

Abstract
Recent reports have demonstrated beneficial effects of proinsulin C-peptide in the diabetic state, including improvements of kidney and nerve function. To examine the background to these effects, C-peptide binding to cell membranes has been studied by using fluorescence correlation spectroscopy. Measurements of ligand–membrane interactions at single-molecule detection sensitivity in 0.2-fl confocal volume elements show specific binding of fluorescently labeled C-peptide to several human cell types. Full saturation of the C-peptide binding to the cell surface is obtained at low nanomolar concentrations. Scatchard analysis of binding to renal tubular cells indicates the existence of a high-affinity binding process with Kass > 3.3 × 109 M−1. Addition of excess unlabeled C-peptide is accompanied by competitive displacement, yielding a dissociation rate constant of 4.5 × 10−4 s−1. The C-terminal pentapeptide also displaces C-peptide bound to cell membranes, indicating that the binding occurs at this segment of the ligand. Nonnative d-C-peptide and a randomly scrambled C-peptide do not compete for binding with the labeled C-peptide, nor were crossreactions observed with insulin, insulin-like growth factor (IGF)-I, IGF-II, or proinsulin. Pretreatment of cells with pertussis toxin, known to modify receptor-coupled G proteins, abolishes the binding. It is concluded that C-peptide binds to specific G protein-coupled receptors on human cell membranes, thus providing a molecular basis for its biological effects.
Since the discovery in 1967 of the mode of insulin biosynthesis (1, 2), it has generally been held that C-peptide, the connecting segment of proinsulin, does not possess biological activity of its own. However, recently, several studies have raised doubts concerning this view. Short-term C-peptide replacement in animals with experimental diabetes and in patients with type 1 diabetes is accompanied by improved renal function (3, 4), augmented glucose utilization (3, 5, 6), increased blood flow in muscle and skin (5, 7), and improved autonomic nerve function (8). Prolonged C-peptide administration (1–3 months) in type 1 diabetes patients results in improvements of renal function and amelioration of autonomic and sensory nerve dysfunction (9, 10). In vitro studies have confirmed that C-peptide stimulates glucose transport in skeletal muscle and that this effect is mediated via pathways other than the insulin receptor (11). C-peptide also improves RBC deformability in type 1 diabetes patients (12). The different effects correlate with stimulation of both Na+,K+-ATPase and endothelial NO synthase activities by C-peptide [refs. 13–15; and T. Kunt (Johannes Gutenberg University Hospital, Mainz, Germany), personal communication]. In renal tubular segments, this stimulation is compatible with activation of a G protein-coupled receptor with subsequent activation of Ca2+-dependent intracellular signaling pathways (13). However, a C-peptide receptor has not yet been demonstrated, and it has even been suggested that C-peptide effects may be mediated by nonchiral membrane interaction (14, 15). In the present study, we examine the occurrence and nature of C-peptide binding to cell membranes by using fluorescence correlation spectroscopy (FCS).
In FCS, the thermodynamic fluctuations of single dye-labeled molecules are observed after excitation by a sharply focused laser beam (Fig. 1). From the autocorrelation function of the fluctuations in fluorescence intensity, the average number of molecules in the volume of measurement can be calculated. This method was introduced in the early 1970s (16–18), but it is only recently that it has become applicable to bioscience because of a substantial increase in sensitivity (19), allowing single-molecule analysis (20, 21). The small volume elements (0.2 fl) in which the measurements are performed make it possible to evaluate molecular processes at the cell membrane. We have now applied FCS to examine the existence of membrane-linked binding sites specific for C-peptide and compared the results with those obtained for insulin. To provide a fluorescence signal, the peptides were labeled with the fluorophore tetramethylrhodamine (Rh). Evidence is presented that C-peptide is bound to specific cell membrane receptors of several different cell types.
Figure 1.

FCS experimental setup. Light from an argon ion laser is focused by means of a dichroic mirror and a lens to form a small volume element (0.2 fl). The laser beam is projected from below into a well containing a monolayer of cultured cells and tetramethylrhodamine (Rh)-labeled ligand (see magnified diagram at the top). After excitation of the labeled ligand, emitted light is transmitted via the dichroic mirror, a bandpass filter, and a pinhole to a photodetector. The volume element is positioned onto the cell surface with a microscope for detection of ligand binding. The dimensions of the laser beam focus and the pinhole together define the confocal volume element. The detector signal is fed into a digital signal correlator, which calculates the autocorrelation function of the detected intensity fluctuations.
Materials and Methods
Cell Culture.
Human renal tubular cells were cultured from the outer cortex of renal tissue obtained from nondiabetic patients undergoing elective nephrectomy for renal cell carcinomas (22). The cells were cultured in RPMI 1640 (Life Technologies, Grand Island, NY) supplemented with 10% FCS, 2 mM l-glutamine, 10 mM Hepes, benzylpenicillin (100 units/ml), and streptomycin (100 μg/ml) and passaged at near confluence by trypsinization. Growing cells exhibited epithelial morphology with a central nucleus, a granular cytoplasm, and cobblestone appearance on light microscopy. The culture could be maintained for 8–10 passages. Cells from the second and third passages were used for experiments.
Fibroblasts were obtained from explant cultures of the dermal part of biopsies from the upper arm of healthy subjects and insulin-dependent diabetes mellitus patients. The cells were cultured in DMEM containing 10% new-born calf serum and antibiotics (penicillin 50 units/ml and streptomycin 50 μg/ml) at 37°C in 5% CO2 humidified atmosphere. The cells were passaged once every week, and the medium was changed every second day. Cells of the third to fifth passages were used for experiments. The cells were plated on chamber slides 48 h before the studies, allowed to adhere for 36 h, and were then serum-starved until the start of the experiments.
Fresh sections of human great saphenous veins and umbilical cords were rinsed with PBS [50 mM phosphate buffer (pH 7.4) with 150 mM NaCl] and subsequently filled with collagenase (0.1% in PBS). After 20 min of incubation at 37°C, endothelial cells were harvested and centrifuged at 800 × g for 5 min and resuspended in DMEM supplemented with 40% human serum, antibiotics (penicillin 50 units/ml and streptomycin 50 μg/ml), isobutylmethylxanthine (33 μM), and cholera toxin (10−10 M) (saphenous vein cells) or medium 199 supplemented with 20% new-born calf serum and antibiotics (umbilical vein cells). Cells were cultured in gelatin-coated culture flasks at 37°C in 5% CO2 humidified atmosphere and subsequently passaged with trypsin/EDTA (0.05%/0.01% in PBS). For experiments, secondary or tertiary cell cultures were used.
All biopsies or tissue collections were undertaken with informed consent of the subjects and approval by the institutional ethics committee.
Labeling of Peptides.
Human C-peptide (Eli Lilly) and human insulin (Novo-Nordisk, Copenhagen) were labeled with Rh (absorption 555 nm, emission 580 nm), using a succinimidyl ester derivative (FluoReporter, F-6163; Molecular Probes) at 5- to 10-fold molar excess of the reagent. Labeled C-peptide and insulin were separated from unreacted dye by loading the reaction mixture on NAP-5 columns (Amersham Pharmacia) followed by elution with PBS. The fluorescently labeled C-peptide and insulin were separated from unlabeled material by reverse-phase chromatography (Hewlett–Packard 1090 HPLC) on a Kromasil C8 column 4.6 × 250 mm (particle size, 7 μm; pore size, 10 nm) (Eka Nobel, Surte, Sweden). Elution was performed at 1 ml/min with a gradient of 20–40% acetonitrile containing 0.1% trifluoroacetic acid. The absorbance was monitored simultaneously at 555 and 220 nm. Fractions containing fluorescently labeled C-peptide and insulin were pooled, adjusted to pH 8 by addition of NH3, and lyophilized. The identities of the purified labeled peptides were confirmed by matrix-assisted laser desorption ionization MS.
Procedure.
All binding studies with FCS were carried out on cells cultured in eight-well Nunc chambers (Nalge Nunc) at 20°C. Prior to the experiments, cells were washed five times with PBS and incubated with binding buffer [20 mM Hepes (pH 7.4)/115 mM NaCl/24 mM NaHCO3/4.7 mM KCl/1.26 mM CaCl2/1.2 mM KH2PO4/1.2 mM MgSO4/11.1 mM glucose/5 mg/ml BSA]. Binding of C-peptide was measured after 60 min of incubation of the cells in the presence of 5 nM Rh-labeled C-peptide (Rh-CP). Specificity of C-peptide binding was demonstrated by competitive displacement of bound Rh-CP from the cell surfaces after 3 h of incubation of 5 μM nonlabeled C-peptide added to the cell incubations (postincubation). Specific binding was also demonstrated by the inability of Rh-CP to bind to cell surfaces that had been pretreated with nonlabeled C-peptide for 1–3 h. In a similar fashion, competitive displacement was evaluated after pre- and postincubations with insulin, insulin-like growth factor (IGF)-I, IGF-II, proinsulin, d-C-peptide, scrambled C-peptide (a peptide with the same residues as human C-peptide but assembled in random order), or a segment of the C-terminal part of the C-peptide (EGSLQ). Binding curves for C-peptide and insulin were obtained by addition of various concentrations of Rh-CP or Rh-insulin to the incubations, allowing 60 min before determination of binding. Renal tubular cells were pretreated with pertussis toxin (1 μg/ml) at 37°C for 4 h under 95% O2/5% CO2 before binding studies were carried out. Human insulin was obtained from Novo-Nordisk, IGF-II from Eli Lilly, IGF-I from Pharmacia–Upjohn, and human proinsulin from Sigma. d-C-peptide, randomly scrambled C-peptide, and the C-terminal pentapeptide of C-peptide were synthesized by Genosys (Cambridge, U.K.). Pertussis toxin was from Sigma.
FCS.
FCS was performed with confocal illumination of a volume element of 0.2 fl in a ConfoCor instrument from Zeiss Evotec (Fig. 1) (19). As focusing optics, a Zeiss Neofluar 40× numerical aperture 1.2 objective for water immersion was used in an epiillumination setup. Separation of exciting from emitted radiation was achieved by dichroic (Omega 540 DRL PO2; Omega Optical, Brattleboro, VT) and bandpass (Omega 565 DR 50) filters. The Rh-CP was excited with the 514.5-nm line of an argon laser. The intensity fluctuations were detected by an avalanche photodiode (SPCM 200, EG & G, Quebec, Canada) and processed with a digital correlator (ALV 5000; ALV, Langen, Germany). The measurements were performed in solution and on cells cultured in Nunc chambers. With the objective used, a 0.2-fl volume element was illuminated with dimensions of w = 0.25 μm and z = 1.25 μm. To avoid photobleaching with the diffusion times observed (τmax = 100 ms), the exciting intensity was adjusted such that the detected photon count rate did not exceed 3,000 per molecule and s (23).
Evaluation of FCS Data.
Fluorescence intensity fluctuations 〈δI〉 around the mean fluorescence intensity 〈I〉 occurring in a volume element with half-axes w = 0.25 μm and z = 1.25 μm are correlated (16–19), and for calculation of parameters of the autocorrelation function G(t), nonlinear least-square minimization was used (24). The autocorrelation function for three-dimensional diffusion of the unbound Rh-CP in solution and two-dimensional diffusion of bound Rh-CP to membranes on the cell surface is given by:
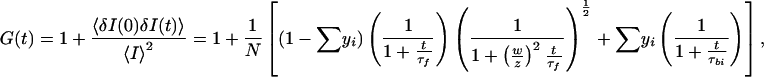 |
1 |
where diffusion time τ and diffusion coefficient D are related as τ = w2/4D; yi is the fraction of membrane-bound C-peptide diffusing with diffusion time τbi, and (1 − Σyi) is the fraction of unbound C-peptide with diffusion time τf. The relative amount of cell-surface bound C-peptide ligand (Σyi) is obtained at increasing concentration of Rh-CP. The Scatchard representation of the mass action law is obtained from:
 |
2 |
where Ki and ni are the association constant and the number of ligand binding sites per receptor molecules, respectively. Lo is the total ligand concentration. Ro, the total receptor concentration, cannot be determined independently but was calculated from the maximum number of bound ligand molecules at saturation, assuming one binding site for one receptor molecule (n = 1).
Instead of evaluating for distinct species (yi) and their characteristic diffusion times (τi), one can determine from the correlation function G(t) a distribution P(τi) describing the fraction of species yi:
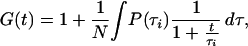 |
3 |
with P(τi) = Σyi dy. This represents a model-independent analysis of the diffusion processes observed by the correlation function. For the evaluations of the distribution functions, the contin program using restrained regularization (25) was applied.
Results
Binding of Rh-CP to Cell Membranes.
Fluorescence intensity fluctuations and autocorrelation functions of Rh-CP free in solution and bound to human renal tubular cell membranes are presented in Fig. 2. Examination of the Brownian motion of unbound Rh-CP in the buffer medium above the cell surface exhibits typical fluctuations (Fig. 2A) and a diffusion time (τ) of 0.15 ms (Fig. 2B). With the volume element positioned at the level of the cell membrane, an increase and a broadening of the fluctuation peaks are observed (Fig. 2D). Correlation analysis of the intensity fluctuations then shows a diffusion process of the cell-bound C-peptide with at least two components characterized by diffusion times of τ1 = 80 ms and τ2 = 1 ms, respectively, and corresponding weight factors (fractions) of y1 = 0.75 and y2 = 0.15 (Fig. 2E). Because the volume element extends into the space above the cell membrane, a small fraction of unbound Rh-CP, τ3 = 0.15 ms, y3 = 0.1, is also observed. A model-independent analysis of the diffusion processes (Eq. 3) is presented in Fig. 3.
Figure 2.

C-peptide binding and displacement to the membranes of cultured renal tubular cells. Fluorescence intensity fluctuations (A) and autocorrelation function (B) for Rh-CP (5 nM) free in solution, τ = 0.15 ms. Fluorescence intensity fluctuations (D) and autocorrelation function (E) for Rh-CP bound to membranes on the cell surface. Diffusion times (τ) and corresponding fractions (y): τ1 = 80 ms, y1 = 0.75; τ2 = 1 ms, y2 = 0.15; τ3 = 0.15 ms, y3 = 0.1. Autocorrelation functions of displacement of membrane bound Rh-CP by postincubation of a thousandfold molar excess of nonlabeled C-peptide (F) and nonlabeled C-terminal pentapeptide (C). The observed and calculated data points are completely overlapping (B, C, E, and F).
Figure 3.

contin distributions of diffusion times P(τi) of C-peptide binding and displacement to the membranes of cultured renal tubular cells. Rh-CP free in the incubation medium (A), binding of Rh-CP to the cell membranes (B), displacement of membrane-bound Rh-CP by incubation with a thousandfold molar excess of nonlabeled C-peptide (C), and inhibition of membrane binding of Rh-CP after pretreatment of the cells with pertussis toxin (D).
Increasing concentrations of Rh-CP in the buffer medium leads to an increased proportion of membrane-bound labeled C-peptide. Saturation of the binding process occurs at about 0.9 nM Rh-CP, and 50% binding is found at 0.3 nM Rh-CP for renal tubular cells (Fig. 4). Scatchard analysis reveals a binding process with an equilibrium association constant, Kass of 3.3 × 109 M−1, assuming one receptor binding site per C-peptide (n = 1). Evidence for the possible existence of a second binding process with a higher affinity and a ligand/receptor ratio of n < 1 is also observed (Fig. 4 Inset). Similar or slightly lower association constants were found for Rh-CP binding to human fibroblasts and endothelial cells (Table 1).
Figure 4.

C-peptide binding curve. Binding of Rh-labeled C-peptide to cell membranes of renal tubular cells. Fractional saturation of the membrane-bound Rh-CP (y) as a function of the ligand concentration (L) in the binding medium. Each data point represents the mean of at least six measurements. The binding curve was simulated with Kass = 3.3 × 109 M−1 and n = 1. Scatchard plot is shown as Inset.
Table 1.
Binding of C-peptide and insulin to different cell types
| Ligand | Cells | Receptors/μm2* | Kass, 109 M−1 |
|---|---|---|---|
| C-peptide | Renal tubular | 75 ± 12 | 3.3 |
| Fibroblasts | 55 ± 10 | 2.5 | |
| Endothelial | 43 ± 4 | 2.0 | |
| Insulin | Renal tubular | 200 ± 10 | 1.2 |
Calculated from the number of Rh-C-peptide or Rh-insulin binding sites in the volume element with an area of 0.196 μm2.
Specificity and Kinetics of Binding.
To determine the specificity of Rh-CP binding, we examined competitive displacement with nonlabeled C-peptide. Cells were incubated with Rh-CP, and, after 60 min, a thousandfold molar excess of nonlabeled C-peptide was added. This results in a reduction of Rh-CP binding, and, after 3 h, about 85% of the total binding is displaced (Fig. 5), as indicated by the autocorrelation function in Fig. 2F and in the model-independent distribution analysis in Fig. 3C. Moreover, Rh-CP binding is prevented when cells are preincubated with 5 μM nonlabeled C-peptide, demonstrating that all binding sites are already occupied by nonlabeled C-peptide. The dissociation curve plotted in log scale (Fig. 5 Inset) indicates that displacement of Rh-CP binding occurred in a monoexponential mode. Analysis of the dissociation curve yields a dissociation time (τdiss) of 2,217 s, and a dissociation rate constant (kdiss) calculated as kdiss = 1/τdiss = 4.5 × 10−4 s−1. From knowledge of the dissociation time and the Kass (3.3 × 109 M−1, see above) the recombination rate constant (krec) can be calculated: krec = Kass × kdiss = 1.5 × 106 M−1⋅s−1. The C-terminal pentapeptide segment of the human C-peptide molecule (EGSLQ), previously shown to stimulate Na+,K+-ATPase activity (14), was tested with regard to its ability to displace bound Rh-CP. A thousandfold molar excess of the pentapeptide was found to be as effective as the intact molecule in displacing C-peptide bound to renal tubular cells (Fig. 2C) and showing the same distribution pattern as for C-peptide.
Figure 5.

Time course of displacement of Rh-CP by nonlabeled C-peptide. After incubation of cells with 5 nM Rh-CP for 60 min, 5 μM nonlabeled C-peptide was added, and FCS measurements were carried out at given time intervals. Each data point represents the mean of at least six measurements. Log scale for the binding displacement process is shown as Inset.
To determine whether the binding of Rh-CP to cell membranes is stereospecific, we incubated cells preexposed to Rh-CP with 5 μM of nonlabeled all-d-amino acid (enantio) C-peptide for 3 h. The Rh-CP binding was not displaced by the d-enantio C-peptide. Cells preincubated with Rh-CP were also exposed to 5 μM scrambled C-peptide. Exposure to scrambled C-peptide did not result in competitive displacement of the Rh-CP binding. In addition, Rh-CP binding was not displaced by a thousandfold molar excess of insulin, proinsulin, IGF-I, or IGF-II. We also carried out binding studies with Rh-labeled insulin and renal tubular cells (data not shown). Rh-insulin binding was observed in the nanomolar range and was displaceable after addition of a thousandfold molar excess of nonlabeled insulin. The insulin–receptor interaction, when characterized by a Scatchard plot, is compatible either with two independent binding processes with different affinities or with negative cooperativity.
Effect of Pertussis Toxin on the C-Peptide Binding.
To test whether C-peptide binding is accompanied by G protein involvement, we pretreated cells with pertussis toxin. This resulted in complete loss of the slowly diffusing C-peptide receptor component (Fig. 3D). A small component of rapidly diffusing (1 ms) complexes (10–15%) remained after pertussis toxin pretreatment. This component was found to represent nonspecific binding, because it was not displaced by addition of excess of unlabeled C-peptide. However, when pertussis toxin-pretreated cells were exposed to Rh-CP concentrations of 50–100 nM, the C-peptide 1-ms binding complex increased to 50%, and this component could be displaced very rapidly (within 10 min) by addition of unlabeled C-peptide.
Discussion
The current results demonstrate specific binding of human C-peptide to membrane-bound receptors in several human cell types. Binding of C-peptide to cultured renal tubular cells, skin fibroblasts, and saphenous vein endothelial cells could be shown, and the number of binding sites per unit cell surface area was highest for renal tubular cells (Table 1). In contrast, endothelial cells from umbilical cord veins failed to show binding of C-peptide, in agreement with the observation that C-peptide stimulates NO synthase activity in aortic endothelial cells but not in umbilical vein cells (T. Kunt, personal communication).
The present binding data are compatible with a specific ligand–receptor interaction, as suggested for C-peptide and transplantable rat islet tumor cells (26) and for C-peptide and rat renal tubular cells (13, 14). The specificity of the binding is attested to by the consistent displacement of bound Rh-CP after addition of a 1,000-fold molar excess of unlabeled C-peptide. Likewise, preincubation of cells with excess unlabeled C-peptide results in failure of Rh-CP to bind. Further evidence for the binding specificity is obtained from the fact that a 31-residue peptide with the same amino acid composition as human C-peptide but in random sequence (scrambled C-peptide) failed to displace bound Rh-CP. Addition of d-enantio C-peptide did not result in displacement of bound Rh-CP, demonstrating the stereospecific nature of the binding. The C-terminal pentapeptide segment of C-peptide has previously been shown to possess 100% of the intact molecule’s ability to stimulate Na+,K+-ATPase (14). We now show that addition of excess unlabeled C-terminal pentapeptide was accompanied by displacement of bound Rh-CP in the same manner as after addition of intact C-peptide, indicating that the C-terminal segment is involved in the binding process. The free C terminus end of the segment is required because proinsulin, which includes the pentapeptide segment, failed to displace bound Rh-CP.
No binding of C-peptide to receptors of other peptide hormones was observed. Thus, insulin, IGF-I, IGF-II, and proinsulin, when added in excess, all failed to elicit displacement of bound Rh-CP. This is in contrast to the recent report that C-peptide binds with low affinity to a proinsulin receptor (27). When Rh-labeled insulin was bound to cell membranes, addition of unlabeled C-peptide did not result in displacement of the labeled insulin. Crossreactions with insulin or the other tested peptide hormones thus appear unlikely.
The C-peptide binding isotherm indicates that half saturation occurs at a C-peptide concentration of 0.3 nM, and that full saturation is reached at approximately 0.9 nM (Fig. 4). This finding has clinical relevance, because it has been a consistent finding in previous studies that no effects of C-peptide could be demonstrated in healthy subjects or animals. It is only in C-peptide-negative type 1 diabetes patients or animals with experimentally induced diabetes that several physiological effects can be elicited by C-peptide (3–10). This seeming discrepancy can now be explained by the present demonstration that saturation of C-peptide receptors occurs at very low C-peptide levels. Consequently, in healthy subjects or animals, receptor saturation is obtained at the ambient C-peptide level, and no additional biological activity can be expected when the concentration is increased further. The fact that physiological effects of C-peptide can be demonstrated only in C-peptide-deficient patients or animals has most likely contributed to the delay in the recognition of C-peptide’s biological activity.
In the Scatchard analysis of the binding process (Fig. 4), we have assumed the existence of one C-peptide binding site per receptor molecule. The receptor concentration cannot be determined independently, but only by the interaction with the labeled C-peptide. The Scatchard plot shows a binding process with a Kass of 3 × 109 M−1, for n = 1. There is also evidence for an additional, higher binding affinity, extrapolating to n < 1. A possible interpretation of the latter finding is that one C-peptide molecule may interact with two or more receptor molecules. This is in agreement with the slowly diffusing C-peptide/receptor complex in Fig. 2B, giving rise to large intensity fluctuations (Fig. 2D). In the Scatchard analysis, the existence of independent binding sites is assumed. However, binding sites interfering with each other directly or indirectly (negative cooperativity) can result in a nonlinear Scatchard plot, as is well established in the case of the insulin–receptor interaction (28).
The possible existence of different forms of the C-peptide/receptor complexes stems from the observation that rapidly (τ = 1 ms) and slowly (τ = 80 ms) diffusing complexes are both found on the cell surface, and that both can be competitively displaced by unlabeled C-peptide. Exposure of cells to physiological concentrations of C-peptide leads to the appearance of slowly diffusing complexes after 60 min (Figs. 2E and 3B). Addition of excess unlabeled C-peptide abolishes all of these complexes but only part of the rapidly diffusing complexes. This indicates that, in addition to the specific binding, there is also a component of nonspecific interaction with the cell membranes. A consistent observation is that the relative amount of the slowly diffusing ligand/receptor complex increases with time. This may reflect a ligand-induced receptor aggregation and/or immobilization of the ligand/receptor complexes as a result of intracellular interactions. A similar behavior has been observed for the interaction of Rh-labeled galanin and its receptor (29).
The binding of C-peptide to its specific binding site was tested by the time-dependent displacement of Rh-CP after addition of excess unlabeled C-peptide. In the time interval tested (20–120 min) the dissociation kinetics follow a single-exponential function characteristic of a relatively slowly dissociating process, with a kdiss = 4 × 10−4 s−1. This value is in the same range as those observed for insulin (28) and galanin (30) dissociations from their receptors or for α-bungarotoxin from its acetylcholine receptor (31).
The recorded variations in fluorescence intensity in a FCS experiment are because of the fluctuating number of emitting molecules in the volume element and can be used to determine the average number of molecules present in the volume element. Because bound and unbound ligands can be distinguished by their different diffusion times (Eq. 1), the number of membrane-bound Rh-CP molecules can be estimated. From the distribution analysis of membrane-bound Rh-CP before and after displacement with unlabeled C-peptide (Fig. 3 B and C), we estimate the background of nonspecifically bound Rh-CP (15%) to be significantly below the specific signal. Because the calculations are based on the number of diffusing complexes, which may be in different states of aggregation, it is possible that the values given in Table 1 may represent underestimates.
The highest receptor concentration among the cells investigated was found for renal tubular cells (75 per μm2, Table 1), while the corresponding number for insulin receptors was 3 times higher. Assuming a cell surface of ca. 20 μm2, the total number of C-peptide-specific receptors calculates to about 1,000–1,500 receptors per cell. These values are in line with or slightly lower than those obtained for the interaction of C-peptide with β-cells obtained from transplantable rat islet cell tumors (26).
The observation that exposure of the cells to pertussis toxin interferes with the C-peptide-induced stimulation of Na+,K+-ATPase provides evidence for the involvement of G proteins in the signal transduction pathway. With detailed structural (32) and functional (33) data on G proteins accumulating, the hypothesis may be considered that a G protein interacting with a ligand-activated receptor can constitute an allosteric system, involving alternative conformational states of both the receptor and the G protein. Pertussis toxin is known to affect a cysteine residue in the C-terminal chain of the α-subunit of the Gi protein, thereby interfering the interaction between the G protein and the loop regions of the membrane-spanning receptor (32). Preincubation of cells with pertussis toxin in the present study was found to abolish the binding of C-peptide to the membrane at physiological conditions. This finding may be explained as the result of allosteric actions according to the principle of detailed balance (34). Addition of Rh-CP at 50 nM and higher concentrations to cells pretreated with pertussis toxin reveals a rapidly diffusing C-peptide/receptor complex (τ = 1 ms), which, in part, can be displaced within a few minutes with an excess (5 μM) of unlabeled C-peptide (data not shown). These observations are compatible with the existence of at least two different C-peptide/receptor complexes, one with low affinity and high mobility and another with high affinity and low mobility.
Interaction of Rh-labeled insulin with renal tubular cells was investigated, and results were similar to those obtained for C-peptide. Evidence was found for a high-affinity binding to the cell membranes, which can be displaced with insulin but not with C-peptide. The insulin binding yields a curvilinear plot of the Scatchard analysis, indicating several binding processes. A binding constant of about 1.2 × 109 M−1 was obtained on extrapolation to one binding site per receptor (n = 1). Additional binding processes with higher affinities extrapolating to smaller n values were observed. These results are in agreement with data published for insulin (28) in which binding of 125I-labeled insulin to baby hamster kidney cells with overexpressed insulin receptors was studied. Those data have been explained by the interaction of insulin molecules with both a monomeric and a dimeric receptor. Our data are fully compatible with this notion. As in the case of C-peptide, slowly diffusing insulin/receptor complexes are observed after addition of Rh-insulin.
The conventional radioligand methods are usually employed for analysis of ligand–receptor interactions. For this reason, it was of interest to test the binding to renal tubular cells of C-peptide with an additional 125I-iodinated tyrosine at the N terminus end. Under conditions where significant interactions of Rh-CP were observed with FCS, only marginal effects were found in the radioligand assay. This result is in contrast to a similar study with galanin in which the interaction of the Rh-labeled ligand with its membrane-bound receptor was studied (29) and could be compared with radioligand binding in cells overexpressing the galanin receptor (35). It is concluded that the receptor density present in the cells used in our study is sufficient for successful FCS analysis but not for radioligand studies.
It has recently been suggested that effects of human C-peptide on vascular and neural dysfunction in diabetic rats might be explained by nonchiral interactions with membrane lipids (15). This effect resembles that of antimicrobial and surfactant peptides, which result in the formation of ion channels and inhibition of phospholipase A2 activity (36, 37). The present results, however, demonstrate that the C-peptide signal transduction follows the normal rules of ligand and receptor biochemistry; the binding is stereospecific and occurs within the physiological concentration range (0.3–2 nM). Moreover, the fact that bound Rh-CP is readily displaced by excess unlabeled C-peptide argues against a nonspecific membrane interaction. In addition, displacement of bound Rh-CP by the C-terminal pentapeptide fragment indicates that the binding involves this part of the C-peptide, which is consistent with the previous observation that the pentapeptide segment is as active as the intact C-peptide with regard to its ability to stimulate Na+,K+-ATPase (14). Nevertheless, the current findings do not preclude the possibility that C-peptide can possess channel-forming properties at a higher than physiological C-peptide concentration (15).
The progressive destruction of the β-cells of the pancreas that is characteristic of diabetes mellitus type 1 results in deficiency and eventually total lack of both circulating insulin and C-peptide. Insulin replacement therapy, even when carefully adjusted with multiple daily injections, cannot prevent the development of long-term complications, involving kidneys, nerves, and eyes (38). The present study shows that C-peptide binds specifically to cell membranes. This observation, coupled with previous findings of physiological effects (3–11) relating to Na+,K+-ATPase (13–15) and endothelial NO synthase activities (T. Kunt, personal communication), provides a basis for the hypothesis that C-peptide is a biologically active peptide hormone whose effects are targeted to the microcirculation of renal, nerve, and retinal tissues. Whether C-peptide, when administered together with insulin to type 1 diabetes patients on a long-term basis can retard or prevent the development of microvascular complications will henceforth be an important question to address.
Acknowledgments
The study was supported by grants from the Marianne and Marcus Wallenberg Foundation, the Swedish Medical Research Council, the Swedish Natural Science Research Council, the Swedish Technical Science Research Council, Schwarz Pharma AG (Monheim, Germany), and Karolinska Institutet.
Abbreviations
- FCS
fluorescence correlation spectroscopy
- Rh
tetramethylrhodamine
- Rh-CP
Rh-labeled C-peptide
- IGF
insulin-like growth factor
References
- 1.Steiner D F, Oyer P E. Proc Natl Acad Sci USA. 1967;57:473–480. doi: 10.1073/pnas.57.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steiner D F, Cunningham D, Spigelman L, Aten B. Science. 1967;157:697–700. doi: 10.1126/science.157.3789.697. [DOI] [PubMed] [Google Scholar]
- 3.Johansson B-L, Sjöberg S, Wahren J. Diabetologia. 1992;35:121–128. doi: 10.1007/BF00402543. [DOI] [PubMed] [Google Scholar]
- 4.Sjöquist M, Huang W, Johansson B-L. Kidney Int. 1998;54:758–764. doi: 10.1046/j.1523-1755.1998.00074.x. [DOI] [PubMed] [Google Scholar]
- 5.Johansson B-L, Linde B, Wahren J. Diabetologia. 1992;35:1151–1158. doi: 10.1007/BF00401369. [DOI] [PubMed] [Google Scholar]
- 6.Wu W, Oshida Y, Yang W-P, Ohsawa I, Sato J, Iwao S, Johansson B-L, Wahren J, Sato Y. Acta Physiol Scand. 1996;157:253–258. doi: 10.1046/j.1365-201X.1996.489236000.x. [DOI] [PubMed] [Google Scholar]
- 7.Forst T, Kunt T, Pohlmann T, Goitom K, Engelbach M, Beyer J, Pfützner A. J Clin Invest. 1998;101:2036–2041. doi: 10.1172/JCI2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johansson B-L, Borg K, Fernqvist-Forbes E, Odergren T, Remahl S, Wahren J. Diabetologia. 1996;39:687–695. doi: 10.1007/BF00418540. [DOI] [PubMed] [Google Scholar]
- 9.Johansson B-L, Kernell A, Sjöberg S, Wahren J. J Clin Endocrinol Metab. 1993;77:976–981. doi: 10.1210/jcem.77.4.8408474. [DOI] [PubMed] [Google Scholar]
- 10.Johansson, B.-L., Borg, K., Fernqvist-Forbes, E., Kernell. A., Odergren, T. & Wahren, J. (1999) Diabet. Med., in press. [DOI] [PubMed]
- 11.Zierath J R, Handberg A, Tally M, Wallberg-Henriksson H. Diabetologia. 1996;39:306–313. doi: 10.1007/BF00418346. [DOI] [PubMed] [Google Scholar]
- 12.Kunt T, Schneider S, Pfützer A, Goitom K, Engelbach M, Schauf B, Beyer J, Forst T. Diabetologia. 1999;42:465–471. doi: 10.1007/s001250051180. [DOI] [PubMed] [Google Scholar]
- 13.Ohtomo Y, Aperia A, Sahlgren B, Johansson B-L, Wahren J. Diabetologia. 1996;39:199–205. doi: 10.1007/BF00403963. [DOI] [PubMed] [Google Scholar]
- 14.Ohtomo Y, Bergman T, Johansson B-L, Jörnvall H, Wahren J. Diabetologia. 1998;41:287–291. doi: 10.1007/s001250050905. [DOI] [PubMed] [Google Scholar]
- 15.Ido Y, Vindigni A, Chang K, Stramm L, Chance R, Heath W, DiMarchi R, Cera E D, Williamsson J. Science. 1997;277:563–566. doi: 10.1126/science.277.5325.563. [DOI] [PubMed] [Google Scholar]
- 16.Magde D, Elson E L, Webb W W. Phys Rev Lett. 1972;29:705–711. [Google Scholar]
- 17.Elson E L, Magde D. Biopolymers. 1974;13:1–27. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 18.Ehrenberg M, Rigler R. Chem Phys. 1974;4:390–401. [Google Scholar]
- 19.Rigler R, Mets Ü, Widengren J, Kask P. Eur Biophys J. 1993;22:169–175. [Google Scholar]
- 20.Rigler R, Mets Ü. Soc Photo-Opt Instrum Eng. 1992;1921:239–248. [Google Scholar]
- 21.Eigen M, Rigler R. Proc Natl Acad Sci USA. 1994;91:5740–5747. doi: 10.1073/pnas.91.13.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Söderhäll M, Bergerheim U S, Jacobson S H, Lundahl J, Mollby R, Normark S, Winberg J. J Urol. 1997;157:346–350. [PubMed] [Google Scholar]
- 23.Wennmalm S, Rigler R. J Phys Chem. 1999;103:2516–2519. [Google Scholar]
- 24.Marquardt D W. J Soc Ind Appl Math. 1963;11:431–441. [Google Scholar]
- 25.Provencher S W. Comput Phys Commun. 1982;27:213–227. [Google Scholar]
- 26.Flatt P R, Swanston-Flatt S K, Hampton S M, Bailey C J, Marks V. Biosci Rep. 1986;6:193–199. doi: 10.1007/BF01115006. [DOI] [PubMed] [Google Scholar]
- 27.Jehle P M, Fussgänger R D, Angelus N K O, Jungwirth R J, Saile B, Lutz M P. Am J Physiol. 1999;276:E262–E268. doi: 10.1152/ajpendo.1999.276.2.E262. [DOI] [PubMed] [Google Scholar]
- 28.DeMeyts P. Diabetologia. 1994;37,Suppl. 2:125–148. [Google Scholar]
- 29.Pramanik A, Juréus A, Langel U, Bartfai T, Rigler R. Biomed Chromatogr. 1999;13:119–120. [Google Scholar]
- 30.Land T, Langel Ü, Fisone G, Bedecs K, Bartfai T. Methods Neurosci. 1991;5:225–234. [Google Scholar]
- 31.Rauer B, Neumann E, Widengren J, Rigler R. Biophys Chem. 1996;58:3–12. doi: 10.1016/0301-4622(95)00080-1. [DOI] [PubMed] [Google Scholar]
- 32.Hamm H E. J Biol Chem. 1998;273:669–672. doi: 10.1074/jbc.273.2.669. [DOI] [PubMed] [Google Scholar]
- 33.Farfel Z, Bourne H R, Iiri T. N Engl J Med. 1999;340:1012–1020. doi: 10.1056/NEJM199904013401306. [DOI] [PubMed] [Google Scholar]
- 34.Monod J, Wyman J, Changeux J P. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 35.Kask K, Berthold M, Kahl U, Nordvall G, Bartfai T. EMBO J. 1996;15:236–244. [PMC free article] [PubMed] [Google Scholar]
- 36.Hugosson M, Andreu D, Boman H G, Glaser E. Eur J Biochem. 1994;223:1027–1033. doi: 10.1111/j.1432-1033.1994.tb19081.x. [DOI] [PubMed] [Google Scholar]
- 37.Johansson J, Curstedt T. Eur J Biochem. 1997;244:675–693. doi: 10.1111/j.1432-1033.1997.00675.x. [DOI] [PubMed] [Google Scholar]
- 38.The Diabetes Control and Complications Trial Research Group. N Eng J Med. 1993;329:977–983. [Google Scholar]