

Abstract
Vascular endothelial growth factor (VEGF) expression is elevated in a wide variety of solid tumours. Inhibition of VEGF activities is able to reduce angiogenesis and tumour growth. We have recently shown in vitro that carboxymethyl dextran benzylamide (CMDB7) prevents the binding of VEGF165 to its cell surface receptors and thus inhibits VEGF activities on endothelial cells. In the present study, we explored the effects of CMDB7 on highly aggressive human epidermoid carcinoma A431 cells known to overexpress epidermal growth factor receptors (EGFRs) and produce a high amount of VEGF and a minor quantity of bFGF. In vitro, CMDB7 blocked the mitogenic activity of A431-conditioned medium on endothelial cells. Concerning A431 cells, CMDB7 inhibited their proliferation and the VEGF165 binding to them. In vivo, administration of CMDB7 (10 mg kg−1) three times per week for 2 weeks inhibited the growth of A431 xenografts in nude mice by 73% as compared to the control group. Immunostaining of endothelial cells with mouse-specific GSL-1 lectin in tumour sections revealed that CMDB7 also inhibited the density of intratumour endothelial cells by 66%. These findings demonstrate that CMDB7 has an efficient antiangiogenic and antitumour action in vivo even when tumour cells produce a high level of VEGF and EGFRs.
Keywords: dextran derivative, vascular endothelial growth factor (VEGF), epidermoid carcinoma A431 cells, angiogenesis
Neovascularisation is critical for supporting the rapid growth of solid tumours (Folkman, 1990). Tumour angiogenesis appears to be achieved by the overexpression of angiogenic agents within solid tumours that stimulate host vascular endothelial cell mitogenesis and possibly chemotaxis. It is well established now that the induction of vascular endothelial growth factor (VEGF) expression, including via tumour hypoxia (Hlatky et al, 1994), plays a major role in tumour angiogenesis (Dvorak et al, 1991; Millauer et al, 1994; Goldman et al, 1998). In recent years, it has been widely shown that VEGF activity is a key feature during tumour growth and angiogenesis, and that blocking of this signal transduction pathway may inhibit tumour progression (Cheng et al, 1996; Relf et al, 1997). In vivo, VEGFs act as potent mitogenic factors for endothelial cells and as blood vessel permeabilising agents (Senger et al, 1983; Plouët et al, 1989; Klagsbrun and Soker, 1993; Yuan et al, 1996). The VEGF gene family currently includes six members: VEGF-A (prototype VEGF), placenta growth factor (PlGF), VEGF-B, VEGF-C, VEGF-D and VEGF-E, (produced by Orf virus), (reviewed by Veikkola et al, 2000). VEGF is a homodimeric glycoprotein that exists in six isoforms containing 121, 145, 162, 165, 189 and 206 amino-acid residues as a result of alternative splicing from a single gene (Ferrara and Davis-Smith, 1997; Lange et al, 2003). The predominant and the best characterised VEGF species is the heparin-binding 165-amino acid-long form VEGF165 (reviewed by Neufeld et al, 1999).
Three receptors with tyrosine kinase activities have been identified as VEGF receptors: VEGFR-1 (Flt-1) (deVries et al, 1992), VEGFR-2 (KDR/Flk-1) (Terman et al, 1992), proteins with apparent molecular weights of 180 and 230 kDa, respectively, and VEGFR-3 (Kaipainen et al, 1995). They have been shown to bind VEGF with high affinity. Recently, an additional binding site, neuropilin-1 (NP-1), was identified and shown to be expressed on the surface of endothelial and tumour cells (Soker et al, 1998). NP-1 modulates the binding of VEGF165 to VEGFR-2 acting as a coreceptor that enhances VEGF165-induced activities mediated by VEGFR-2 (Whitaker et al, 2001; Soker et al, 2002).
Carboxymethyl dextran benzylamide (CMDB7) is a noncytotoxic substituted dextran. We have recently shown in vitro that it prevents the binding of VEGF165 to human umbilical vein endothelial cell surface and thus inhibits VEGF165-induced phosphorylation of VEGFR-2 and consequently endothelial cell proliferation (Hamma-Kourbali et al, 2001). In the present study, we explored in vitro and in vivo the effects of CMDB7 on human epidermoid carcinoma A431 cells known to produce a high amount of VEGF and a minor quantity of bFGF (Myoken et al, 1991). The other peculiarity of A431 cells is the production of a newly identified splice form of VEGF, VEGF-162, which binds more efficiently than VEGF-165 to a natural basement membrane of endothelial cells (Lange et al, 2003). Moreover, A431 cells express a high level of epidermal growth factor receptors (EGFRs) activated by EGF, a nonheparin-binding growth factor, which does not interact with CMDB7 (Bagheri-Yarmand et al, 1998b). Interestingly, the resistance of A431 tumours to treatment with EGF receptor-blocking antibodies is associated with an elevated expression of VEGF (Viloria-Petit et al, 2001). Such cells, xenografted in nude mice, provide a model of highly VEGF-dependent (Melnyk et al, 1996) and aggressive tumour growth in an in vivo system. Since A431 cells have been recently described to express the VEGF165-binding sites (Li et al, 2001), we explored also the possible effect of CMDB7 on radiolabelled VEGF binding. We demonstrate that CMDB7 acts on both tumour and endothelial cells, decreasing in a potent manner the tumour growth and angiogenesis in vivo.
MATERIALS AND METHODS
Dextran derivative preparation
A water-soluble dextran derivative (CMDB7) was prepared as previously described (Chaubet et al, 1995). Its chemical composition, determined by acidimetric titration and elementary analysis of nitrogen, is 0% dextran, 70% carboxymethyl and 30% benzylamide. Average molecular weight was estimated as 80 000 g mol−1.
Cell lines and cell culture
Human epidermoid carcinoma cell line (A431) and human umbilical vein endothelial cell line (HUV-EC-C) were purchased from the American Type Culture Collection (ATCC, Rockville, MD, USA). A431 cells were routinely grown in DMEM (Life Technologies, Inc., Gaithersburg, MD, USA) and HUV-EC-Cs in M199 (Life Technologies, Inc.) and were cultured at 37°C in a 5% CO2-humidified atmosphere. Both culture media were supplemented with 10% foetal calf serum (FCS), 2 mM L-glutamine, 1 mM sodium pyruvate, 50 U ml−1 penicillin and 50 μg ml−1 streptomycin (all obtained from Life Technologies, Inc.). The cells were free of mycoplasma, bacteria and viruses.
Preparation of conditioned media (CMs)
To assess the production of VEGF-A, the A431 cells at three different density were seeded into a 24-well culture plate (Falcon, Strasbourg, France) in DMEM supplemented with 10% FCS for 24 h. To obtain CM containing only the growth factors secreted by A431, the cells were washed twice with PBS, and incubated in 1 ml of serum-free DMEM containing 0.1% BSA (Sigma, St Louis, MO, USA). At the indicated time, the media were collected, cleared by centrifugation, and stored at −80°C before use. For other experiments, the cells were grown in 150 mm-diameter Petri dishes (Falcon) to 80% confluence in DMEM/10% FCS, washed and incubated in 10 ml dish−1 of serum-free medium.
Determination of VEGF-A concentration in the A431-CMs by radioimmunoassay
The surface of flat-bottomed polystyrene wells (Disposable Immulon 1 Remowawell, Dynatech, Cambridge, MA, USA) were coated overnight at 4°C with 200 μl of PBS buffer containing 50 ng polyclonal neutralising anti-VEGF IgG (R&D Systems, Abingdon, UK). The nonspecific interactions were saturated with PBS containing 0.1% BSA and 0.01% Tween-20 (PBT buffer) for an additional overnight at 4°C. After blocking, the wells were washed three times with 300 μl of PBT buffer. Then, A431-CM or VEGF165 (R&D Systems) as standard at increasing concentrations (0–250 ng ml−1) and 50 pM 125I-VEGF165 (Amersham Pharmacia Biotech, Orsay, France) were added to a final volume of 200 μl in PBT buffer. After an overnight incubation at 4°C, wells were washed three times with 300 μl of PBT buffer and the radioactivity remaining in each well was measured in a γ-counter (LKB 1261 Multigamma).
A431-CM effects on HUV-EC-C proliferation
HUV-EC-Cs were seeded at a density of 2 × 104 well−1 into 24-well tissue culture plates (Falcon) in M199-10% FCS. After 24 h, the cells were growth arrested by serum starvation for another 24 h. Then, the cells were incubated for 48 h with A431-CM diluted in a serum-free medium to a final VEGF concentration of 10 ng ml−1 (concentration at which VEGF165 has the maximal mitogenic effect on HUV-EC-Cs (Hamma-Kourbali et al, 2001) in the presence or absence of 5 μM CMDB7 (optimal concentration at which CMDB7 completely prevents the VEGF165 mitogenic effect on HUV-EC-Cs (Hamma-Kourbali et al, 2001) or 1 μg ml−1 anti-human VEGF neutralising antibody (Sigma) characterised by neutralisation dose50=0.01–0.1 μg ml−1. Cells were washed with PBS, dissociated with 0.025% trypsin-EDTA (Life Technologies) and counted using a Coulter counter (Coultronics, Margency, France). All experiments were performed in triplicate and data illustrate the mean cell numbers±s.e. provided from one representative of three independent experiments.
A431 proliferation assay
A431 cells were seeded at a density of 104 cells well−1 into 24-well tissue culture plates (Falcon) in DMEM–10% FCS and allowed to adhere to the plastic for 24 h. After washing with DMEM, the cells were incubated with CMDB7 at the indicated concentrations (day zero) in DMEM–1% FCS. At different times, cells were washed with PBS, dissociated with 0.025% trypsin-EDTA (Life Technologies, Inc.) and counted using a Coulter counter (Coultronics). In each case, samples were performed in triplicate, and data illustrate mean cell numbers±s.e. of one representative of three independently performed experiments.
VEGF165 binding to A431 cells
For displacement binding assays, A431 cells were grown until confluence on 24-well tissue culture plates (Falcon). After an overnight incubation in serum-free medium and two washings with ice-cold binding buffer (PBS/0.1% BSA), the cells were incubated with 7 pM 125I-VEGF165 (Amersham, Pharmacia Biotech) and CMDB7 at various concentrations from 8 × 10−9 to 4 × 105 M at 4°C for 2 h. Incubation was terminated by gently aspirating the medium and washing the cell monolayer three times with ice-cold binding buffer. After cell solubilisation in 0.3 ml of 0.5 N NaOH, the bound radioactivity was measured in a γ-counter (LKB 1261 Multigamma). Nonspecific binding was determined in the presence of an excess (5000 pM) of unlabelled VEGF165 (R&D Systems).
Tumour cell inoculation in nude mice
All in vivo experiments were carried out with ethical committee approval and met the standards required by the UKCCCR guidelines (Workman et al, 1998). A431 cells (5 × 106) were inoculated s.c. in the right flank of 4-week-old athymic nude mice (nu/nu) (Charles River Laboratory, Aubin-les-Elbeuf, France). The animals (n=20) were kept in a temperature-controlled room on a 12 h : 12 h light–dark schedule with food and water ad libitum. All mice developed single s.c. palpable tumours of approximately 50 mm3 6 days after cell inoculation. Then, mice were placed in control (n=10) and CMDB7-treated groups (n=10). Mice were treated by subcutaneous (s.c.) injection of 0.1 ml PBS alone (control) or containing 10 mg kg−1 CMDB7 close to the tumour, three times a week for 2 weeks. Tumours were measured along two major axes with a calliper. Tumour volume was calculated as follows:
| V = (4/3)πR12R2 |
where R1 is radius 1, R2 is radius 2, and R1 <R2.
Tissue preparation and immunohistochemical staining
Immediately after surgical resection, the tumour specimens were fixed with 4% paraformaldehyde and processed to paraffin inclusion. The intratumour mice endothelial cells were specifically stained with GSL-1 lectin (Vector Laboratories, Burlingame, CA, USA) in 5-μm sections as previously described (Bagheri-Yarmand et al, 1999). The GSL-1 lectin binds specifically to galactosyl residues present on vascular endothelium in mice (Alroy et al, 1987; Mattsson et al, 2002). The proliferative index of tumour xenograft was determined by human Ki-67 staining with monoclonal mouse antibody (MIB-1; 1 : 50; Dako, Trappes, France). The epitope retrieval was performed in 10 mM citrate buffer pH=6.0 for 40 min at 98°C. Specific reactions were visualised with 3,3′-diaminobenzidine (DAB) as chromogen.
Image analysis
For each GSL-1- or MIB-1-labelled section of control or CMDB7-treated tumour, five fields containing exclusively viable tumour cells, as indicated by the haematoxylin staining, were selected randomly for analysis. Image analysis was performed using the NIH programme (developed at NIH and available on the Internet at http://rsb.info.nih.gov/nih-image/). The endothelial cell density in each field was expressed as the ratio of endothelial cell area and the total viewed area × 100 (%). To determine the proliferative index, we estimated the percentage of tumour cell nuclei positive for Ki-67 marker. These values were then averaged for untreated (control) and treated-CMDB7 tumours.
Statistical analysis
Multiple statistical comparisons were performed using ANOVA in a multivariate linear model. Statistical comparisons were conducted using the Mann–Whitney t-test. P<0.05 was considered statistically significant.
RESULTS
CMDB7 inhibits, like neutralising anti-VEGF165 antibody, mitogenic effect of A431-CM on HUV-EC-Cs
According to previous studies (Melnyk et al, 1996), we found that A431 cells secrete in the culture medium large amounts of VEGF-A. Moreover, we showed here that VEGF production is cell number- and time-dependent (Table 1).
Table 1. Concentration of VEGF in the conditioned media of A431 tumour cells.
| Cell number | 24 h | 48 h |
|---|---|---|
| 2.5 × 104 | <0.4 | 1 |
| 5 × 104 | 2 | 4 |
| 105 | 8 | 16 |
A431 cells were seeded at the indicated density into 24-well plate in DMEM–10% FCS medium for 24 h. After washing, they were incubated in serum-free medium. At the indicated time, the media were collected. VEGF-A concentration was asessed by radio immunoassay as described in Materials and Methods. Values are in ng ml−1. The limit of sensitivity of the assay was 0.4 ng ml−1.
As expected, A431-CM stimulated the in vitro proliferation of HUV-EC-Cs by 2.5-fold after 48 h of incubation (Figure 1). This mitogenic effect is, at least in part, VEGF-specific since the neutralising antibodies against recombinant VEGF inhibited the A431-CM-induced proliferation of HUV-EC-Cs by 45% after 48 h treatment. A431-CM, used in this experiment, contained 10 ng ml−1 of VEGF165 as revealed by specific radioimmunoassay. At the same concentration, recombinant VEGF165 has a similar mitogenic effect on HUV-EC-Cs (Hamma-Kourbali et al, 2001), as described above the addition of 5 μM CMDB7 prevented the stimulatory effect of A431-CM on HUV-EC proliferation (Figure 1). When HUV-EC-Cs were cultivated in serum-free medium, CMDB7 or neutralising anti-VEGF165 antibodies had no effect.
Figure 1.

CMDB7 inhibits A431-CM mitogenic effect. Quiescent HUV-EC cells were incubated with A431-CM with or without 5 μM CMDB7 or 1 μg ml−1 anti-human VEGF neutralising antibody. After 48 h, the cells were trypsinised and counted using a Coulter counter. The values represent mean cell numbers±s.e. (bars), obtained in triplicate in one of the three independent experiments.
CMDB7 inhibits A431 cell proliferation in vitro
Next, we tested CMDB7 for its ability to affect the in vitro growth of A431 tumour cells. We demonstrated that treatment with CMDB7 at increasing concentrations, ranging from 0.1 to 20 μM, resulted in a concentration- and time-dependent inhibition of A431 cell number (Figure 2).
Figure 2.

Inhibition of A431 cell growth in vitro. A431 cells were seeded at 104 cells well−1 in 24-well plates in DMEM containing 10% FCS. On the following day (day 0), the medium was changed to DMEM containing 1% serum (x) and 0.1 μM (○), 1 μM (•), 5 μM (□), 10 μM (▪), 15 μM (▵), or 20 μM (▴) CMDB7. At the indicated time, cells were trypsinised and counted. The inset shows the percentages of inhibition of the A431 cell growth by CMDB7 at increasing concentrations at day 6. The values represent mean cell numbers±s.e. (bars), obtained in triplicate in one of the three independent experiments.
In contrast, 1 μg ml−1 anti-VEGF antibody had no effect on A431 proliferation in vitro (data not shown) as reported by others (Melnyk et al, 1996).
CMDB7 inhibits VEGF165 binding to A431 tumour cells
Since A431 cells produce VEGF-A and binds VEGF165 on the surface (Li et al, 2001), we explored if CMDB7 is able to compete for VEGF165-specific binding (Figure 3). CMDB7 decreased the 125I-VEGF165-specific binding to A431 cells at concentrations ranging from 0.1 to 50 μM with a half-maximum inhibitory effect (IC50) at concentration 2 μM.
Figure 3.

CMDB7 inhibits VEGF165 binding to A431 cells. Confluent A431 cells were incubated for 2 h at 4°C in the presence of 7 pM 125I-VEGF165 and CMDB7 at the indicated concentrations (logarithmic scale). Nonspecific binding was determined in the presence of 5000 pM unlabelled VEGF165. Results are expressed as the mean±s.e. (bars) of experiments done in duplicates and repeated at least twice.
CMDB7 inhibits the growth of A431 cells xenografted in nude mice
The tumours appeared in 100% of mice 6 days after A431 cell inoculation. CMDB7 inhibited the growth of A431 tumours by 73% (P<0.001) after 2 weeks of treatment (Figure 4).
Figure 4.

CMDB7 inhibits primary tumour growth. A431 carcinoma cells (5 × 106) were inoculated s.c. into the right flank of female nude mice. When tumour volume reached 100 mm3 (6 day), CMDB7 (10 mg kg−1) was administrated s.c. three times a week for 2 weeks. Tumours were measured and the results are presented as the mean tumour volume ±s.e. (bars) obtained from 10 mice in each group, P<0.001; CMDB7-treated group vs controls.
No apparent toxicity was noticed during treatment with CMDB7. No signs of toxicity such as diarrhoea, infection, weakness or lethargy were observed. The body weight of the inoculated mice was not affected by CMDB7 after 2 weeks of treatment. All treated mice were alive at the end of treatment.
CMDB7 decreases the proliferative index of A431 xenografts
The specific Ki-67 staining was less intense in CMDB7-treated tumours as compared to control (nontreated) ones. The proliferative index for treated and control xenografts were significantly (P=0.05) diffferent, 26±8 and 34±10%, respectively (mean±s.e.m). These data suggest that CMDB7 inhibited directly in vivo the proliferation of tumour cells. In all xenografts, treated as well as nontreated, the areas of necrosis/apoptosis were large, but localised in the centre of tumour. There did not appear to be obvious differences in the degree of necrosis observed in both cases. We had no difficulties in obtaining five fields of viable cells in all tumours.
CMDB7 inhibits the intratumour endothelial cell density
Selective GSL-1 staining showed that CMDB7 treatment reduced the endothelial cell quantity in tumour tissue (Figure 5B) as compared to control (Figure 5A). The mean percentage of endothelial cell area (endothelial cell density) in viable fields of CMDB7-treated tumours (2.9 ± 0.6; 50 fields in 10 tumours) was inhibited by 66% (P<0.001) as compared to control tumour value (8.6±0.7; 50 fields in 10 tumours) (Figure 5C).
Figure 5.
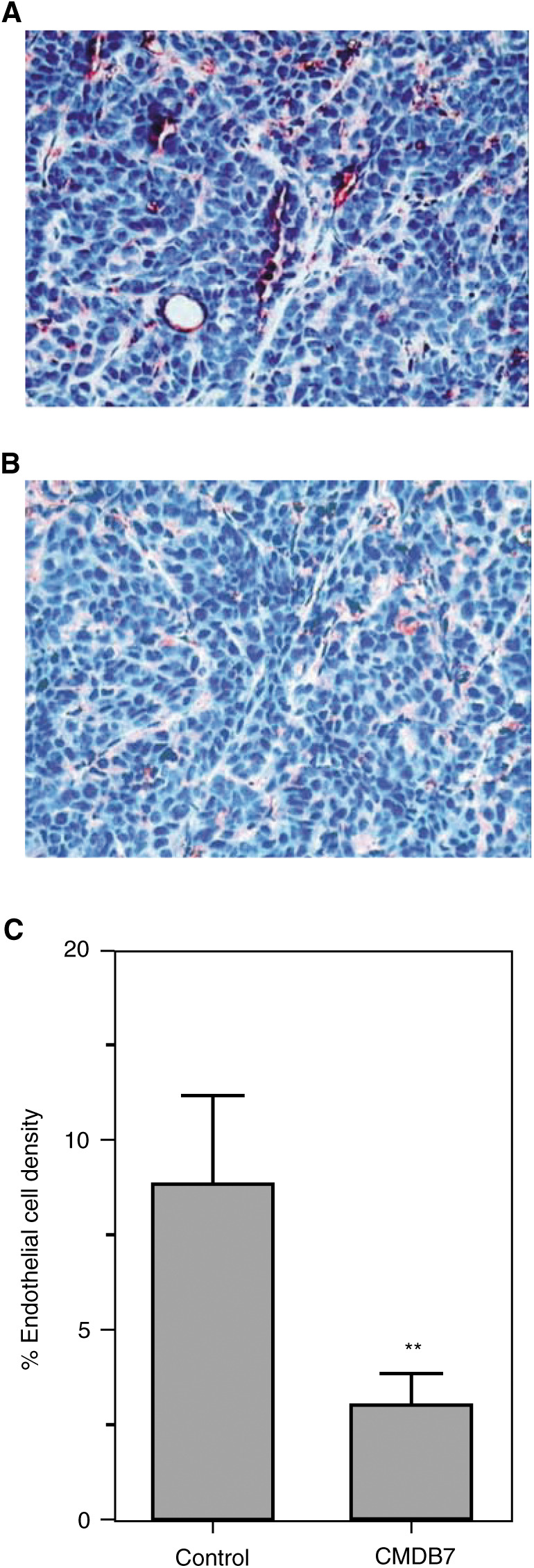
CMDB7 inhibits the tumour angiogenesis. Intratumour vascularisation was analysed immunohistologically after labelling of endothelial cells with a specific marker GSL-1. (A) Section of saline-treated (control) tumour and (B) section of tumour treated with 30 mg kg week−1 of CMDB7. (C) Endothelial cell density was evaluated by image analysis of GSL-1-labelled endothelial cells and the results are presented as the mean areas±s.e. (bars) of endothelial cells in the CMDB7-treated and control tumour sections obtained from 10 mice for each group. **, significantly different from control (P<0.001).
DISCUSSION
Antiangiogenesis is a promising therapeutic approach for the treatment of cancer (Folkman, 1995; Schweigerer, 1995). VEGF plays a crucial role in tumour angiogenesis and the inhibition of VEGF action decreases tumour growth in vivo (Kim et al, 1993; Goldman et al, 1998; Lin et al, 1998). Since the human A431 carcinoma cells secrete high amounts of VEGF (Myoken et al, 1991) and develop in nude mice tumours whose growth is highly VEGF-dependent (Melnyk et al, 1996), they provide a good model to test the availability of molecules that inhibit VEGF bioactivity. In this study, we assessed the anti-VEGF activity of CMDB7, described recently in vitro (Hamma-Kourbali et al, 2001), on A431 xenografted in nude mice, an extremely aggressive tumour model. CMDB7 is, to our knowledge, the only one of heparin analogues reported to be efficient in A431 xenograft model.
This study demonstrated that the s.c. injection of viable A431 cells yielded a 100% tumour uptake rate. The 2 week treatment with CMDB7 resulted in a 73% tumour growth inhibition associated with a 66% decrease in endothelial cell density. Compared to the human breast MDA-MB-435 (Bagheri-Yarmand et al, 1999) and MCF-7ras (Bagheri-Yarmand et al, 1998b) tumours, the magnitude of response of the A431 tumours to CMDB7 treatment was greater. Here, we observed that rapid A431 tumour growth was associated with high intratumour endothelial cell density, suggesting a direct relation between vascularisation within the primary tumour and the tumour growth rate. In 3-week-old A431 control (untreated) tumours, the endothelial cell density was 8.6%, while in 12-week-old MDA-MB-435 and MCF-7ras xenografts this value was 4.9% (Bagheri-Yarmand et al, 1999) and 6.1% (Bagheri-Yarmand et al, 1998b), respectively. Our observations are in agreement with results of Kim (1993), which demonstrated that the inhibitory effect of anti-VEGF antibody on tumour growth was more pronounced in the case of human A673 rhabdomyosarcoma secreting the highest quantity of VEGF and giving the most rapidly growing tumours as compared to G55 glioblastoma and SK-LMS-myosarcoma. In the CMDB7-treated tumours, a reduction of 66% in the density of endothelial cells indicates that this treatment attenuated the rate of neovascularisation, but did not completely reverse the initial activation of angiogenesis. The augmentation of CMDB7 dose did not result in increased efficiency of the drug in vivo (data not shown). Our results demonstrate that CMDB7 inhibited A431 tumour growth by, at least in part, decreasing intratumour endothelial cell density. The mechanism of CMDB7 action on endothelial cells is probably not direct and involves, as we recently described in vitro (Hamma-kourbali et al, 2001), a direct interaction of the drug with VEGF165 that becomes unavailable for specific receptors. In agreement, we demonstrate here that CMDB7 inhibits the A431-CM stimulation of endothelial cell proliferation.
The other mechanism by which CMDB7 reduced the A431 tumour growth is direct inhibition of A431 cell proliferation as evidenced by a decrease of proliferative index in treated xenografts compared to nontreated ones. In this study, we demonstrated that CMDB7 inhibited, like VEGF, the binding of 125I-VEGF165 to A431 cells with IC50 similar to the concentration at which CMDB7 inhibits efficiently the A431 proliferation in vitro. These findings could argue for the possible autocrine mitogenic action of VEGF on A431 cells. However, the depletion of VEGF amount in A431-conditioned medium by anti-VEGF antibody did not affect the A431 proliferation, although it did inhibit endothelial cell growth. It suggests that VEGF binding sites on the A431 cell surface are not involved in classical, KDR-dependent transmission of mitogenic signal. The A431 growth decrease by CMDB7 in vitro could involve the inhibition of other mitogenic growth factors. This interpretation can be strengthened by our previous studies demonstrating that CMDB7 inhibited the activity of heparin-binding PDGF and TGFβ by altering their conformation, but did not change the activity of EGF and IGF1, which are not heparin-binding growth factors (Bagheri-Yarmand et al, 1997,1998a,1998b). Independently, the possible VEGF autocrine pathway in A431 could mediate tumour cell survival by protecting them from apoptosis as it was recently reported for breast cancer MDA-MB-231 cells (Bachelder et al, 2001). Further studies are necessary to understand the mechanisms of direct CMDB7 inhibitory action on A431 proliferation in vitro.
Altogether, our findings demonstrate that CMDB7 has a strong antiangiogenic and antitumour action in vivo, also when tumour cells produce a high level VEGF and EGFRs. CMDB7 acts directly on both tumour and endothelial cells, decreasing in a potent manner the tumour growth by increasing the proliferation of tumour cells and especially angiogenesis in vivo. The development of resistance to antiangiogenic drugs is becoming apparent (Kerbel et al, 2001). It is very important, now, to enlarge the diversity of molecular targets for antiangiogenic drugs and to use a combination of antiangiogenic therapies. One of the possible mechanisms of this resistance may be due to redundancy of different proangiogenic growth factors made by tumour cells. When one angiogenic factor is targeted, the cancer cells increase production of other angiogenic factors. In this context, we believe that the ability of CMDB7 to interact with several angiogenic factors, including VEGF (Hamma-Kourbali et al, 2001), bFGF (Bagheri-Yarmand et al, 1997,1998a), TGF-β and PDGF (Bagheri-Yarmand et al, 1998b), will permit to oppose or at least put off the development of resistance. Recently, it was reported that the resistance of tumours to treatment with EGF receptor-blocking antibodies can be associated with an elevated expression of VEGF (Viloria-Petit et al, 2001). Since we show in this study that CMDB-7 efficiently blocks in vivo the effects of VEGF produced at high level, we can speculate that this drug could be useful in the case of failure to anti-EGFR treatment. It is believed now that because angiogenesis is a complex and multistage process, treatment with more than one antiangiogenic agent may be beneficial (Cherrington et al, 2000). Also, the neutralisation of angiogenic growth factors, especially VEGF, in tumour with CMDB7 may increase the effects of a variety of antiangiogenic inhibitors (Kerbel et al, 2001). For example, the reduced ability of Taxotere to induce apoptosis of endothelial cells in the presence of VEGF (Sweeney et al, 2001) could be restored by combined treatment with CMDB7. CMDB7 can be used not only as monotherapy but also especially in combination with other antiangiogenic and anticancer drugs to cause acute tumour regression by delaying development of resistance and by enhancing the effects of other drugs.
Acknowledgments
This work was supported by ARC (Association pour la Recherche sur le Cancer, Paris, France). We thank S Duflot, B Lejeune and O Saint-Catherine for excellent technical assistance.
References
- Alroy J, Goyal V, Skutelsky E (1987) Lectin histochemistry of mammalian endothelium. Histochemistry 68: 603–607 [DOI] [PubMed] [Google Scholar]
- Bachelder RE, Crago A, Chung J, Wendt MA, Shaw LM, Robinson G, Mercurio AM (2001) Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res 61: 5736–5740 [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Kourbali Y, Mabilat C, Morère JF, Martin A, Lu H, Soria C, Jozefonvicz J, Crépin M (1998a) The suppression of fibroblast growth factor 2/fibroblast growth factor 4-dependent tumour angiogenesis and growth by the anti-growth factor activity of dextran derivative (CMDB7). Br J Cancer 78: 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Kourbali Y, Morère JF, Jozefonvicz J, Crépin M (1998b) Inhibition of MCF-7ras tumor growth by carboxymethyl benzylamide dextran: blockage of the paracrine effect and receptor binding of transforming growth factor β1 and platelet-derived growth factor-BB. Cell Growth Differ 9: 497–504 [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Kourbali Y, Rath AM, Vassy R, Martin A, Jozefonvicz J, Soria C, Lu H, Crépin M (1999) Carboxymethyl benzylamide dextran blocks angiogenesis of MDA-MB435 breast carcinoma xenografted in fat pad and its lung metastases in nude mice. Cancer Res 59: 507–510 [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Liu JF, Ledoux D, Morère JF, Crépin M (1997) Inhibition of human breast epithelial HBL100 cell proliferation by a dextran derivative (CMDB7): interference with the FGF2 autocrine loop. Biochem Biophys Res Commun 239: 424–428 [DOI] [PubMed] [Google Scholar]
- Chaubet F, Champion J, Maîga R, Maurey S, Jozefonvicz J (1995) Synthesis and structure–anticoagulant property relationships of functionalized dextrans: CMDBS. Carbohydr Polym 28: 145–152 [Google Scholar]
- Cheng SY, Huang H-JS, Nagane M, Ji X-D, Wang D, Shih CC, Arap W, Huang CM, Cavenee WK (1996) Suppression of glioblastoma angiogenicity and tumorigenicity by inhibition of endogenous expression of vascular endothelial growth factor. Proc Natl Acad Sci USA 93: 8502–8507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrington JM, Strawn LM, Shawver LK (2000) New paradigms for the treatment of cancer: the role of anti-angiogenesis agents. Adv Cancer Res 79: 1–38 [DOI] [PubMed] [Google Scholar]
- DeVries C, Escobedo JA, Ueno H, Houck K, Ferrara F, Williams LT (1992) The fms-like tyrosine kinase flt; a receptor for vascular endothelial growth factor. Science 255: 989–991 [DOI] [PubMed] [Google Scholar]
- Dvorak HF, Sioussat TM, Brown LF, Berse B, Nagy JA, Sotrel A, Manseau EJ, Van de Water L, Senger DR (1991) Distribution of vascular permeability factor (vascular endothelial growth factor) in tumors: concentration in tumor blood vessels. J Exp Med 174: 1275–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Davis-Smith (1997) The biology of vascular endothelial growth factor. Endocrinol Rev 18: 4–25 [DOI] [PubMed] [Google Scholar]
- Folkman J (1990) What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst 82: 4–6 [DOI] [PubMed] [Google Scholar]
- Folkman J (1995) Clinical applications of research on angiogenesis. N Engl J Med 333: 1757–1763 [DOI] [PubMed] [Google Scholar]
- Goldman CK, Kendall RL, Cabrera G, Soroceanu L, Heike Y, Gillespie GY, Siegal GP, Mao X, Bett AJ, Huckle WR, Thomas KA, Curiel DT (1998) Paracrine expression of a native soluble vascular endothelial growth factor receptor inhibits tumor growth, metastasis, and mortality rate. Proc Natl Acad Sci USA 9: 8795–8800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamma-Kourbali Y, Vassy R, Starzec A, Le Meuth-Metzinger V, Oudar O, Bagheri-Yarmand R, Perret G, Crépin M (2001) Vascular endothelial growth factor 165 (VEGF165) activities are inhibited by carboxymethyl benzylamide dextran that competes for heparin binding to VEGF165 and VEGF165. KDR complexes. J Biol Chem 276: 39748–39754 [DOI] [PubMed] [Google Scholar]
- Hlatky L, Tsionou C, Hahnfeldt P, Coleman CN (1994) Mammary fibroblasts may influence breast tumor angiogenesis via hypoxia-induced vascular endothelial growth factor up-regulation and protein expression. Cancer Res 54: 6083–6086 [PubMed] [Google Scholar]
- Kaipainen A, Korhonen J, Mustonen T, Van Hinsbergh VW, Fang G-H, Dumont D, Breitman M, Alitalo K (1995) Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci USA 92: 3566–3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerbel RS, Yu J, Tran J, Man S, Viloria-Petit A, Klement G, Coomber BL, Rak J (2001) Possible mechanisms of acquired resistance to anti-angiogenic drugs: implications for the use of combination therapy approaches. Cancer Metast Rev 20: 79–86 [DOI] [PubMed] [Google Scholar]
- Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N (1993) Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362: 841–844 [DOI] [PubMed] [Google Scholar]
- Klagsbrun M, Soker S (1993) VEGF/VPF: the angiogenic factor found? Curr Biol 3: 699–712 [DOI] [PubMed] [Google Scholar]
- Lange T, Gutman-Raviv N, Baruch L, Machluf M, Neufeld G (2003) VEGF162: a new heparin binding VEGF splice form that is expressed in transformed human cells. J Biol Chem, May 9; 278 (19): 17164–17169 [DOI] [PubMed] [Google Scholar]
- Li S, Peck-Radosavljevic M, Koller E, Koller F, Kaserer K, Kreil A, Kapiotis S, Hamwi A, Weich HA, Valent P, Angelberger P, Dudczak R, Virgolini I (2001) Characterization of 123I-vascular endothelial growth factor-binding sites expressed on human tumour cells : possible implication for tumour scintigraphy. Int J Cancer 91: 789–796 [DOI] [PubMed] [Google Scholar]
- Lin P, Sankar S, Shan S-Q, Dewhirst MW, Polverini PJ, Quinn TQ, Peters KG (1998) Inhibition of tumor growth by targeting tumor endothelium using a soluble vascular endothelial growth factor receptor. Cell Growth Differ 9: 49–58 [PubMed] [Google Scholar]
- Mattsson G, Carlsson PO, Olausson K, Jansson L (2002) Histological markers for endothelial cells in endogenous and transplanted rodent pancreatic islets. Pancreatology 2: 155–162 [DOI] [PubMed] [Google Scholar]
- Melnyk O, Shuman MA, Kim KJ (1996) Vascular endothelial growth factor promotes tumor dissemination by a mechanism distinct from its effect on primary tumor growth. Cancer Res 56: 921–924 [PubMed] [Google Scholar]
- Millauer B, Shawver LK, Plate KH, Riseau W, Ullrich A (1994) Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature 367: 576–579 [DOI] [PubMed] [Google Scholar]
- Myoken Y, Kayada Y, Okamoto T, Kan M, Sato GH, Sato D (1991) Vascular endothelial cell growth factor (VEGF) produced by A-431 human epidermoid carcinoma cells and identification of VEGF membrane binding sites. Proc Natl Acad Sci USA 88: 5819–5823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z (1999) Vascular endothelial growth factor (VEGF) and its receptors. FASEB J 13: 9–22 [PubMed] [Google Scholar]
- Plouët J, Schilling J, Gospodarowicz D (1989) Isolation and characterization of a newly identified endothelial cell mitogen produced by AtT-20 cells. EMBO J 8: 3801–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relf M, LeJeune S, Scott PAE, Fox S, Smith K, Leek R, Moghaddam A, Whitehouse R, Bicknell R, Harris AL (1997) Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor β-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res 57: 963–969 [PubMed] [Google Scholar]
- Schweigerer L (1995) Antiangiogenesis as a novel therapeutic concept in pediatric oncology. J Mol Med 73: 497–508 [DOI] [PubMed] [Google Scholar]
- Senger DR, Dvorack HF, Perruzzi CA, Feder J (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219: 983–985 [DOI] [PubMed] [Google Scholar]
- Soker S, Miao HQ, Nomi M, Takashima S, Klagsbrun M (2002) VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165- receptor binding. J Cell Biochem 85: 357–368 [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M (1998) Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 92: 735–745 [DOI] [PubMed] [Google Scholar]
- Sweeney CJ, Miller KD, Sissons SE, Nozaki S, Heilman DK, Shen J, Sledge GWJ (2001) The antiangiogenic property of docetaxel is synergistic with a recombinant humanized monoclonal antibody against vascular endothelial growth factor or 2-methoxyestradiol but antagonized by endothelial growth factors. Cancer Res 61: 3369–3372 [PubMed] [Google Scholar]
- Terman BI, Dougher-Vermazen M, Carrion ME, Dimitrov D, Armellino DC, Gospodarowicz D, Böhlen P (1992) Identification of the KDR tyrosine kinase as a receptor for vascular endothelial growth factor. Biochem Biophys Commun 187: 1579–1586 [DOI] [PubMed] [Google Scholar]
- Veikkola T, Karkkainen M, Claesson-Welsh L, Alitalo K (2000) Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res 60: 203–212 [PubMed] [Google Scholar]
- Viloria-Petit A, Crombet T, Jothy S, Hicklin D, Schlaeppi JM, Rak J, Kerbel RS (2001) Acquired resistance to the tumour effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res 61: 5090–5101 [PubMed] [Google Scholar]
- Whitaker GB, Limberg BJ, Rosenbaum JS (2001) Vascular endothelial growth factor receptor-2 and neuropilin-1 form a receptor complex that is responsible for the differential signaling potency of VEGF165 and VEGF121. J Biol Chem 276: 25520–25531 [DOI] [PubMed] [Google Scholar]
- Workman P, Twentyman P, Balkwill F, Balmain A, Chaplin D, Double J, Embleton J, Newell D, Raymond R, Stables J, Stephens T, Wallace J (1998) United Kingdom co-ordinating committee on cancer research (UKCCCR) guidelines for the welfare of animals in experimental neoplasia (Second Edition). Br J Cancer 77: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK (1996) Time-dependent vascular regression and permeability changes in established human tumor xenografts induced by an anti-vascular endothelial growth factor/vascular permeability factor antibody. Proc Natl Acad Sci USA 93: 14765–14770 [DOI] [PMC free article] [PubMed] [Google Scholar]