

Abstract
To understand the underlying mechanisms leading to such cardiac defects, the functional domains of mXinα and its interacting proteins were investigated. Interaction studies using co-immunoprecipitation, pull-down, and yeast two-hybrid assays revealed that mXinα directly interacts with β-catenin. The β-catenin-binding site on mXinα was mapped to amino acids 535–636, which overlaps with the known actin-binding domains composed of the Xin repeats. The overlapping nature of these domains provides insight into the molecular mechanism for mXinα localization and function. Purified recombinant glutathione S-transferase- or His-tagged mXinα proteins are capable of binding and bundling actin filaments, as determined by co-sedimentation and electron microscopic studies. The binding to actin was saturated at an approximate stoichiometry of nine actin monomers to one mXinα. A stronger interaction was observed between mXinα C-terminal deletion and actin as compared with the interaction between full-length mXinα and actin. Furthermore, force expression of green fluorescent protein fused to an mXinα C-terminal deletion in cultured cells showed greater stress fiber localization compared with force-expressed GFP-mXinα. These results suggest a model whereby the C terminus of mXinα may prevent the full-length molecule from binding to actin, until the β-catenin-binding domain is occupied by β-catenin. The binding of mXinα to β-catenin at the adherens junction would then facilitate actin binding. In support of this model, we found that the actin binding and bundling activity of mXinα was enhanced in the presence of β-catenin.
The striated muscle-specific Xin genes encode proteins containing several proline-rich regions, a highly conserved sequence homologous to the Myb-A and Myb-B DNA-binding domain, and a region with 15–28 16-amino acid (aa)4 repeating units (called the Xin repeats) (1-3). In the mouse, two Xin genes, mXinα and mXinβ, exist, whereas only one cXin gene is found in the chick. The expression of both cXin and mXinα is regulated by the muscle transcription factor, MEF2C, and the homeodomain transcription factor, Nkx2.5 (1, 2). Similarly, the expression of mXinβ (also termed myomaxin) is also under the control of MEF2A (4). Treatment of chick embryos with cXin antisense oligonucleotides results in abnormal cardiac morphogenesis and a disruption in cardiac looping, suggesting that Xin plays an essential role in cardiac development (2). Embryonic lethality was expected based on this antisense oligonucleotide experiment in chicks; however, viable and fertile mXinα knock-out mice were observed. This viability probably results from functional compensation through the up-regulation of mXinβ at both message and protein levels (5). Consistent with the compensatory role of mXinβ, we have previously shown that mXinβ, like mXinα (2, 6), localizes to the intercalated disc of the adult heart (5). Despite this compensation, the adult mXinα-deficient mouse hearts are hypertrophied and exhibit cardiomyopathy with conduction defects (5). This suggests that each of the mXin proteins may have a unique function in the heart. However, the molecular mechanism behind mXinα functions remains to be elucidated. The first step toward answering this question is to characterize the functional domains on mXinα and its interacting partners.
Study with the human homologs (hXinα, also termed Cmya1 and hXinβ, also termed Cmya3) of mXinα and mXinβ reveals that the Xin repeats bind actin filaments in vitro and that a minimum of three Xin repeats is required for the detectable binding (3). Therefore, Xin proteins should have multiple independent actin-binding sites, which could then cross-link actin filaments into a loosely packed meshwork. This cross-linking activity has been demonstrated only with recombinant protein containing 3∼16 Xin repeats from hXinα (3, 7) but not with full-length hXinα protein. Additional studies with hXin demonstrated the ability of hXin to directly bind to filamin c, Mena/VaSP, as well as colocalize at the intercalated discs, suggesting that Xin may play a role in remodeling of the actin cytoskeleton (8). However, it is unclear why in the heart, mXinα does not associate with actin thin filaments but rather colocalizes with β-catenin and N-cadherin at the intercalated disc. In the chicken heart, cXin is also associated with the N-cadherin-β-catenin complex, as demonstrated by co-immunoprecipitation (co-IP) experiments (6). Thus, the molecular mechanisms underlying the role of Xin in cardiac morphogenesis and myofibrillogenesis remain to be elucidated.
In this study, we gained further insight into the function of mXinα through the identification of mXinα binding partners, including β-catenin and actin. Additionally, we have mapped the β-catenin-binding domain to aa residues 535–636 of mXinα, which overlaps with the actin-binding domain. To gain further insight into the involvement of Xin with the actin cytoskeleton, we further studied the significance of Xin actin binding ability. We have found that recombinant full-length mXinα protein aggregates actin filaments into ordered actin bundles, as observed by negative staining electron microscopy. Full-length mXinα interacts with actin more weakly than its C-terminal deletion mutant, which lacks the β-catenin-binding domain and the C terminus, but retains most of the Xin repeats. The binding of mXinα to actin filaments can be further enhanced by the presence of β-catenin. From these results, we propose a model in which the β-catenin-binding domain and the C terminus of mXinα prevent an interaction between full-length mXinα and actin (an autoinhibited state), until the β-catenin-binding domain is occupied by β-catenin. The binding of mXinα to β-catenin at the adherens junction of the intercalated disc would then enable subsequent actin binding and bundling (an open state).
EXPERIMENTAL PROCEDURES
Yeast Two-hybrid Assay and Library Screening
Protein-protein interactions between mXinα and either β-catenin or N-cadherin were tested utilizing the Matchmaker two-hybrid system 3 (Clontech, Palo Alto, CA) in yeast strain AH109. Full-length mXinα cDNA (1) was subcloned into the SalI/SmaI sites of pEGFP-C2 (Clontech). The resulting plasmid pEGFP-mXinα was used for constructing various deletions by PCR-based mutagenesis. The mXinα cDNA and several deletion fragments were subcloned into the SalI/SmaI sites of the pGBKT7 vector. The full-length construct (pGBKT7-mXinα) encodes aa 1–1129 of mXinα. The mXinαRΔ-1, -2, and -3 constructs (pGBKT7-mXinαRΔ-1, -2, and -3) represent mutants with a deletion of the first half (aa 73–361), the second half (aa 362–746), and all (aa 73–746), respectively, of the Xin repeats. The mXinαCΔ is a C-terminal deletion construct (pGBKT7-mXinαCΔ) encoding aa 1–532. Another construct, pGBKT7-β-catBR, encoding aa 533–746, was PCR-amplified using the following primers: 5′-AGTACCATCGATGTGGTACG-3′ and 5′-AGCCCATGGGACAGTTTTC-3′. The resulting product, after ClaI/NcoI digestion and fill in with the Klenow fragment, was subcloned into the SmaI site of pGBKT7. This β-catenin binding region was further divided into four fragments (fragments CA, AP, PN, and CP), each of them flanking with a pair of either ClaI, ApaI, PstI, or NcoI digestion sites. Primer pairs used to generate these fragments were 5′-AGTACCATCGATGTGGTACG-3′ and 5′-CAGAGAGATTGGGGCCCTTTCAT-3′ for the CA fragment, 5′-ATGTTTGGGCCCCAATCTCTG-3′ and 5′-CACCCGGCTGCAGTACCTTAC-3′ for the AP fragment, 5′-GTAAGGTACTGCAGCCGGGTG-3′ and 5′-AGCCCATGGGACAGTTTC-3′ for the PN fragment, and 5′-AGTACCATCGATGTGGTACG-3′ and 5′-CACCCGGCTGCAGTACCTTAC-3′ for the CP fragment. The individual PCR-amplified fragment was then digested with the respective enzymes, Klenow-filled in, and subcloned into the SmaI site of the pGBKT7 vector. The insert sequences of all constructs were confirmed with DNA sequencing at the Roy J. Carver Center for Comparative Genomics, Department of Biological Sciences, University of Iowa.
To construct pGADT7-β-catenin, pGEX-KG-β-catenin (a generous gift from Dr. Janne Balsamo, University of Iowa) was digested with BamHI, filled in, and subcloned into the NdeI site of pGADT7. To construct pGADT7-N-cadherin, the N-cadherin cytoplasmic domain was PCR-amplified from the pSP72-N-cadherin (a gift from Dr. Janne Balsamo) with the primers 5′-ggaattcATGAAGCGCCGTGATAAGG-3′ and 5′-ccatcgatAATAAAAGCAATGCGATGTAAC-3′. This PCR fragment was digested and subcloned into the EcoRI/ClaI sites of pGADT7. These prey constructs were separately transformed into AH109, which had been previously transformed with either full-length mXinα or one of the deletion constructs. Direct interaction between the N-cadherin or β-catenin prey and the mXinα bait was determined using growth on selective media and a β-galactosidase expression (X-gal assay) as described in the Clontech Two-Hybrid System user manual (40). A positive control was constructed to validate the yeast two-hybrid assay using p53 and the large T antigen. A negative control was constructed replacing β-catenin with the large T antigen.
In order to identify novel mXinα-interacting proteins, pGBKT7-mXinα was further used as bait to screen a custom Matchmaker cDNA library prepared from 5-week-old rat hearts and constructed in pGAD10 vector by Clontech. After recovery of the positive prey plasmids and retransformation to confirm the true interaction, complete insert sequences were determined. Among 20 known and novel positive clones, two independent clones, pL1.192 and pL2P6, encoding aa 1–258 and 1–215, respectively, of rat cardiac α-actin as well as a clone, pL2P10, encoding aa 497–780 of rat gelsolin were obtained and reported here as mXinα-interacting proteins. In another screening using a pretransformed mouse 17-day embryo Matchmaker cDNA library constructed in the pACT2 prey vector and a custom adult mouse heart Matchmaker cDNA library constructed in the pGADT7-RecAB prey vector, two independent clones, p4Q39 and p4Q79, each encoding full-length mouse cardiac α-tropomyosin, and another clone, pL3Q8, encoding aa 2,533–2,603 of mouse filamin b were obtained and reported here. In addition, full-length cDNAs for p120 catenin (a generous gift from Dr. Janne Balsamo), talin (a generous gift from Dr. Richard Hynes (Massachusetts Institute of Technology)) and vinculin (a generous gift from Dr. Wolfgang Goldman (University of Erlangen, Germany)) were individually subcloned into pGADT7 plasmids and used as preys in a yeast two-hybrid assay to test whether these proteins interact with mXinα.
Constructions of Plasmids and Purification of Recombinant Proteins
Expression plasmid pGEX-mXinα for GST-mXinα fusion protein was constructed by ligating an EcoRI mXinα cDNA insert from pGBKT7-mXinα with EcoRI-digested pGEX4T-1 vector. Another expression plasmid, pET30-mXinα, for His-mXinα fusion protein was derived from the SalI/SmaI mXinα fragment (5.6 kb) of pGEM3ZmXin3 subcloned into the XhoI (fill-in)/SalI sites of pET30a vector. pGEX-KG-β-catenin was used for the production of GST-β-catenin fusion protein. Recombinant proteins were expressed in Escherichia coli BL21(DE3)pLysS cells and purified by a glutathione-Sepharose 4B column for GST-tagged proteins or His GraviTrap column for His-tagged proteins (GE Healthcare) according to the manufacturer's protocols.
Co-IP, Pull-down Assay, and Western Blot Analysis
Adult mouse hearts were homogenized in IB buffer (20 mm phosphate buffer, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 0.1% SDS, and protease inhibitor mixture; Roche Applied Science). The homogenate (150 μg of total protein/immunoprecipitation) was cleared by centrifugation at 12,000 × g for 15 min and incubated with anti-β-catenin, anti-N-cadherin, anti-p120 catenin, anti-plakoglobin, anti-filamin c, anti-vinculin, or control mouse serum for 2 h, followed by protein G-Sepharose beads (GE Healthcare) for 1 h at 4 °C. The beads were washed three times with IB buffer and once with PBS. The bound proteins were eluted in SDS-PAGE sample buffer, fractionated by 7.5% SDS-PAGE, and immunoblotted with anti-mXinα U1013 antibody or anti-β-catenin as described previously for Western blot analysis (5).
For the pull-down assay, various amounts (5–60 nm) of recombinant His-mXinα were mixed with 30 nm GST-β-catenin in binding buffer containing 20 mm HEPES, buffer pH 7.5, 100 mm KCl, 1 mm dithiothreitol, 0.1% Triton X-100, and 2.5 mm phenylmethanesulfonyl fluoride for 2 h at 4 °C. The mixture was immunoprecipitated with a monoclonal anti-β-catenin antibody. The immunoprecipitate was further analyzed by Western blot with polyclonal anti-mXinα antibody as described above.
Actin Binding Assay
Rabbit skeletal muscle actin (>99% pure) was purchased from Cytoskeleton, Inc. (Denver, CO) and used in a slightly modified actin binding assay as described previously (9). Briefly, actin (9.3 μm) and various amounts of His-mXinα (0–7.64 μm) or GST-mXinα were mixed in 100 μl of 10 mm HEPES buffer, pH 7.5, 100 mm KCl, 0.05% Triton X-100, 0.1 mm dithiothreitol, 0.1 mm ATP, and 1.5 mm MgCl2 in the absence or presence (1.95 μm) of GST-β-catenin. The mixtures were incubated at room temperature for 30 min and centrifuged for 30 min in a Beckman Airfuge at 26 p.s.i. high speed centrifugation (100,000 × g) to separate bound and unbound fractions. To test the cross-linking or bundling activity of recombinant mXinα, the protein mixed with actin was centrifuged at low speed centrifugation (10,000 × g) for 15 min. Under this condition, actin filaments remain in the supernatant except for cross-linked or bundled filaments formed by binding proteins. Aliquots of the supernatant and pellets were analyzed by 7.5% SDS-PAGE, and the protein bands after stain were quantified as described (9).
Cell Culture, DNA Transfection, and Fluorescence Microscopy
The mouse skeletal muscle cell line, C2C12, was grown on glass coverslips in Dulbecco's modified Eagle's medium plus 5% fetal bovine serum and 15% defined supplemented calf serum in a humidified incubator at 37 °C with 5% CO2. Chinese hamster ovary (CHO) cells were grown on glass coverslips in Dulbecco's modified Eagle's medium plus 10% fetal bovine serum. Myoblasts or CHO cells were transfected with pEGFP-C2 (empty vector control), pEGFP-mXinα, pEGFP-mXinαRΔ-1, -2, and -3, or pEGFP-mXinαCΔ using Lipofectamine PLUS reagent (Invitrogen) as previously described (6). After 24 h, cells on coverslips were fixed in 3.7% formaldehyde and either processed for immunofluorescence microscopy with a monoclonal anti-vinculin antibody (Sigma) and a rhodamine-conjugated goat anti-mouse secondary antibody or directly mounted onto glass slides and observed under a Zeiss epifluorescence photomicroscope III. The fluorescence and phase-contrast images were collected with a Leica digital camera and processed using Adobe Photoshop.
Electron Microscopy
Small aliquots (10 μl) of actin and recombinant mXinα mixtures in the binding condition used for the actin-binding assay were applied to carbon-coated Formvar grids and negatively stained with 1.0% uranyl acetate. Samples were then observed under a JEOL 1230 transmission electron microscope at an accelerating voltage of 100 kV (Central Microscopy Research Facility, University of Iowa). The images were collected with Gatan CCD digital camera attached to the electron microscope. The thickness of the actin bundles was measured from images against a stained catalase resolution standard (Polysciences, Inc., Warrington, PA).
RESULTS
mXinα Is Associated with N-cadherin, β-Catenin, and p120 Catenin in Adult Mouse Heart
The cXin gene is believed to play a vital role in cardiac morphogenesis (2), probably through an association with N-cadherin and β-catenin (6) and the intracellular signaling at the adherens junctions of the intercalated discs. In the mouse heart, co-localization of mXinα with N-cadherin and β-catenin at the intercalated discs has been demonstrated throughout embryogenesis and adulthood (6). Targeted deletion of mXinα in the mouse results in cardiac hypertrophy and cardiomyopathy with abnormal intercalated disc ultrastructure (5). Although the underlying mechanism leading to cardiac defects remains unclear, a significant down-regulation of N-cadherin, β-catenin, and p120 catenin is observed in the mXinα-null mouse heart (5). This suggests an association of mXinα with these adherens junctional components. To test this association, co-IP experiments were carried out using anti-N-cadherin, anti-β-catenin, and anti-p120 catenin antibodies with adult mouse heart extracts. Western blot analysis with U1013 anti-mXinα antibody identified that both mXinα and the alternatively spliced isoform mXinα-a were present in the total extract as well as in the anti-β-catenin immunoprecipitate, the anti-N-cadherin immunoprecipitate, and the anti-p120 catenin immunoprecipitate but not in the control mouse serum immunoprecipitate (Fig. 1A, left). As expected, anti-N-cadherin immunoprecipitate but not anti-plakoglobin immunoprecipitate also contains β-catenin (Fig. 1A, right). These data suggest that mXinα and mXinα-a are indeed components of the N-cadherin-β-catenin-p120 catenin complex and, therefore, support our finding of simultaneous down-regulation of these components in mXinα-null mouse heart (5). It should be noted that relatively more of the mXinα-a isoform was consistently found in these immunoprecipitates than mXinα, suggesting that these two isoforms may have slightly different mechanisms for association with the immunocomplexes. As we have previously showed, the minor isoform, mXinα-a, is encoded by an alternatively spliced mRNA with the inclusion of intron 2 from the mXinα gene (5). Therefore, the aa sequences of both mXinα-a and mXinα are identical, except in the C terminus, where the last two residues of mXinα are replaced with an additional 683 aa residues. This extra C-terminal sequence was also found in one (called Xin A) of the hXinα isoforms (8). A filamin c-binding domain was previously mapped to the last 158 residues of this hXinα isoform, Xin A (8). When the co-IPs were further performed with anti-filamin c or anti-vinculin antibody, the resulting immunoprecipitates contained both mXinα-a and mXinα (Fig. 1A, left), suggesting that both of these mXinα isoforms are associated with focal adhesion components in addition to the N-cadherin-catenin complex. The additional association between filamin c and mXinα-a in the extra C-terminal region may account for the observation of more mXinα-a associated with these immunoprecipitates, since mXinα lacks this region.
FIGURE 1. Co-IP of mXinα and β-catenin from adult mouse heart and from purified recombinant proteins.
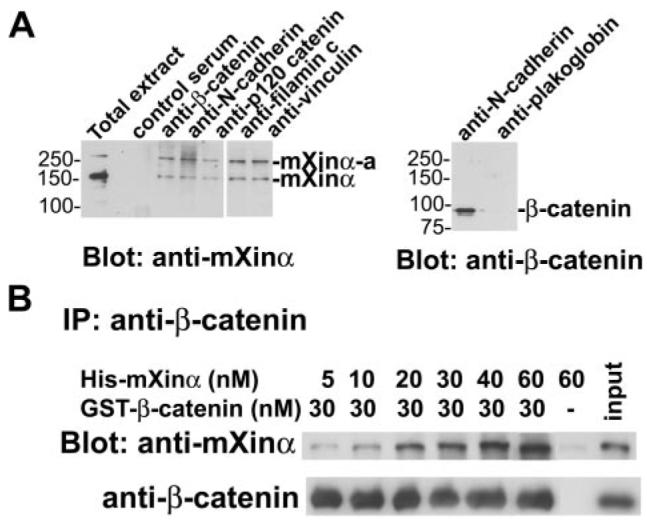
A, total extract prepared from adult mouse hearts was immunoprecipitated with mouse control serum, monoclonal anti-β-catenin, anti-N-cadherin, anti-p120 catenin, anti-filamin c, anti-vinculin, or anti-plakoglobin. Western blots on immunoprecipitates were probed with the indicated antibody. Both mXinα and its isoform, mXinα-a, are detected in the anti-β-catenin, anti-N-cadherin, anti-p120-catenin, anti-filamin c, and anti-vinculin immunoprecipitates, but not in the control serum immunoprecipitate. β-Catenin is detected in the anti-N-cadherin immunoprecipitate but not the anti-plakoglobin immunoprecipitate. B, increasing amounts of purified recombinant His-mXinα were mixed with GST-β-catenin in binding buffer and subjected to IP by anti-β-catenin antibody. Western blots (Blot)on the immunoprecipitate were probed with polyclonal anti-mXinα U1013 antibody to detect co-immunoprecipitated mXinα or anti-β-catenin antibody to demonstrate equal amounts of β-catenin in the immunoprecipitate. An increasing amount of mXinα directly binds to β-catenin. When an mXinα (60 μm) to β-catenin molar ratio of 2:1 was used, the co-pelleted mXinα still increased. On the other hand, the mXinα alone (control) at this mXinα concentration could not be co-pelleted. This result suggests that a molecule of β-catenin can bind at least two molecules of mXinα.
mXinα Directly Interacts with β-Catenin
To examine whether a direct interaction between mXinα and β-catenin exists, co-IPs with purified proteins (Fig. 1B) and yeast two-hybrid assays (Fig. 2) were carried out. Increasing amounts of purified, recombinant His-mXinα were mixed with GST-β-catenin in solution, followed by immunoprecipitation with monoclonal anti-β-catenin and Western blot analysis of the immunoprecipitate with U1013 anti-mXinα polyclonal antibody. As can be seen in Fig. 1B, a 30 nm concentration of GST-β-catenin was able to co-precipitate increasing amounts (5–60 nm) of His-mXinα protein. On the other hand, the immunoprecipitation in the absence of GST-β-catenin could not bring down His-mXinα protein. These results clearly suggest a direct interaction between mXinα and β-catenin. The amount of HismXinα brought down by 30 nm GST-β-catenin did not reach a plateau when 30 nm His-mXinα was added, further suggesting that multiple mXinα-binding sites could be present in the β-catenin molecule if mXinα functions as a monomer.
FIGURE 2. Determination of the β-catenin-binding domain on mXinα.

A, schematic representation of mXinα and various deletion constructs used in the yeast two-hybrid assay. The mXinα protein contains a putative DNA-binding domain (DBD), a putative nuclear localization signal (NLS), a region with 15 Xin repeats, and a proline-rich region. The deletion constructs include mXinαRΔ-1, mXinαRΔ-2, mXinαRΔ-3 (which represent deletions of the first eight repeats, the second half, and all 15 Xin repeats, respectively) and mXinαCΔ (a C-terminal deletion containing only the first 10 repeats), and β-catBR (β-catenin-binding region). Their relative strengths of interaction with β-catenin, as determined by an X-gal assay, are indicated by the numbers of plus signs, and the minus sign represents weak or no interaction. B, results of β-galactosidase filter assay for the interaction between mXinα and β-catenin. After co-transformation of bait and prey into yeast AH109 cells, colonies grown on selective media were transferred to a membrane for the X-gal assay. The constructs that lack aa 533–746, including mXinαRΔ-2 (c), mXinαRΔ-3 (d), and mXinαCΔ (e), show low β-galactosidase activity, indicating lack of interaction. In contrast, the mXinα (a), mXinαRΔ-1 (b), and β-catBR (f) constructs, which retain this region, demonstrate a high level of β-galactosidase activity, suggesting a strong interaction. The region represented by amino acids 533–746 is both necessary and sufficient for the binding of mXinα to β-catenin. C, the β-catenin-binding domain resides within the CA fragment of β-catBR. The β-catBR region of aa 533–746 was further divided into four fragments (CA, AP, PN, and CP). Each fragment was subcloned and used as bait in the yeast two-hybrid assay. Only CA and CP containing a common region of aa 535–636 showed strong interaction with β-catenin in the X-gal filter assay. Therefore, the β-catenin-binding domain locates within aa 535–636. D, comparison of the β-catenin-binding domain of Xin proteins from mouse, human, and chick. The β-catenin-binding domain (aa 535–636) defined on mXinα was used to align all predicted Xin proteins from mouse (mXinβ, AY775570–775572), human (hXinα/Cmya1, AJ626899; hXinβ/Cmya3, XM_496606), and chick (cXin, AF051944) using the ClustalW program (DNASTAR, Inc., Madison, WI). The dashes indicate gaps introduced for optimal alignment. Yellow, green, and red, 5 of 5, 4 of 5, and 3 of 5 identity, respectively. This β-catenin-binding domain represents a highly conserved amino acid sequence among these Xin proteins, and the sequence identity between mXinα and cXin, mXinβ, or hXinα is 49.02, 53.92, or 57.84%, respectively.
A yeast two-hybrid assay was also used to determine whether mXinα directly interacts with N-cadherin and β-catenin and map the domain of these potential interactions, since we have shown that mXinα is part of the N-cadherin-β-catenin-p120 catenin complex (Fig. 1A). Full-length mXinα cDNA and several deletions were constructed into pGBKT7 vector and used as baits (Fig. 2A). β-Catenin or the cytoplasmic domain of N-cadherin served as the “prey” and was constructed into pGADT7. A direct protein-protein interaction was not observed for mXinα and N-cadherin (data not shown). However, as shown in Fig. 2B, mXinα, mXinαRΔ-1, and β-catBR, but no other deletions, directly interact with β-catenin and give rise to positive X-gal stain. These results demonstrate that the β-catenin binding region (β-catBR) on mXinα is localized within the region from aa 533 to 746. This region was shown to be both necessary and sufficient for the interaction with β-catenin. Interestingly, this β-catenin binding region is located within the last four Xin repeats of mXinα. As shown in Fig. 2C, this β-catenin binding region was further divided into four fragments (CA, AP, PN, and CP) and used as baits in yeast two-hybrid assay to map the minimal region for binding to β-catenin. Only CA and CP baits showed positive interaction with β-catenin prey (Fig. 2C). Thus, the minimal β-catenin-binding domain resides in the CA fragment, located within aa 535–636. The aa sequence alignment among Xin proteins from chick, mouse, and human reveals high sequence identity within this β-catenin-binding domain (Fig. 2D): 57.8% between mXinα and hXinα, 53.9% between mXinα and mXinβ, and 49.0% between mXinα and cXin. The secondary structure of this β-catenin-binding domain predicted by the Chou-Fasman method (10, 11) is composed of 39.2 and 35.3% of β-sheet and α-helix amino acids, respectively. Each of them forms two stretches, organizing into a β-sheet (15 aa)-α-helix (26 aa)-β-sheet (21 aa)-α-helix (14 aa) structure.
mXinα Binds and Bundles Actin Filaments
It has been shown that recombinant Xin repeats from hXinα are capable of binding and cross-linking actin filaments (3, 7). To test whether full-length mXinα also binds to actin filaments, recombinant GST-tagged or His-tagged mXinα was used in an actin binding assay as described under “Experimental Procedures.” After high speed centrifugation (100,000 × g for 30 min), both GST-mXinα and His-mXinα are co-sedimented with actin filaments into the pellet fraction (P in Fig. 3A, tubes 1-6). Under the same conditions, GST-mXinα or His-mXinα alone remains in the supernatant (S in Fig. 3A, tubes 7 and 8). It is noted that GST-mXinα co-sedimented with actin filaments appears to be more efficient than His-mXinα. Although the exact mechanism for the higher efficiency remains unknown, the fact that GST alone is able to form dimers in solution (12) probably provides some explanation. The binding of His-mXinα to actin filaments appears to be saturable (Fig. 3B). The estimated molar ratio of actin to bound mXinα is 9.1 at saturation. It is known that purified actin filaments in solution could not be pelleted by low speed centrifugation (10,000 × g for 15 min) unless the filaments become aggregates by actin cross-linking or bundling proteins. Fig. 4 shows the results of such a low speed co-sedimentation assay. An increased amount of recombinant GST-mXinα can proportionally aggregate actin filaments (Fig. 4, lanes 2P-6P). Under this condition, GST-mXinα alone remained in the supernatant (Fig. 4, lane 7S), actin filaments alone could not be sedimented (lane 1P), and recombinant GST by itself (lane 8P) or bovine serum albumin (lane 9P) also could not aggregate actin filaments into the pellet (Fig. 4). Similarly, we also found that His-mXinα was able to aggregate actin filaments, as determined by a low speed co-sedimentation assay (supplemental Fig. 1). These results suggest that full-length mXinα is capable of either cross-linking or bundling the actin filaments.
FIGURE 3. Actin binding of purified recombinant His-mXinα and GST-mXinα.
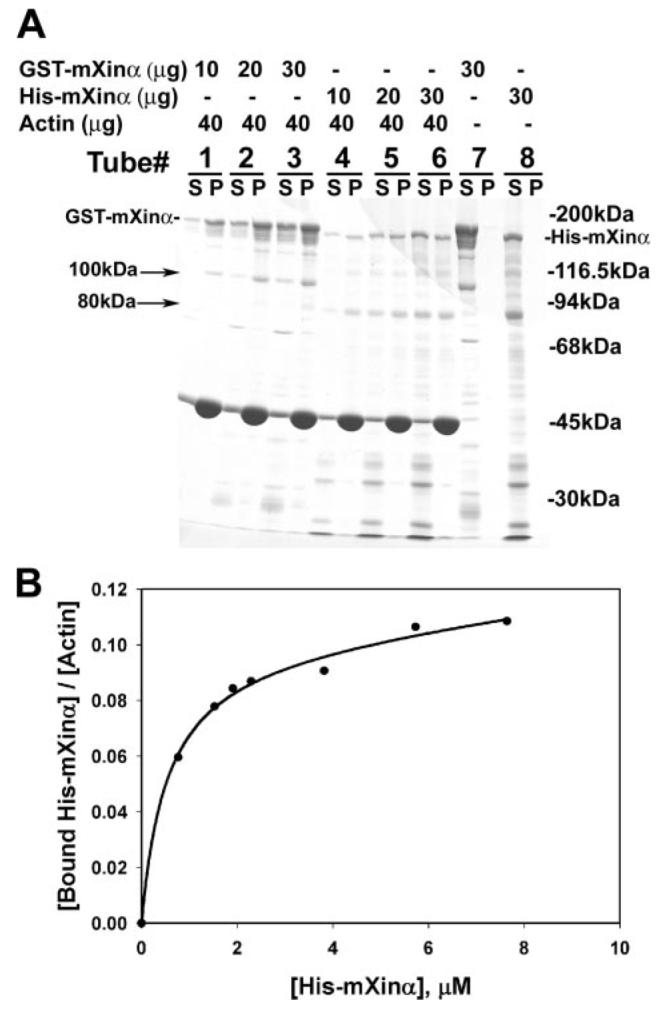
A, SDS-PAGE analysis of GST-mXinα and His-mXinα. An actin binding assay was performed based on the co-sedimentation method at 100,000 × g centrifugation. Under this condition, both GST-mXinα (tube 7) and His-mXinα (tube 8) alone remained in the supernatant (S). In the presence of muscle actin filaments, an increasing amount of GST-mXinα (tubes 1–3) and His-mXinα (tubes 4–6) was co-sedimented in the pellet (P). Numerous minor fragments recognized by anti-mXinα antibody (data not shown) represent degraded products during purification. Particularly, a 100-kDa band from GST-mXinα and a 80-kDa band from His-mXinα are also co-pelleted with actin filaments. The apparent difference in binding affinity between GST-mXinα and HismXinα may be due to the ability of GST by itself to form dimers (12). B, actin binding curve of His-mXinα. The binding of His-mXinα to actin was determined by quantifying stained gels of both supernatants and pellets. The amounts of bound His-mXinα/mol of actin were calculated and plotted against total concentrations of His-mXinα using the SigmaPlot 9.0 computer program. The binding reached saturation at an actin to His-mXinα molar ratio of 9.1:1.
FIGURE 4. SDS-PAGE analysis of actin aggregates by GST-mXinα.
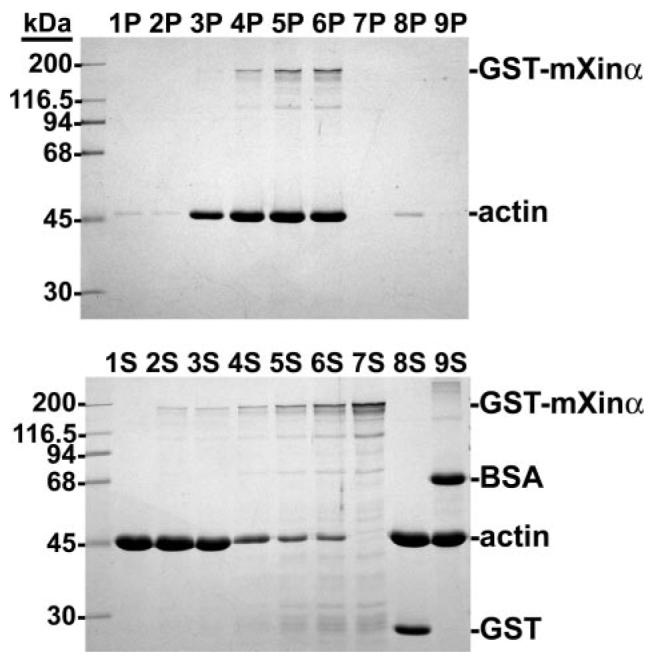
An actin binding assay was performed with increasing amounts of GST-mXinα as described in the legend to Fig. 3, except that low speed centrifugation (10,000 × g for 15 min) was used. Under this centrifugation condition, a trace amount of actin was pelleted from the tubes containing actin alone (lane 1P), actin plus 2 μg of GST-mXinα (lane 2P), actin plus GST (lane 8P), or actin plus bovine serum albumin (BSA)(lane 9P). However, GST-mXinα at 5 μg(lane 3P) started to bring down a detectable amount of actin aggregates. The more GST-mXinα (10 μg in lane 4P and 20 μg in lane 5P) that was added, the more actin aggregates were present. GST-mXinα at 30 μg (lane 6P) did not bring down more actin aggregates, suggesting that a saturation point was reached. Under this assay condition, 30 μg of GST-mXinα alone remained in the supernatant (lane 7S).
To distinguish these two activities, negatively stained actin filaments in the absence and presence of recombinant mXinα were examined under the electron microscope. Actin filaments alone are thin and long with an average diameter of 6–8 nm (Fig. 5A). At a ratio of actin to recombinant His-mXinα of 5:1, actin filaments were aggregated into side-by-side bundles (Fig. 5B). Similar bundling activity was also observed with GST-mXinα (Fig. 5C). Furthermore, the size of the bundles that form paralleled the concentration of His-mXinα used. At the molar ratio of actin to His-mXinα of 10:1 (black bars in Fig. 5D), the average bundle sizes formed after 13 and 48 h of incubation were significantly smaller than that formed at the ratio of 5:1 (hatched bars in Fig. 5D, p < 0.05), suggesting that the bundling reaction is mXinα concentration-dependent. Although there was no transverse band observed in these actin bundles, at higher concentrations of His-mXinα (actin/mXinα ratio of 1:2), individual actin filaments appeared to be decorated by mXinα with a periodicity of 36.0 ± 0.4 nm (n = 129) (Fig. 5E). The nature and significance of this periodicity remain to be determined.
FIGURE 5. Characterization of actin bundles formed by His-mXinα and GST-mXinα.
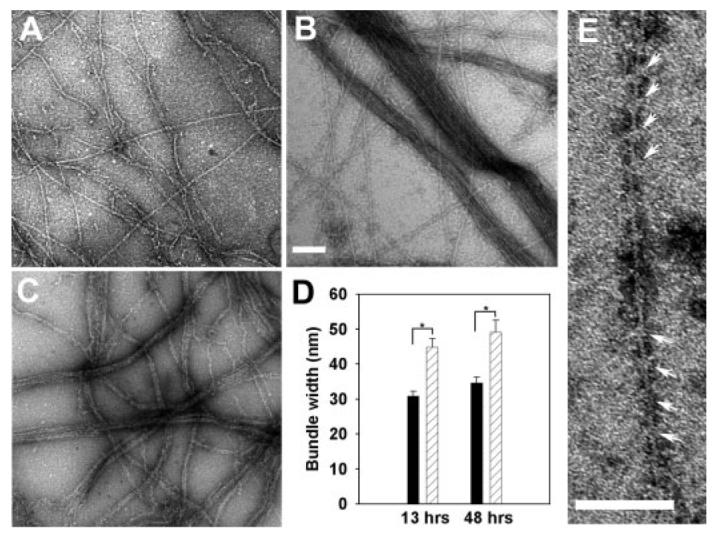
Electron microscopic images of actin alone (A), actin bundles formed by His-mXinα (B), and actin bundles formed by GST-mXinα (C). A–C, actin alone (2.4 μm) or actin mixed with recombinant mXinα (0.48 μm) was applied onto a grid, negatively stained by 1% uranyl acetate, and then observed under an electron microscope. Bar, 100 nm. D, the sizes of actin bundles formed by two different concentrations of His-mXinα were measured from randomly selected micrographs and compared. The average bundle size (width in nm) was affected by the concentration of mXinα in the mixture, although actin concentration was kept the same (2.4 μm). Black bar, 0.24 μm His-mXinα; hatched bar, 0.48 μm His-mXinα;*, p < 0.05 by rank sum test; error bar, S.E. E, actin filaments that had not been included into the bundles were decorated by His-mXinα, forming 36-nm periodicity marked by arrows. Bar, 100 nm.
The β-Catenin-binding Domain and the C-terminal Half of mXinα Prevents Ectopically Expressed mXinα from Localizing to Stress Fibers within C2C12 Myoblasts
Recombinant mXinα and its Xin repeat region bind and aggregate actin filaments in vitro; however, in the adult mouse heart, the mXinα protein does not associate with actin thin filaments; instead, mXinα preferentially localizes to the intercalated discs. Undifferentiating C2C12 myoblasts do not express mXinα (13); however, upon differentiation, mXinα expression in myotubes is localized to a few stress fibers and near the periphery of the cell (6). The protein domain responsible for its localization was investigated in the C2C12 cells by transient transfections with plasmids expressing GFP fused to mXinα or to its various deletion mutants. Control C2C12 myoblasts transfected with pEGFP-C2 vector alone showed diffusely distributed GFP at the perinuclear region and in the nucleus (Fig. 6A). In contrast, myoblasts transfected with pEGFP-mXinα exhibited some GFP-mXinα fusion protein localized to stress fibers (Fig. 6B), whereas cells transfected with pEGFP-mXinαRΔ-3, which lacks all 15 Xin repeats and the β-catenin-binding domain, showed very little stress fiber localization and mostly a diffuse distribution of GFP-mXinαRΔ-3 (Fig. 6C). When the C-terminal deletion construct was used, pEGFP-mXinαCΔ, which also lacks the β-catenin-binding domain as well as the C-terminal proline-rich region, transfected cells demonstrated increased stress fiber localization and peripheral localization of GFP-mXinαCΔ (Fig. 6D). Nearly 100% of pEGFP-mXinαCΔ-transfected myoblasts had clearly visible stress fiber and peripheral staining, whereas only <50% of myoblasts transfected with other plasmids exhibited such localization. All transfected proteins associated with stress fibers colocalized with either vinculin or phalloidin within myoblasts (data not shown). Interestingly, cells transfected with pEGFP-mXinαRΔ-3, lacking all 15 Xin repeats and β-catenin-binding domain, appeared to have inhibited cell spreading ability and a smaller cell size. The differences in cell size and shape were further characterized in transiently transfected CHO cells using the two-dimensional dynamic image analysis software program (14-16). Transfection of the full-length mXinα (pEGFP-mXinα) did not appear to affect cell size and shape compared with the control pEGFP-C2 vector transfection, as measured through mean cell area (μm2), perimeter (μm), or roundness (percentage) (Table 1). However, transfection of pEGFP-mXinαRΔ-3 significantly reduced cell area and roundness as compared with the full-length or control vectors (Table 1). These results suggest that the β-catenin-binding domain together with the C terminus of mXinα may inhibit stress fiber localization of expressed GFP-mXinα. The Xin repeats appeared to be important for cell shape and size determination.
FIGURE 6. Immunofluorescence microscopy of transiently transfected C2C12 myoblasts.
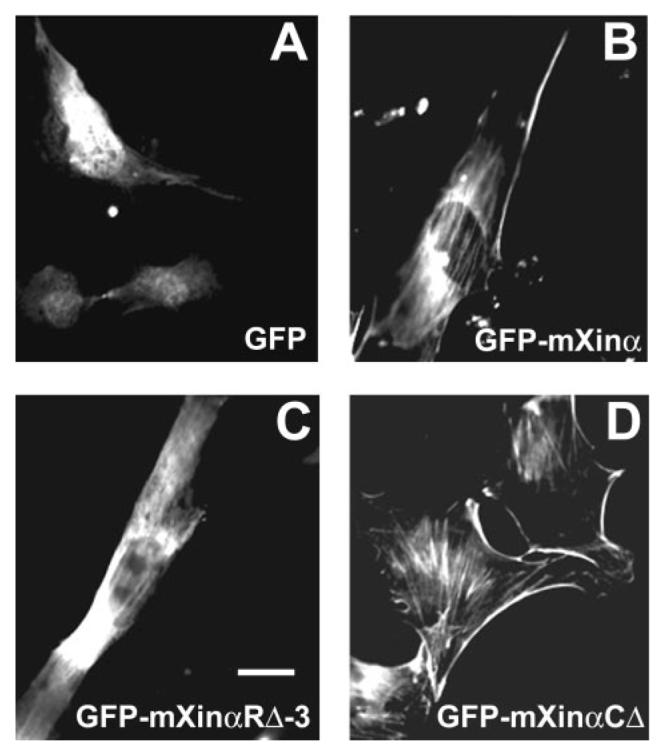
Myoblasts maintained under growth conditions were transiently transfected with pEGFP-C2 vector alone (A), pEGFP-mXinα (B), pEGFP-mXinαRΔ-3 (C), or pEGFP-mXinαCΔ (D). Twenty-four hours post-transfection, cells were fixed and processed for fluorescence microscopy to view GFP-tagged protein expression. Force-expressed GFP-mXinα showed diffuse staining, peripheral, and stress fiber localizations (B), whereas most of force-expressed GFP-mXinαRΔ-3 (lacking all 15 Xin repeats) were diffusely distributed within the cells with a very few stress fiber localization (C). On the other hand, cells transfected with pEGFP-mXinαCΔ (having 10 Xin repeats but lacking β-catenin-binding domain and the C terminus) showed much more GFP signal associated with stress fibers and cell periphery than wild type GFP-mXinα (D), suggesting a possible inhibition of stress fiber localization by the β-catenin-binding domain and the C terminus. The GFP signal in the cells transfected with empty vector was observed mainly in the nucleus and perinuclear regions (A). Bar, 50 μm.
TABLE 1. Computer assisted measurements of cell size and shape in transfected CHO cells.
Student's t test was performed to calculate the p value if the samples passed the test of normality and equal variance. Otherwise, a rank sum test was used. NS, not significant (p > 0.05).
| Transfected plasmid | Number of cells | Mean area | Mean perimeter | Roundnessa |
|---|---|---|---|---|
| μm2 | μm | % | ||
| pEGFP-C2 (a) | 134 | 1,176.8 ± 676.8 | 172.4 ± 55.8 | 49.5 ± 16.1 |
| pEGFP-mXinα(b) | 115 | 1,162.7 ± 712.8 | 173.3 ± 50.7 | 47.1 ± 14.2 |
| pEGFP-mXinαRΔ-3 (c) | 126 | 858.9 ± 538.7 | 159.1 ± 46.1 | 42.3 ± 16.2 |
| p value: a versus b | NS | NS | NS | |
| p value: a versus c | 0.0001 | NS | 0.0001 | |
| p value: b versus c | 0.0001 | 0.03 | 0.004 |
Roundness is a percentage measure of how efficiently a given amount of perimeter encloses the area. A circle has the largest area and has a roundness of 100%.
Function of the Xin Repeats in Stress Fiber Localization
In order to determine the function of the Xin repeats in mXinα, CHO cells expressing GFP fused to mXinα or to its various deletion mutants were counterstained with monoclonal anti-vinculin antibody and rhodamine-conjugated secondary antibody and then analyzed under the fluorescence microscope. A representative GFP image and its merged image with vinculin localization from each transfected cell line are shown in Fig. 7. The vinculin staining at the focal adhesion site was used to confirm the stress fiber localization of GFP fusion proteins. It is clear that an increasing association of GFP fusion protein with stress fibers is seen in the following order: GFP-mXinαRΔ-3 (Fig. 7, A and B) < GFP-mXinαRΔ-1 (Fig. 7, C and D) < GFP-mXinαRΔ-2 (Fig. 7, E and F) < GFP-mXinα (Fig. 7, G and H) < GFP-mXinαCΔ (Fig. 7, I and J). These results are consistent with that obtained from transfected C2C12 myoblasts (Fig. 6).
FIGURE 7. Immunofluorescence microscopy of CHO cells transfected with plasmids for GFP fused to either full-length mXinα or various mXinα deletion mutants (mXinαRΔ-1, mXinαRΔ-2, mXinαRΔ-3, and mXinαCΔ).
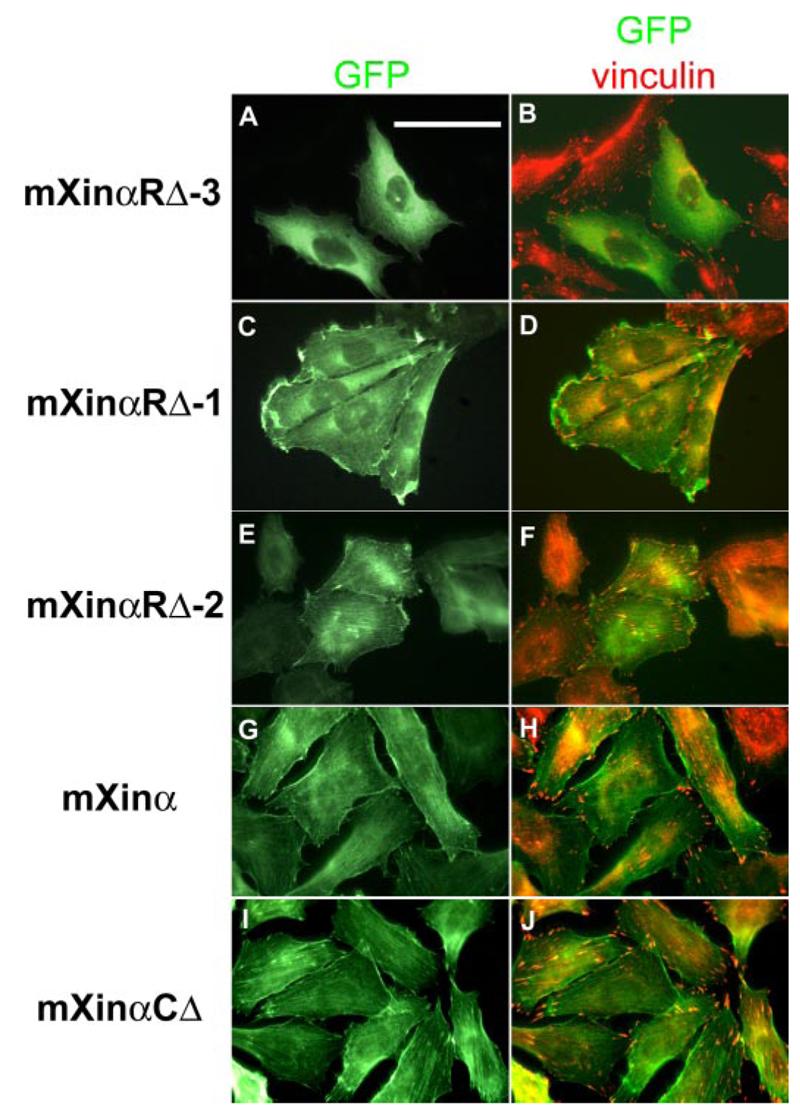
Transfected cells were processed for immunofluorescence microscopy by counterstaining with anti-vinculin primary antibody and rhodamine-conjugated secondary antibody. Because vinculin is known to localize to the focal adhesion, anti-vinculin stains and GFP signals highlight the presence of the stress fibers (B, D, F, H, and J). Cells expressing either the GFP-mXinα (G and H) or the GFP-mXinαCΔ (I and J) contained many observable stress fibers. In contrast, although cells expressing the GFP-mXinαRΔ-1 (C and D) or the GFP-mXinαRΔ-2 (E and F) exhibited a moderate decrease in the number of stress fibers observed. Most notably, cells expressing the GFP-mXinαRΔ-3 (A and B) were much smaller in size and exhibited a significant reduction in the number of observable stress fibers. These data are consistent with the quantitative data presented in Table 2. Bar, 25 μm.
For quantification, randomly selected cells from each transfected cell line were scored for the frequency of force-expressed GFP signal associated with detectable stress fibers and then grouped into four categories: group I–IV (cells with 0, 3–10, 10–20, and >20 stress fibers, respectively). As shown in Table 2, deletion of the last seven Xin repeats in mXinαRΔ-2 leads to a weaker stress fiber association (a significant increase in group II) compared with the wild-type mXinα construct with the 15 Xin repeats (p < 0.01, χ2 test). However, deletion of the first eight Xin repeats (mXinαRΔ-1) results in an even weaker association with stress fibers (a significant decrease in group II, p < 0.025, and a significant increase in group III, p < 0.0001). Force-expressed mXinαRΔ-3, which completely lacks the Xin repeats as well as the overlapping β-catenin-binding domain, totally abolishes the stress-fiber association (almost all cells are categorized into group I). Thus, the extent of stress fiber association is roughly proportional to the number of the Xin repeats present in these force-expressed proteins. However, the mXinαCΔ construct, which contains 10 Xin repeats but lacks the β-catenin-binding domain and the C terminus of the mXinα protein, exhibits an even stronger stress fiber association than the wild type mXinα with 15 Xin repeats (increased group IV cells, p < 0.025). This result again suggests that the β-catenin-binding domain and the C terminus of mXinα may play an inhibitory effect on actin association.
TABLE 2. Scoring the population of transfected CHO cells showing GFP fused to mXinα and to various deletion mutants associated with stress fibers.
CHO cells transfected with expressing plasmids for GFP fused to full-length mXinα or various deletion mutants were scored under a fluorescence microscope by counting numbers of detectable stress fibers with GFP signal per cell. Transfected cells were then classified into group I with no detectable stress fiber, group II with 3–10 stress fibers, group III with 10–20 stress fibers, or group IV with more than 20 stress fibers. The χ2 test was performed to calculate p value. NS, not significant difference; p value a, pairwise comparisons between cells transfected with plasmid for full-length mXinα and with plasmids for various deletion mutants; p value b, comparison between cells transfected with plasmid for mXinαRΔ-1 and with plasmid for mXinαRΔ-2; p value c, comparison between cells transfected with plasmid for mXinαRΔ-2 and with plasmid for mXinαCΔ.
| Transfected plasmid | Number of cells scored | Percentage of total |
|||
|---|---|---|---|---|---|
| Group I | Group II | Group III | Group IV | ||
| % | % | % | % | ||
| pEGFP-mXinα | 934 | 0.43 | 17.02 | 76.12 | 6.42 |
| pEGFP-mXinαRΔ-1 | 516 | 5.23 | 88.57 | 4.07 | 2.13 |
| p value a | NS | <0.0001 | <0.0001 | NS | |
| pEGFP-mXinαRΔ-2 | 1,066 | 0.66 | 47.65 | 50.09 | 1.60 |
| p value a | NS | <0.01 | NS | NS | |
| p value b | NS | <0.025 | <0.0001 | NS | |
| pEGFP-mXinαRΔ-3 | 1,043 | 95.78 | 4.22 | 0 | 0 |
| p value a | <0.0001 | <0.05 | <0.0001 | NS | |
| pEGFP-mXinαCΔ | 1,107 | 0 | 11.02 | 61.34 | 27.64 |
| p value a | NS | NS | NS | <0.025 | |
| p value c | NS | <0.0001 | NS | <0.0001 | |
It is known that a minimum of three Xin repeats is required for actin binding (3). Therefore, between force-expressed mXinαRΔ-1 with seven Xin repeats and mXinαRΔ-2 with eight repeats, or between mXinαRΔ-2 with eight Xin repeats and mXinαCΔ with 10 repeats, the number of Xin repeats should play a very minimal role in their stress-fiber association. The major difference between mXinαRΔ-1 and mXinαRΔ-2 or between mXinαRΔ-2 and mXinαCΔ is in the presence or absence of the β-catenin-binding domain and the C terminus, respectively. The observed, significant differences in stress fiber association for these two comparisons (p value b and p value c in Table 2) are further consistent with the inhibitory role of the β-catenin-binding domain and the C terminus in the ability of mXinα to bind actin.
In a separate yeast two-hybrid assay to identify novel mXinα interaction partners, a rat heart cDNA yeast two-hybrid library was screened using mXinα as bait. From this screen, we obtained two independent clones, pL1.192 and pL2P6, encoding aa 1–258 and 1–215, respectively, of cardiac α-actin. Interestingly, the interaction between actin and mXinαCΔ appeared to be stronger, as observed with more β-galactosidase activity (stronger blue), than that between actin and mXinα in yeast cells grown on nutritionally selective plates containing X-gal (Fig. 8A). Using liquid culture assay with O-nitrophenyl β-d-galactopyranoside as substrate for β-galactosidase to quantify the amounts of reporter gene expression after bait and prey interaction in yeasts (40), we found about 3.6-fold higher β-galactosidase expression in the cells with mXinαCΔ and actin interaction as compared with that in the cells with mXinα and actin interaction. These results are also in a good agreement with the idea that the β-catenin-binding domain and the C terminus of mXinα prevent the binding of mXinα to actin filaments.
FIGURE 8. Yeast two-hybrid assay to demonstrate the interaction of mXinα with mXinα-interacting proteins.
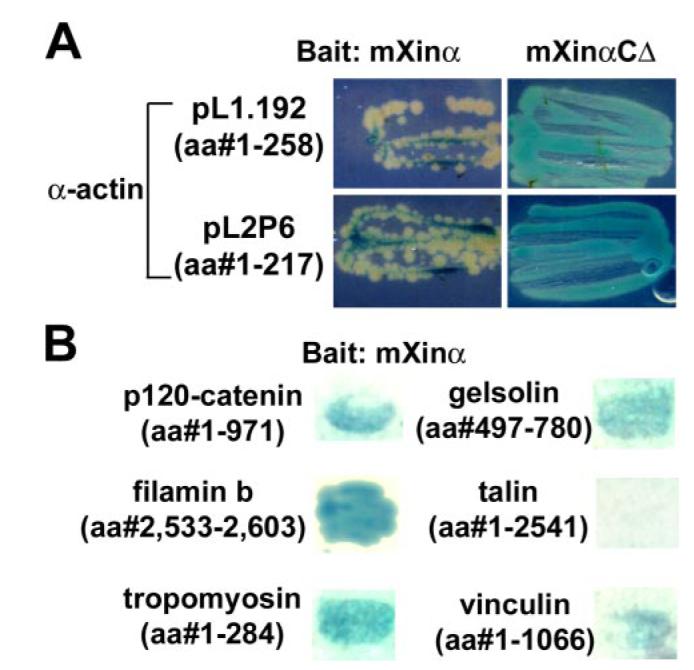
Using mXinα as bait to screen yeast two-hybrid cDNA library, cardiac α-actin and several actin-binding proteins were identified as mXinα-interacting proteins. A, the interaction between C-terminal deletion mutant mXinαCΔ and actin is stronger than that between full-length mXinα and actin. Yeast cells retransformed with either mXinα or mXinαCΔ as bait and two independent actin preys were grown on X-gal-containing selective plates for the same period of time. A stronger blue color was developed from yeast cells containing mXinαCΔ bait and either actin prey, as compared with the lighter color associated with yeast cells containing mXinα bait and either actin prey. These results imply a stronger interaction between mXinαCΔ and actin than that between mXinα and actin. B, results of a β-galactosidase filter assay for the interaction between mXinα and several mXinα-interacting proteins. After retransformation of mXinα bait and various preys into yeast cells, colonies grown on selective media were transferred to membranes for X-gal assay. It appears that mXinα interacts with the extreme C termini of filamin b (aa 2,533–2,603) and gelsolin (aa 497–780) as well as tropomyosin, p120 catenin, and vinculin but not talin, all of which contain full protein coding regions.
In addition to two cardiac α-actin clones obtained from yeast two-hybrid library screening, we have also identified several positive clones, encoding fragments of known actin-binding proteins, including filamin b (aa 2,533–2,603), muscle α-tropomyosin (full-length, aa 1–294), and gelsolin (aa 497–780). Retransformation of these prey plasmids with the mXinα bait in yeast confirmed the interactions, since all turned blue in colony lift β-galactosidase filter assays (Fig. 8B). Under the same condition, full-length p120 catenin and full-length vinculin preys also gave the positive interaction with mXinα, whereas full-length talin prey as a negative control did not interact with mXinα (Fig. 8B).
The Presence of β-Catenin Enhances mXinα Binding to Actin Filaments in Vitro
We have shown that the β-catenin-binding domain overlaps with the 12th and 13th Xin repeats and that the β-catenin-binding domain and the C terminus of mXinα together prevent mXinα from binding to actin stress fibers. These results led us to ask whether the binding of β-catenin to mXinα would release the inhibition and then enhance the actin binding. To address this question, we performed an actin binding co-sedimentation assay and a quantitative measurement of sizes of actin bundles formed by His-mXinα in the absence and presence of GST-β-catenin. As expected, in the absence of GST-β-catenin, the amounts of His-mXinα co-sedimented with actin filaments increased as increasing amounts of His-mXinα were used in the co-sedimentation assay (Fig. 9). In the presence of β-catenin, the amounts of His-mXinα co-pelleted with actin appeared to increase about 2-fold (Fig. 9). The effects of β-catenin on the actin-bundling activity of mXinα were further analyzed by negative staining electron microscopy. In this experiment, His-mXinα alone or an equal molar mixture of His-mXinα and GST-β-catenin were preincubated on ice for 2 h and then mixed with actin filaments. At different time points (1.5, 8, and 30 min), aliquots from the mixture were applied onto Formvar grids and processed for negative staining. Samples were observed under a JEOL-1230 electron microscope, and 50 micrographic pictures were randomly taken from each sample. The width of all the bundles from the micrographs was quantified using the ImageJ program. Substitution of GST-β-catenin in the mixture by GST was used as a negative control. With an increase in the incubation time, actin bundles formed by mXinα alone or a combination of mXinα and β-catenin increased in both numbers and sizes (Fig. 10). GST did not have significant effects on the actin-bundling activity of mXinα (data not shown). However, it is clear that in the presence of β-catenin, many more bundles were formed at a given time point, and also bigger bundles appeared more frequently (Fig. 10). Thus, these results together support the idea that β-catenin binding to mXinα releases the inhibition of actin binding imposed by the β-catenin-binding domain and the C terminus and then facilitates the actin binding/bundling activity of mXinα.
FIGURE 9. Effect of GST-β-catenin on the binding of His-mXinα to actin filaments.
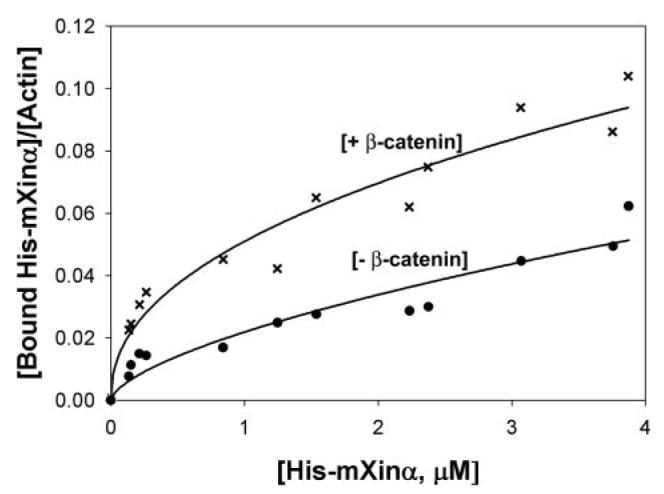
An actin binding assay was performed with increasing amounts of His-mXinα in the absence (−) or presence (+) of GST-β-catenin (1.95 μm). After quantification of His-mXinα in the pellet and supernatant fractions, data were plotted using SigmaPlot 9.0. The presence of β-catenin appears to enhance the binding of mXinα to actin filaments. The experiments were repeated three times, and representative data are shown here.
FIGURE 10. Actin bundle formation was accelerated by the presence of GST-β-catenin.
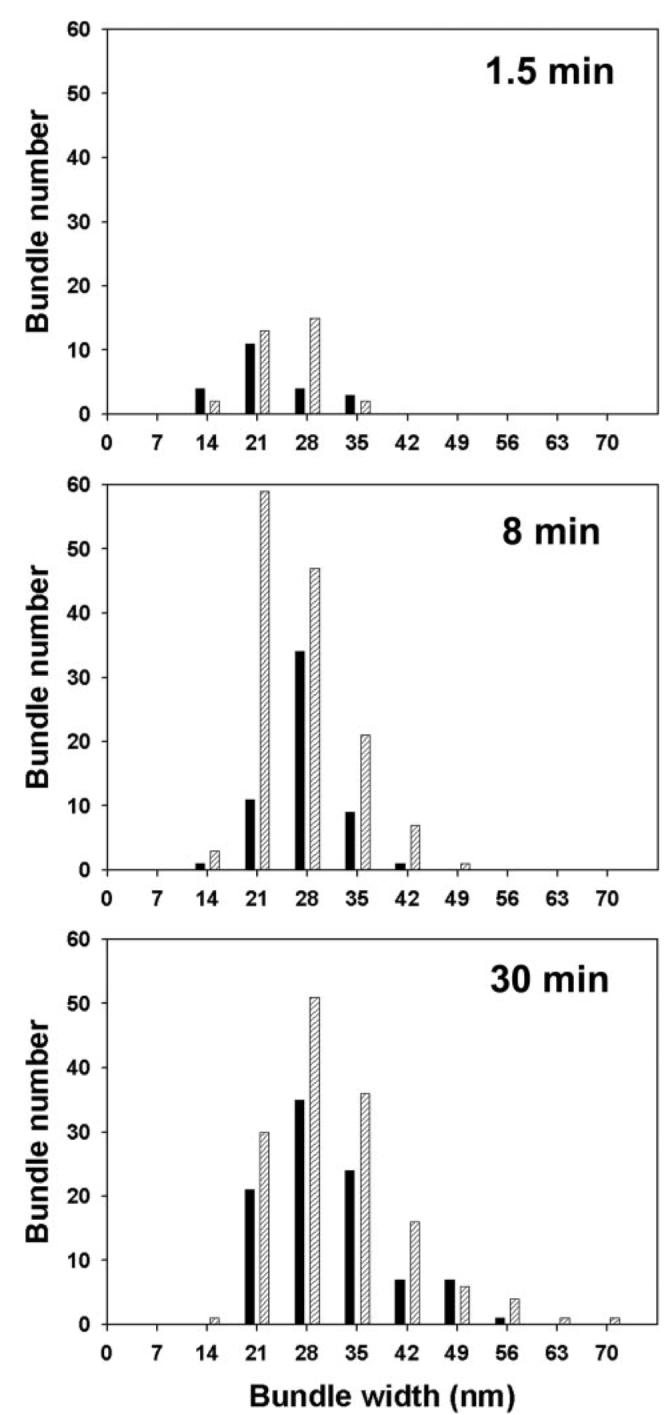
His-mXinα alone or mixed with an equal molar amount of GST-β-catenin (0.24 μm) was incubated on ice for 2 h. The proteins were then mixed with actin (2.4 μm) and incubated at room temperature. At the time points indicated in the figure, 10-μl aliquots were taken and applied onto grids for negative staining. 50 electron micrographs were randomly captured from each grid. The widths of all of the bundles in the micrograph were measured and presented by histograms. Black bar, without GST-β-catenin; hatched bar, with GST-β-catenin. During the assay period, the number and size of bundles formed are time-dependent. The bundles formed by mXinα in the presence of β-catenin appear to be more numerous and bigger than that formed by mXinα alone.
DISCUSSION
Model for How mXinα Functions at the Adherens Junction of the Heart
In the present study, we have shown that β-catenin is capable of directly interacting with aa 535–636 of mXinα. Through this interaction, mXinα can subsequently enhance its ability to bind and to bundle actin filaments. Furthermore, through this interaction, mXinα can then associate with the N-cadherin-β-catenin-p120 catenin adhesion complex and play a pivotal role in vivo, as evidenced by the cardiac defects observed in the mXinα-null mice (5). The finding that β-catenin-binding domain on mXinα overlaps with actin binding Xin repeats provides a plausible explanation why mXinα is not associated with the actin filaments of sarcomeres, although it contains 15 Xin repeats, which constitute multiple actin binding sites. Instead, mXinα is preferentially localized to the intercalated disc of the heart. The results presented here further imply that newly synthesized mXinα may be present in an autoinhibited state, as far as actin binding is concerned, until the β-catenin-binding domain is occupied by the β-catenin. The binding of mXinα to β-catenin at the adherens junction would then change its confirmation into an open state, which would enable subsequent actin binding and bundling (Fig. 11). In this regard, mXinα is an integral component of adherens junctions at the intercalated disc, where it links the N-cadherin-mediated adhesion complex to the actin cytoskeleton. In the present study, we have also provided strong evidence to support the proposed two-state model for mXinα interaction with actin. First, a deletion of the β-catenin-binding domain and the C terminus found in mXinαCΔ mutant allows the Xin repeats to be more readily exposed to bind actin and relieves the inhibition imposed by the deleted C-terminal fragment. As a result, mXinαCΔ strongly enhances its ability to interact with actin in yeast and to associate with actin stress fibers in both transfected C2C12 and CHO cells. Second, in the presence of β-catenin, both actin binding and bundling activities are significantly increased.
FIGURE 11. Schematic model for how mXinα functions at the adherens junction.
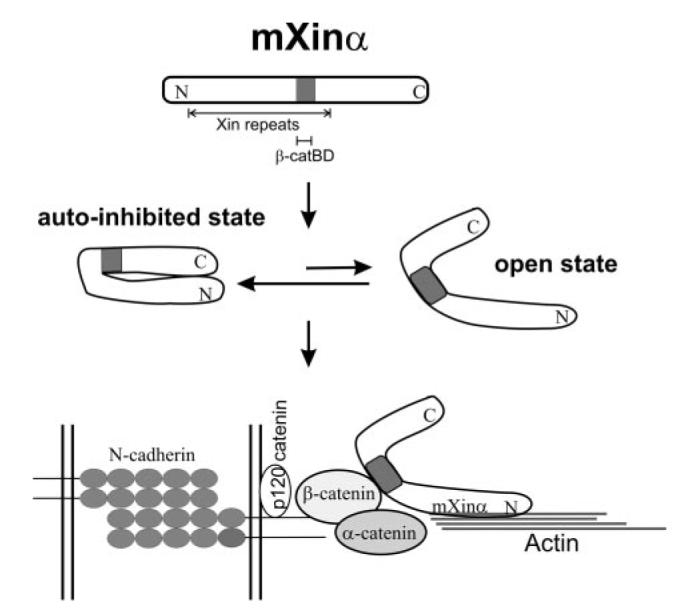
The full-length mXinα molecule containing 15 Xin repeats exists in equilibrium between an autoinhibited state and open state, favoring the autoinhibited state. β-Catenin located at the adherens junction region binds to the β-catenin-binding domain (β-catBD) overlapping with the 12th and 13th Xin repeats, and then the equilibrium shifts toward the open state of the mXinα molecule, which facilitates subsequent binding/bundling of actin filaments. Thus, mXinα is an integral component that links N-cadherin-mediated adhesion to the actin cytoskeleton.
Adherens junctions at the intercalated disc function to connect the mature myocytes and provide the attachment sites at the membrane for myofibrils (17). This function is dependent upon the assembly of the cadherin-catenin complex (18). The highly conserved cytoplasmic domain of N-cadherin binds to β-catenin and/or plakoglobin (γ-catenin). p120-catenin, a distant relative of β-catenin, binds to the juxtamembrane region of cadherin and regulates cadherin turnover (19). In a classic view, an actin-bundling protein, α-catenin, then binds β-catenin to organize the adhesion complex that links to the actin cytoskeleton, either via a direct association with actin filaments or through an indirect association with actin-binding proteins, including vinculin and α-actinin (20). This stable linkage role for α-catenin has been recently proven to not exist in epithelial cells; instead, compelling evidence suggests that α-catenin is a molecular switch that binds E-cadherin-β-catenin and regulates actin dynamics at the adherens junctions of epithelial cells (21-23). The component that then in fact makes this connection in epithelial cells remains unclear. In this study, we have shown that the mXinα protein is a potent actin-bundling protein, which can additionally interact with β-catenin. Furthermore, β-catenin effectively enhances the binding of mXinα to actin filaments. mXinα localizes to the adherens junction in the intercalated disc of the heart (6). Thus, it is likely that mXinα provides a link between N-cadherin and actin cytoskeleton in cardiomyocytes. Supporting this linkage role of mXinα, mXinα-null mouse hearts begin with abnormal intercalated disc ultrastructure as early as 3 months of age and exhibit cardiac hypertrophy and cardiomyopathy (5). This structural alteration is accompanied by a disorganization of myofibrils at the intercalated disc and by a significant decrease in the expression of N-cadherin, β-catenin, and p120-catenin (5), suggesting that hypertrophy may be due to impaired organization of the intercalated disc and instability of cell-cell adhesion. Although the lack of mXinα protein in the intercalated discs is the most straightforward explanation for the observed cardiac defects, there might still be other possibilities, such as the up-regulation of mXinβ in mXinα-null heart for partially contributing to the observed phenotype. We have shown here that overexpression of mutant mXinαRΔ-3 protein but not the wild-type mXinα protein in CHO cells altered cell size and shape. Therefore, it is unlikely that the up-regulation of wild-type mXinβ in the mXinα-null heart would be the major factor causing the observed cardiac defects.
mXinα Contains a Novel β-Catenin-binding Domain
The amino acid sequence from 613 to 685 of mXinα, which overlaps slightly with the identified β-catenin-binding domain (aa 535–636), shows a 30% sequence identity (39% similarity) to aa 2237–2305 of adenomatous polyposis coli, a known β-catenin-binding protein involved in canonical Wnt signaling (24). Despite this similarity, adenomatous polyposis coli does not use this region to bind β-catenin. Instead, the β-catenin-binding domain on adenomatous polyposis coli has been mapped to aa 1014–1210, containing three 15-aa repeats (25-27). Moreover, the β-catenin-binding domain of mXinα is different from the β-catenin-binding domains found in axin (28-30), α-catenin (31), N-cadherin (32), and Tcf (33, 34). Therefore, mXinα possesses a novel β-catenin-binding domain. As shown in Fig. 2D, this β-catenin-binding domain on mXinα is highly conserved among all Xin proteins (49–57.8% identity). In the chicken heart, cXin has been shown to localize to the intercalated disc (13) and to associate with the N-cadherin-β-catenin complex (6). We have also shown that the mXinβ messages and proteins localize to the intercalated disc of the mouse heart (5). Moreover, an up-regulation of mXinβ at the message and protein levels associated with the targeted deletion of mXinα suggests a functional compensation between mXinα and mXinβ in the heart (5). Thus, it is likely that the β-catenin-binding domains on cXin and mXinβ exist and are also functional.
mXinα Bundles Actin Filaments
A previous study with recombinant His-Xin repeats from hXinα has demonstrated that three Xin repeats are necessary and sufficient to bind actin filaments and that the Xin repeats are capable of aggregating actin filaments into loosely packed meshwork (3, 7). This suggests that the Xin repeats cross-link actin filaments. At saturation conditions, the actin to Xin repeat fragment molar ratio is 4:1 for 16 repeats, 2:1 for six repeats, or 1:1 for three repeats (3). However, using recombinant full-length mXinα in this study, we clearly demonstrate that mXinα aggregates actin filaments into bundles, which can be sedimented at low speed centrifugation (10,000 × g for 15 min). This bundling activity is not attributed to the fusion tags on mXinα. Both GST- and His-mXinα show similar bundling activity, and GST by itself does not have any actin binding/bundling activity. At saturation, one molecule of His-mXinα binds to nine molecules of actin monomers. The bundling reaction of His-mXinα appears to be concentration- and time-dependent, and unlike another actin cross-linking protein, filamin (35), His-mXinα does not cross-link actin filaments into a loosely packed network in all of the concentration ranges we tested (the molar ratios of mXinα to actin tested ranging from 2:1 to 1:200) (supplemental Fig. 2). The differences in observed actin binding properties (stoichiometry and cross-linking versus bundling) between the Xin repeats alone and full-length mXinα suggest that a small portion (aa 1–72) of the N-terminal fragment upstream of the Xin repeats and/or the C-terminal fragment (aa 747–1129) downstream of the Xin repeats are also important in terms of organizing the actin cytoskeleton. Neither actin polymerization activity nor G-actin binding activity is associated with recombinant Xin repeats (3). However, at the present time, we do not know whether or not full-length mXinα has these activities.
In yeast two-hybrid cDNA library screenings, we have obtained two clones encoding cardiac α-tropomyosin, one clone encoding gelsolin, and one clone encoding filamin b, in addition to two clones encoding different fragments of cardiac α-actin. These aforementioned proteins are known to be actin-binding proteins, either functioning to stabilize or regulate actin dynamics and organization in the cells (36). Therefore, it is likely that full-length mXinα may function through these interacting and actin-binding proteins to regulate its bundling activity. In the present study, we have shown that mXinα interacts with filamin b, which is broadly expressed in many tissues and cells, including heart and C2C12 cells (37), as compared with the striated muscle-enriched filamin c (35). Since the mXinα-a isoform has exactly the same sequence of mXinα, except a substitution of the 2 residues at the C terminus with 683 residues (5), we expect that the mXinα-a also interacts with filamin b. The mXinα/mXinα-a binding site on filamin b appears to be located within aa 2,533–2,603 at the end of filamin b, which also has high amino acid sequence identity (70%) to filamin c. Previously, using muscle-specific Ig domain 20 from filamin c as a bait in yeast two-hybrid screens, van der Ven et al. (8) identified a binding partner containing the last 158 amino acid residues from the hXinα-a (human homolog of mXinα-a). However, this sequence is not present in the mXinα isoform. Therefore, mXinα can only interact with filamin b, whereas mXinα-a can use the same site to bind both filamin b and filamin c, as well as use the extra site to bind muscle-specific filamin c.
The Molecular Mechanism of Actin Bundling by Full-length mXinα Remains to be Determined
Using chemical cross-linking assays, it is known that recombinant Xin repeats by themselves do not form polymer to cross-link actin filaments (3). On the other hand, using electron microscopy and the iterative helical real space reconstruction to visualize complexes of actin filaments with recombinant Xin repeats, it is reported that the Xin repeats can bind to actin filaments in two distinct modes; in the side mode, residues from subdomains 3 and 4 of one actin protomer and residues from subdomains 1 and 2 of the adjacent actin protomer were the Xin contact sites, and in the front mode, residues 22–27 and 340–345 of subdomain 1 provide the Xin-binding sites. Thus, each actin molecule contains multiple binding sites for the Xin repeats (7), which together with multiple Xin repeats in mXinα would allow mXinα to bundle actin filaments, in a similar manner as nebulin (38, 39) and nebulette (7). The data presented here provide insight into the role of mXinα in organizing the actin cytoskeleton and in linking between the adherens junction and the actin cytoskeleton in cardiomyocytes. Future studies investigating these and other mXinα-interacting and actin-binding proteins may shed additional light into the precise molecular mechanisms by which mXinα functions in the heart.
Supplementary Material
Acknowledgments
We thank Drs. Janne Balsamo and Jack Lilien (University of Iowa) for providing β-catenin, p120 catenin, and N-cadherin cDNAs; Dr. Wolfgang H. Goldmann (University of Erlangen, Germany) for providing mouse full-length vinculin cDNA; and Dr. Richard O. Hynes (Massachusetts Institute of Technology) for providing mouse full-length talin cDNA. We thank Laura Heise (University of Iowa Summer Research Internship program) for excellent help in immunofluorescence analysis.
Footnotes
This work was supported by National Institutes of Health Grant HL075015.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1 and 2.
The abbreviations used are: aa, amino acid(s); X-gal, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside; IP, immunoprecipitation; GST, glutathione S-transferase; CHO, Chinese hamster ovary; β-catBR, β-catenin binding region.
REFERENCES
- 1.Lin JJ-C, Gustafson-Wagner EA, Sinn HW, Choi S, Jaacks SM, Wang D-Z, Evans SM, Lin JL-C. J. Med. Sci. 2005;25:215–222. doi: 10.1901/jaba.2005.25-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang D-Z, Reiter RS, Lin JL-C, Wang Q, Williams HS, Krob SL, Schultheiss TM, Evans S, Lin JJ-C. Development. 1999;126:1281–1294. doi: 10.1242/dev.126.6.1281. [DOI] [PubMed] [Google Scholar]
- 3.Pacholsky D, Vakeel P, Himmel M, Lowe T, Stradal T, Rottner K, Furst DO, van der Ven PFM. J. Cell Sci. 2004;117:5257–5268. doi: 10.1242/jcs.01406. [DOI] [PubMed] [Google Scholar]
- 4.Huang H-T, Brand OM, Mathew M, Ignation C, Ewen EP, McCalmon SA, Naya FJ. J. Biol. Chem. 2006;281:39370–39379. doi: 10.1074/jbc.M603244200. [DOI] [PubMed] [Google Scholar]
- 5.Gustafson-Wagner E, Sinn HW, Chen Y-L, Wang D-Z, Reiter RS, Lin JL-C, Yang B, Williamson RA, Chen J, Lin C-I, Lin JJ-C. Am. J. Physiol. 2007;293:H2680–H2692. doi: 10.1152/ajpheart.00806.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sinn HW, Balsamo J, Lilien J, Lin JJ-C. Dev. Dyn. 2002;225:1–13. doi: 10.1002/dvdy.10131. [DOI] [PubMed] [Google Scholar]
- 7.Cherepanova O, Orlova A, Galkin VE, van der Ven PFM, Furst DO, Jin J-P, Egelman EH. J. Mol. Biol. 2006;356:714–723. doi: 10.1016/j.jmb.2005.11.082. [DOI] [PubMed] [Google Scholar]
- 8.van der Ven PFM, Ehler E, Vakeel P, Eulitz S, Schenk JA, Milting H, Micheel B, Furst DO. Exp. Cell Res. 2006;312:2154–2167. doi: 10.1016/j.yexcr.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 9.Novy RE, Sellers JR, Liu L-F, Lin JJ-C. Cell Motil. Cytoskel. 1993;26:248–261. doi: 10.1002/cm.970260308. [DOI] [PubMed] [Google Scholar]
- 10.Chou PY, Fasman GD. Adv. Enzymol. 1978;47:45–148. doi: 10.1002/9780470122921.ch2. [DOI] [PubMed] [Google Scholar]
- 11.Chou PY. Prediction of Protein Structural Classes from Amino Acid Composition: Prediction of Protein Structure and the Principles of Protein Conformation. Plenum Press; New York: 1990. pp. 549–586. [Google Scholar]
- 12.Ji X, Zhang P, Armstrong RN, Gilliland GL. Biochemistry. 1992;31:10169–10184. doi: 10.1021/bi00157a004. [DOI] [PubMed] [Google Scholar]
- 13.Lin JJ-C, Wang D-Z, Reiter RS, Wang Q, Lin JL-C, Williams HS. In: Formation of the Heart and Its Regulation. Tomanek RJ, Runyan R, editors. Birkhauser; Boston, MA: 2001. pp. 75–96. [Google Scholar]
- 14.Soll DR. Int. Rev. Cytol. 1995;8:439–454. [PubMed] [Google Scholar]
- 15.Soll DR, Voss E. In: Motion Analysis of Living Cells. Soll D, Wessels D, editors. Wiley-Liss; New York: 1998. pp. 25–52. [Google Scholar]
- 16.Li Y, Lin JJ-C, Reiter RS, Daniels K, Soll DR, Lin JJ-C. J. Cell Sci. 2004;117:3593–3604. doi: 10.1242/jcs.01216. [DOI] [PubMed] [Google Scholar]
- 17.Ong L-L, Kim N, Mima T, Cohen-Gould L, Mikawa T. Dev. Biol. 1998;193:1–9. doi: 10.1006/dbio.1997.8775. [DOI] [PubMed] [Google Scholar]
- 18.Hertig CM, Butz S, Koch S, Eppenberger-Eberhardt M, Kemler R, Eppenberger HM. J. Cell Sci. 1996;109:11–20. doi: 10.1242/jcs.109.1.11. [DOI] [PubMed] [Google Scholar]
- 19.Reynolds AB, Roczniak-Ferguson A. Oncogene. 2004;23:7947–7956. doi: 10.1038/sj.onc.1208161. [DOI] [PubMed] [Google Scholar]
- 20.Pokutta S, Weis WI. Curr. Opin. Struct. Biol. 2002;23:255–262. doi: 10.1016/s0959-440x(02)00318-4. [DOI] [PubMed] [Google Scholar]
- 21.Yamada S, Pokutta S, Drees F, Weis WI, Nelson WJ. Cell. 2005;123:889–901. doi: 10.1016/j.cell.2005.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drees F, Pokutta S, Yamada S, Nelson WJ, Weis WI. Cell. 2005;123:903–915. doi: 10.1016/j.cell.2005.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gates J, Peifer M. Cell. 2005;123:769–772. doi: 10.1016/j.cell.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 24.Nelson WJ, Nusse R. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Science. 1993;262:1731–1734. doi: 10.1126/science.8259518. [DOI] [PubMed] [Google Scholar]
- 26.Shih I-M, Yu J, He T-C, Vogelstein B, Kinzler KW. Cancer Res. 2000;60:1671–1676. [PubMed] [Google Scholar]
- 27.Su L-K, Vogelstein B, Kinzler KW. Science. 1993;262:1734–1737. doi: 10.1126/science.8259519. [DOI] [PubMed] [Google Scholar]
- 28.Behrens J, Jerchow B-A, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Science. 1998;280:586–599. doi: 10.1126/science.280.5363.596. [DOI] [PubMed] [Google Scholar]
- 29.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xing Y, Clements WK, Kimelman D, Xu W. Genes Dev. 2003;17:2753–2764. doi: 10.1101/gad.1142603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pokutta S, Weis WI. Mol. Cell. 2000;5:533–543. doi: 10.1016/s1097-2765(00)80447-5. [DOI] [PubMed] [Google Scholar]
- 32.Sadot E, Simcha I, Shtutman M, Ben-Ze'ev A, Geiger B. Proc. Natl. Acad. Sci. U. S. A. 1998;95:15339–15344. doi: 10.1073/pnas.95.26.15339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Nature. 1996;382:638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- 34.Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. Cell. 1996;86:391–399. doi: 10.1016/s0092-8674(00)80112-9. [DOI] [PubMed] [Google Scholar]
- 35.Stossel TS, Condeelis J, Cooley L, Hartwig JH, Noegel A, Schleicher M, Shapiro SS. Nat. Rev. Mol. Cell. Biol. 2001;2:138–145. doi: 10.1038/35052082. [DOI] [PubMed] [Google Scholar]
- 36.Pollard TD, Blanchoin L, Mullins RD. Annu. Rev. Biophys. Biomol. Struct. 2000;29:545–576. doi: 10.1146/annurev.biophys.29.1.545. [DOI] [PubMed] [Google Scholar]
- 37.van der Flier A, Kuikman I, Kramer D, Geerts D, Kreft M, Takafuta T, Shapiro SS, Sonnenberg A. J. Cell Biol. 2002;136:361–376. doi: 10.1083/jcb.200103037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonsior SM, Guautel M, Hinssen H. J. Muscle Res. Cell Motil. 1998;19:225–235. doi: 10.1023/a:1005372915268. [DOI] [PubMed] [Google Scholar]
- 39.Lukoyanova N, VanLoock MS, Orlova A, Galkin VE, Wang K, Egelman EH. Curr. Biol. 2002;12:383–388. doi: 10.1016/s0960-9822(02)00678-4. [DOI] [PubMed] [Google Scholar]
- 40.Clontech . Matchmakers™ Two-hybrid System 3 & Libraries User Manual. Clontech; Mountain View, CA: 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.