. The limited cell turnover that might compromise the removal of 8-oxoG from the genome of nonproliferating liver also would mean that the mutagenic potential of the large accumulation of 8-oxoG residues is not fully realized by mispairing during DNA replication and produces only the observed modest increase in spontaneous mutagenesis in livers of ogg1 null mice, which remain tumor-free.
These possibilities can be tested directly by generating the relevant double-knockout mice. Experiments with the ogg1 null cell line generated here indicate that, in the absence of the DNA glycosylase, 8-oxoG is preferentially removed from the transcribed strand of DNA by transcription-coupled repair, whereas repair of the nontranscribed strand was not detected (F. le Page and S. Boiteux, personal communication). The acute problem of the presence of miscoding 8-oxoG lesions in the transcribed strand of active genes in ogg1 null cells might be met by transcription-coupled repair, with slower repair of the nontranscribed strand and bulk genome by MutY and global NER. Thus, in the absence of OGG1, the MutY function or transcription-coupled NER could contribute to protecting the cell from an increased load of 8-oxoG residues (44), such that endogenous mutagenic changes are maintained at a level compatible with normal cellular proliferation (45).
Gene knockout mice defective in OGG1 have been constructed independently and found to accumulate similarly high levels of 8-oxoG in liver DNA by T. Noda, S. Nishimura, and their coworkers (personal communication).
Acknowledgments
We thank Magnar Bjørås, Rune Johansen, and Teresa Roldán Arjona for their contributions to particular aspects of this work; Elsebeth Hoff, Mary Ann Jacobs, and Stephen Wilson for technical assistance; Peter Hagger, Gary Martin, and Cheryl Young for animal care; Ruth Peat and Christine Saunders for establishing MEF cell lines; and Lennart Möller and Charles R. Iden for advice on 8-oxoG measurements. This work was supported by the Imperial Cancer Research Fund, the Research Council of Norway, the Norwegian Cancer Society, and the Deutsche Forschungsgemeinschaft (SFB 519). A.K. and E.L. were recipients of travel grants from European Community Concerted Action on DNA Repair and Cancer.
Abbreviations
- 8-oxoG
7,8-dihydro-8-oxoguanine
- faPy
2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine
- HPLC-ECD
HPLC–electrochemical detection
- AP
abasic
- MEF
mouse embryo fibroblast
References
- 1.Ames B N, Shigenaga M K, Hagen T M. Proc Natl Acad Sci USA. 1997;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins A R. BioEssays. 1999;21:238–246. doi: 10.1002/(SICI)1521-1878(199903)21:3<238::AID-BIES8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 3.Grollman A P, Moriya M. Trends Genet. 1993;9:246–249. doi: 10.1016/0168-9525(93)90089-z. [DOI] [PubMed] [Google Scholar]
- 4.Ward J F. In: Advances in DNA Damage and Repair. Dizdaroglu M, Karakaya A E, editors. New York: Plenum; 1998. pp. 431–439. [Google Scholar]
- 5.Michaels M L, Miller J H. J Bacteriol. 1992;174:6321–6325. doi: 10.1128/jb.174.20.6321-6325.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas D, Scott A, Barbey R, Padula M, Boiteux S. Mol Gen Genet. 1996;254:171–178. doi: 10.1007/s004380050405. [DOI] [PubMed] [Google Scholar]
- 7.Beckman K B, Ames B N. J Biol Chem. 1997;272:19633–19636. doi: 10.1074/jbc.272.32.19633. [DOI] [PubMed] [Google Scholar]
- 8.Collins A, Cadet J, Epe B, Gedik C. Carcinogenesis. 1997;18:1833–1836. doi: 10.1093/carcin/18.9.1833. [DOI] [PubMed] [Google Scholar]
- 9.Cadet J, d’Ham C, Douki T, Pouget J-P, Ravanat J-L, Sauvaigo S. Free Radical Res. 1998;29:541–550. doi: 10.1080/10715769800300581. [DOI] [PubMed] [Google Scholar]
- 10.Lindahl T. In: Advances in DNA Damage and Repair. Dizdaroglu M, Karakaya A E, editors. New York: Plenum; 1998. pp. 251–257. [Google Scholar]
- 11.Slupska M M, Baikolov C, Luther W M, Chiang J-H, Wei Y-F, Miller J H. J Bacteriol. 1996;178:3885–3892. doi: 10.1128/jb.178.13.3885-3892.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakumi K, Furuichi M, Tsuzuki T, Kakuma T, Kawabata S, Maki H, Sekiguchi M. J Biol Chem. 1995;268:23524–23530. [PubMed] [Google Scholar]
- 13.Aburatani H, Hippo Y, Ishida T, Takashima R, Matsuba C, Kodama T, Takao M, Yasui A, Yamamoto K, Asano M, et al. Cancer Res. 1997;57:2151–2156. [PubMed] [Google Scholar]
- 14.Arai K, Morishita K, Shinmura K, Kohno T, Kim S-R, Nohmi T, Taniwaki M, Ohwada S, Yokota J. Oncogene. 1997;14:2857–2861. doi: 10.1038/sj.onc.1201139. [DOI] [PubMed] [Google Scholar]
- 15.Bjørås M, Luna L, Johnsen B, Hoff E, Haug T, Rognes T, Seeberg E. EMBO J. 1997;16:6314–6322. doi: 10.1093/emboj/16.20.6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuo F C, Sklar J. J Exp Med. 1997;186:1547–1556. doi: 10.1084/jem.186.9.1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu R, Nash H M, Verdine G L. Curr Biol. 1997;7:397–407. doi: 10.1016/s0960-9822(06)00187-4. [DOI] [PubMed] [Google Scholar]
- 18.Radicella J P, Dherin C, Desmaze C, Fox M S, Boiteux S. Proc Natl Acad Sci USA. 1997;94:8010–8015. doi: 10.1073/pnas.94.15.8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roldán-Arjona T, Wei Y-F, Carter K C, Klungland A, Anselmino C, Wang R-P, Augustus M, Lindahl T. Proc Natl Acad Sci USA. 1997;94:8016–8020. doi: 10.1073/pnas.94.15.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosenquist T A, Zharkov D O, Grollman A P. Proc Natl Acad Sci USA. 1997;94:7429–7434. doi: 10.1073/pnas.94.14.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shinmura K, Kasai H, Sasaki A, Sugimura H, Yokota J. Mutat Res. 1997;385:75–82. doi: 10.1016/s0921-8777(97)00041-4. [DOI] [PubMed] [Google Scholar]
- 22.Barnes D E, Stamp G, Rosewell I, Denzel A, Lindahl T. Curr Biol. 1998;8:1395–1398. doi: 10.1016/s0960-9822(98)00021-9. [DOI] [PubMed] [Google Scholar]
- 23.Tani M, Shinmura K, Kohno T, Shiroishi T, Wakana S, Kim S-R, Nohmi T, Kasai H, Takenoshita S, Nagamachi Y, Yokota J. Mamm Genome. 1998;9:32–37. doi: 10.1007/s003359900675. [DOI] [PubMed] [Google Scholar]
- 24.Todaro G J, Green H. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hofer T, Möller L. Chem Res Toxicol. 1998;11:882–887. doi: 10.1021/tx980041x. [DOI] [PubMed] [Google Scholar]
- 26.Pflaum M, Will O, Epe B. Carcinogenesis. 1997;18:2225–2231. doi: 10.1093/carcin/18.11.2225. [DOI] [PubMed] [Google Scholar]
- 27.Ballmaier D, Briviba K, Sies H, Epe B. Methods Enzymol. 1999;301:311–318. [PubMed] [Google Scholar]
- 28.Boiteux S, O’Connor T R, Lederer F, Gouyette A, Laval J. J Biol Chem. 1990;265:3916–3922. [PubMed] [Google Scholar]
- 29.Will O, Gocke E, Eckert I, Schulz I, Pflaum M, Mahler H-C, Epe B. Mutat Res. 1999;435:89–101. doi: 10.1016/s0921-8777(99)00039-7. [DOI] [PubMed] [Google Scholar]
- 30.Czene S, Harms-Ringdahl M. Mutat Res. 1995;336:235–242. doi: 10.1016/0921-8777(94)00058-e. [DOI] [PubMed] [Google Scholar]
- 31.Pflaum M, Will O, Mahler H C, Epe B. Free Radical Res. 1998;29:585–594. doi: 10.1080/10715769800300631. [DOI] [PubMed] [Google Scholar]
- 32.Young R R, Rogers B J, Provost G S, Short J M, Putnam D L. Mutat Res. 1995;327:67–73. doi: 10.1016/0027-5107(94)00080-o. [DOI] [PubMed] [Google Scholar]
- 33.de Boer J G, Provost S, Gorelick N, Tindall K, Glickman B W. Mutagenesis. 1998;13:109–114. doi: 10.1093/mutage/13.2.109. [DOI] [PubMed] [Google Scholar]
- 34.Andrew S E, Reitmair A H, Fox J, Hsiao L, Francis A, McKinnon M, Mak T W, Jirik F R. Oncogene. 1997;15:123–129. doi: 10.1038/sj.onc.1201180. [DOI] [PubMed] [Google Scholar]
- 35.Andrew S E, McKinnon M, Cheng B S, Francis A, Penney J, Reitmair A H, Mak T W, Jirik F R. Proc Natl Acad Sci USA. 1998;95:1126–1130. doi: 10.1073/pnas.95.3.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krokan H E, Standal R, Slupphaug G. Biochem J. 1997;325:1–16. doi: 10.1042/bj3250001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engelward B P, Weeda G, Wyatt M D, Broekhof J L M, de Wit J, Donker I, Allan J M, Gold B, Hoeijmakers J H J, Samson L D. Proc Natl Acad Sci USA. 1997;94:13087–13092. doi: 10.1073/pnas.94.24.13087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hang B, Singer B, Margison G P, Elder R H. Proc Natl Acad Sci USA. 1997;94:12869–12874. doi: 10.1073/pnas.94.24.12869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson D M, Thompson L H. Proc Natl Acad Sci USA. 1997;94:12754–12757. doi: 10.1073/pnas.94.24.12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chevillard S, Radicella J P, Levalois C, Lebeau J, Poupon M-F, Oudard S, Dutrillaux B, Boiteux S. Oncogene. 1998;16:3083–3086. doi: 10.1038/sj.onc.1202096. [DOI] [PubMed] [Google Scholar]
- 41.Shinmura K, Kohno T, Kasai H, Koda K, Sugimura H, Yokota J. Jpn J Cancer Res. 1998;89:825–828. doi: 10.1111/j.1349-7006.1998.tb00635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reardon J T, Bessho T, Kung H C, Bolton P H, Sancar A. Proc Natl Acad Sci USA. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dianov G, Bischoff C, Piotrowski J, Bohr V A. J Biol Chem. 1998;273:33811–33816. doi: 10.1074/jbc.273.50.33811. [DOI] [PubMed] [Google Scholar]
- 44.Cunningham R P. Curr Biol. 1997;7:R576–R579. doi: 10.1016/s0960-9822(06)00286-7. [DOI] [PubMed] [Google Scholar]
- 45.Drake J W, Charlesworth B, Charlesworth D, Crow J F. Genetics. 1998;148:1667–1686. doi: 10.1093/genetics/148.4.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]